
Transcriptomic and Metabolomics Joint Analyses Reveal the Influence of Gene and Metabolite Expression in Blood on the Lactation Performance of Dual-Purpose Cattle (Bos taurus)

Abstract
1. Introduction
2. Results
2.1. PCA and WGCNA of XJBC and CSC Blood Transcriptome Data
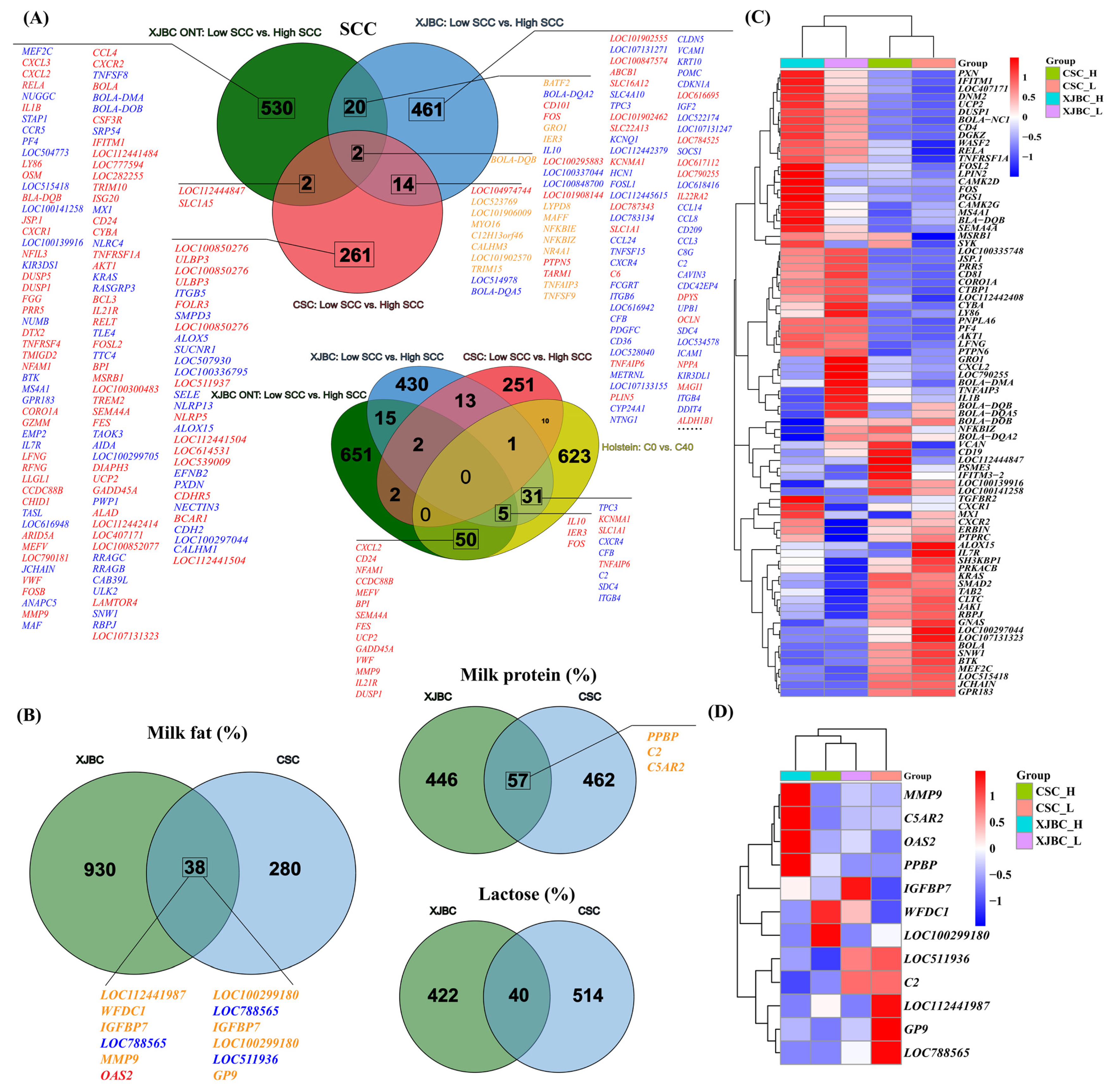
2.2. DEG Analysis
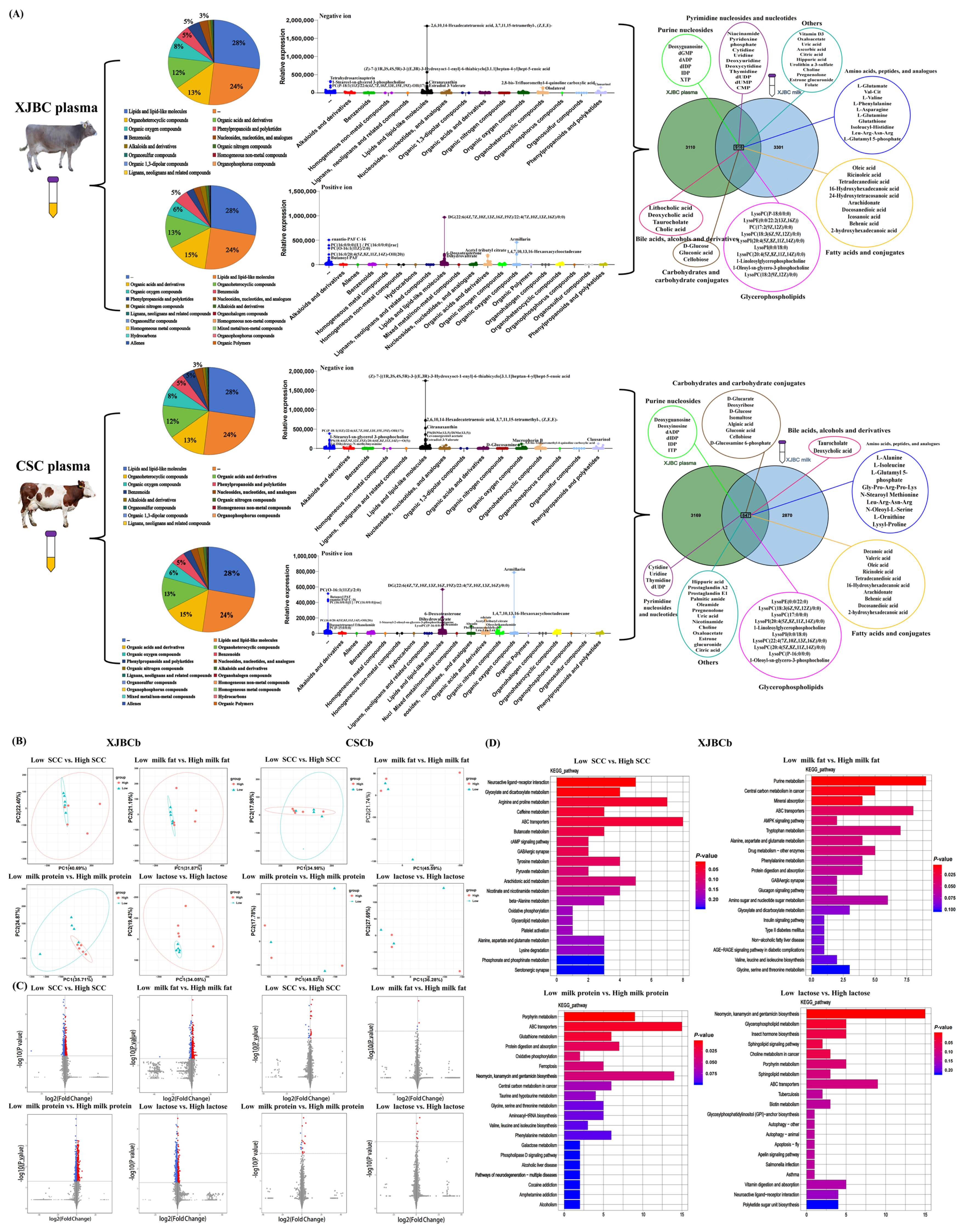
2.3. Review of Plasma Metabolites of XJBC and CSC
2.4. DEM Analysis
2.5. Joint Analysis of DEGs and DEMs
3. Discussion
3.1. The Expression of Genes and Metabolites in Blood Affects the SCC
3.2. Blood Metabolism Affects Milk Fat Percentage, Milk Protein Percentage, and Lactose Percentage
4. Conclusions
5. Materials and Methods
5.1. Sample Collection
5.2. Metabolites and Total RNA Extraction
5.3. Sequencing Library Construction and RNA-Seq
5.4. LC-MS/MS Analysis
5.5. Transcriptome Data Preprocessing
5.6. Metabolome Data Preprocessing
5.7. Bioinformatic Analysis
5.7.1. Bioinformatics Analysis of Transcriptome Data
5.7.2. Bioinformatics Analysis of Metabolome Data
5.7.3. Joint Analysis of Metabolome and Transcriptome Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Reece, W.O.; Swenson, M.J. The composition and functions of blood. In Dukes’ Physiology of Domestic Animals; Comstock Publishing Associates: Ithaca, NY, USA, 2004; ISBN 0-8014-4238-9. [Google Scholar]
- Duarte, R.T.; Carvalho Simões, M.C.; Sgarbieri, V.C. Bovine blood components: Fractionation, composition, and nutritive value. J. Agric. Food Chem. 1999, 47, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.L.; Bauman, D.E. Bioynthesis of Milk. Biosynthesis and Secretion of Milk/Diseases; Academic Press: New York, NY, USA, 2013; Volume 2, Part 1; ISBN 978-1483242316. [Google Scholar]
- Grădinaru, A.C.; Creangă, Ş.; Solcan, G. Milk–a review on its synthesis, composition, and quality assurance in dairy industry. Hum. Vet. Med. 2015, 7, 173–177. [Google Scholar] [CrossRef]
- Davis, S.R.; Collier, R.J. Mammary blood flow and regulation of substrate supply for milk synthesis. J. Dairy Sci. 1985, 68, 1041–1058. [Google Scholar] [CrossRef]
- Cox, D.B.; Owens, R.A.; Hartmann, P.E. Blood and milk prolactin and the rate of milk synthesis in women. Exp. Physiol. Transl. Integr. 1996, 81, 1007–1020. [Google Scholar] [CrossRef]
- Holtenius, K.; Waller, K.P.; Essén-Gustavsson, B.; Holtenius, P.; Sandgren, C.H. Metabolic parameters and blood leukocyte profiles in cows from herds with high or low mastitis incidence. Vet. J. 2004, 168, 65–73. [Google Scholar] [CrossRef]
- Yang, J.; Tang, Y.; Liu, X.; Zhang, J.; Zahoor Khan, M.; Mi, S.; Wang, C.; Yu, Y. Characterization of peripheral white blood cells transcriptome to unravel the regulatory signatures of bovine subclinical mastitis resistance. Front. Genet. 2022, 13, 949850. [Google Scholar] [CrossRef]
- Qayyum, A.; Khan, J.A.; Hussain, R.; Avais, M.; Ahmad, N.; Khan, M.S. Investigation of milk and blood serum biochemical profile as an indicator of sub-clinical mastitis in Cholistani cattle. Pak. Vet. J. 2016, 36, 275–279. Available online: http://www.pvj.com.pk/pdf-files/36_3/275-279.pdf (accessed on 10 September 2024).
- Sadek, K.; Saleh, E.; Ayoub, M. Selective, reliable blood and milk bio-markers for diagnosing clinical and subclinical bovine mastitis. Trop. Anim. Health Prod. 2017, 49, 431–437. [Google Scholar] [CrossRef]
- Wang, D.; Yang, H.; Ma, S.; Liu, T.; Yan, M.; Dong, M.; Zhang, M.; Zhang, T.; Zhang, X.; Chen, H.; et al. Transcriptomic Changes and Regulatory Networks Associated with Resistance to Mastitis in Xinjiang Brown Cattle. Genes 2024, 15, 465. [Google Scholar] [CrossRef]
- Zhong, L.; Ma, S.; Wang, D.; Zhang, M.; Tian, Y.; He, J.; Zhang, X.; Xu, L.; Wu, C.; Tian, K.; et al. Methylation levels in the promoter region of FHIT and PIAS1 genes associated with mastitis resistance in Xinjiang brown cattle. Genes 2023, 14, 1189. [Google Scholar] [CrossRef]
- Luoreng, Z.M.; Yang, J.; Wang, X.P.; Wei, D.W.; Zan, L.S. Expression profiling of microRNA from peripheral blood of dairy cows in response to Staphylococcus aureus-infected mastitis. Front. Vet. Sci. 2021, 8, 691196. [Google Scholar] [CrossRef] [PubMed]
- Dervishi, E.; Zhang, G.; Hailemariam, D.; Dunn, S.M.; Ametaj, B.N. Innate immunity and carbohydrate metabolism alterations precede occurrence of subclinical mastitis in transition dairy cows. J. Anim. Sci. Technol. 2015, 57, 46. [Google Scholar] [CrossRef]
- Dervishi, E.; Zhang, G.; Dunn, S.M.; Mandal, R.; Wishart, D.S.; Ametaj, B.N. GC–MS metabolomics identifies metabolite alterations that precede subclinical mastitis in the blood of transition dairy cows. J. Proteome Res. 2017, 16, 433–446. [Google Scholar] [CrossRef]
- Zandkarimi, F.; Vanegas, J.; Fern, X.; Maier, C.S.; Bobe, G. Metabotypes with elevated protein and lipid catabolism and inflammation precede clinical mastitis in prepartal transition dairy cows. J. Dairy Sci. 2018, 101, 5531–5548. [Google Scholar] [CrossRef]
- Haxhiaj, K.; Li, Z.; Johnson, M.; Dunn, S.M.; Wishart, D.S.; Ametaj, B.N. Blood metabolomic phenotyping of dry cows could predict the high milk somatic cells in early lactation—Preliminary results. Dairy 2022, 3, 59–77. [Google Scholar] [CrossRef]
- Wang, C.; Han, B. Twenty years of rice genomics research: From sequencing and functional genomics to quantitative genomics. Mol. Plant 2022, 15, 593–619. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Liu, X.; Locasale, J.W. Metabolomics: A primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef]
- Mohammadi-Shemirani, P.; Sood, T.; Paré, G. From ‘omics to multi-omics technologies: The discovery of novel causal mediators. Curr. Atheroscler. Rep. 2023, 25, 55–65. [Google Scholar] [CrossRef]
- Zhang, H.W.; Lv, C.; Zhang, L.J.; Guo, X.; Shen, Y.W.; Nagle, D.G.; Zhou, Y.D.; Liu, S.H.; Zhang, W.D.; Luan, X.; et al. Application of omics-and multi-omics-based techniques for natural product target discovery. Biomed. Pharmacother. 2021, 141, 111833. [Google Scholar] [CrossRef]
- Cebron, N.; Maman, S.; Walachowski, S.; Gausserès, B.; Cunha, P.; Rainard, P.; Foucras, G. Th17-related mammary immunity, but not a high systemic Th1 immune response is associated with protection against E. coli mastitis. npj Vaccines 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Yan, M. Analysis of Differential Genes and Alternative Splicing Events in Peripheral Blood of Xinjiang Brown Cattle with High and Low Somatic Cell Count. Master’s Thesis, Xinjiang Agricultural University, Urumqi, China, 2022. [Google Scholar]
- Strucken, E.M.; Laurenson, Y.C.; Brockmann, G.A. Go with the flow—Biology and genetics of the lactation cycle. Front. Genet 2015, 6, 118. [Google Scholar] [CrossRef]
- Capuco, A.V.; Bright, S.A.; Pankey, J.W.; Wood, D.L.; Miller, R.H.; Bitman, J. Increased susceptibility to lntramammary infection following removal of teat canal keratin. J. Dairy Sci. 1992, 75, 2126–2130. [Google Scholar] [CrossRef]
- Lacy-Hulbert, S.J.; Hillerton, J.E. Physical characteristics of the bovine teat canal and their influence on susceptibility to streptococcal infection. J. Dairy Res. 1995, 62, 395–404. [Google Scholar] [CrossRef]
- Neijenhuis, F.; Klungel, G.H.; Hogeveen, H. Recovery of cow teats after milking as determined by ultrasonographic scanning. J. Dairy Sci. 2001, 84, 2599–2606. [Google Scholar] [CrossRef]
- Zhao, X.; Lacasse, P. Mammary tissue damage during bovine mastitis: Causes and control. J. Anim. Sci. 2008, 86, 57–65. [Google Scholar] [CrossRef]
- Ogorevc, J.; Kunej, T.; Razpet, A.; Dovc, P. Database of cattle candidate genes and genetic markers for milk production and mastitis. Anim. Genet. 2009, 40, 832–851. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, R.; Oerlemans, R.; van der Heijden, J.W.; Scheffer, G.L.; de Gruijl, T.D.; Jansen, G.; Scheper, R.J. ABC drug transporters and immunity: Novel therapeutic targets in autoimmunity and cancer. J. Leukoc. Biol. 2009, 86, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Gowane, G.R.; Vandre, R.K.; Nangre, M.; Sharma, A. Major histocompatibility complex (MHC) of bovines: An insight into infectious disease resistance. Livest. Res. Int. 2013, 1, 46–57. [Google Scholar]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef]
- Daneshmand, A.; Kermanshahi, H.; Mesgaran, M.D.; King, A.J.; Ibrahim, S.A.; Klasing, K.C. Combination of purine and pyrimidine nucleosides influences growth performance, gut morphology, digestive enzymes, serum biochemical indices and immune functions in broiler chickens. Anim. Feed Sci. Technol. 2017, 228, 186–193. [Google Scholar] [CrossRef]
- Carver, J.D. Dietary nucleotides: Effects on the immune and gastrointestinal systems. Acta Paediatr. 1999, 88, 83–88. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J. 2000, 16, 534–554. [Google Scholar] [CrossRef]
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, Y.; Song, J.; Zhu, M.; Zhu, G.; Cai, H.; Chen, C.; Jin, M.; Song, Y. Perturbation of arachidonic acid and glycerolipid metabolism promoted particulate matter-induced inflammatory responses in human bronchial epithelial cells. Ecotoxicol. Environ. Saf. 2023, 256, 114839. [Google Scholar] [CrossRef]
- Ruiz, M.; Jové, M.; Schlüter, A.; Casasnovas, C.; Villarroya, F.; Guilera, C.; Ortega, F.J.; Naudí, A.; Pamplona, R.; Pujol, A.; et al. Altered glycolipid and glycerophospholipid signaling drive inflammatory cascades in adrenomyeloneuropathy. Hum. Mol. Genet. 2015, 24, 6861–6876. [Google Scholar] [CrossRef] [PubMed]
- Elaine, A.Y.; He, S.; Jones, D.P.; Sun, Y.V.; Ramirez-Zea, M.; Stein, A.D. Metabolomic profiling demonstrates postprandial changes in fatty acids and glycerophospholipids are associated with fasting inflammation in Guatemalan adults. J. Nutr. 2021, 151, 2564–2573. [Google Scholar] [CrossRef]
- Shen, L.; Shen, Y.; Zhang, Y.; Cao, S.; Yu, S.; Zong, X.; Su, Z. Effects of Anemoside B4 on Plasma Metabolites in Cows with Clinical Mastitis. Vet. Sci. 2023, 10, 437. [Google Scholar] [CrossRef]
- Wang, S.; Tan, K.S.; Beng, H.; Liu, F.; Huang, J.; Kuai, Y.; Zhang, Y.; Tan, W. Protective effect of isosteviol sodium against LPS-induced multiple organ injury by regulating of glycerophospholipid metabolism and reducing macrophage-driven inflammation. Pharmacol. Res. 2021, 172, 105781. [Google Scholar] [CrossRef]
- King, N.E.; Rothenberg, M.E.; Zimmermann, N. Arginine in asthma and lung inflammation. J. Nutr. 2004, 134, 2830S–2836S. [Google Scholar] [CrossRef]
- Satriano, J. Arginine pathways and the inflammatory response: Interregulation of nitric oxide and polyamines. Amino Acids 2004, 26, 321–329. [Google Scholar] [CrossRef]
- Sharma, N.; Singh, N.K.; Bhadwal, M.S. Relationship of somatic cell count and mastitis: An overview. Asian Australas. J. Anim. Sci. 2011, 24, 429–438. [Google Scholar] [CrossRef]
- Pyörälä, S. Indicators of inflammation in the diagnosis of mastitis. Vet. Res. 2003, 34, 565–578. [Google Scholar] [CrossRef]
- Ma, Y.; Ryan, C.; Barbano, D.M.; Galton, D.M.; Rudan, M.A.; Boor, K.J. Effects of somatic cell count on quality and shelf-life of pasteurized fluid milk. J. Dairy Sci. 2000, 83, 264–274. [Google Scholar] [CrossRef]
- Salem Jr, N.; Van Dael, P. Arachidonic acid in human milk. Nutrients 2020, 12, 626. [Google Scholar] [CrossRef]
- Haubert, N.J.B.G.B.; Marchini, J.S.; Cunha, S.F.C.; Suen, V.M.M.; Padovan, G.J.; Junior, A.A.; Alves, C.M.M.M.; Marchini, J.F.; Vannucchi, H. Choline and fructooligosaccharide: Non-alcoholic fatty liver disease, cardiac fat deposition, and oxidative stress markers. Nutr. Metab. Insights 2015, 8, NMI-S24385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tobolski, D.; Zwierzchowski, G.; Mandal, R.; Wishart, D.S.; Ametaj, B.N. Identification of serum-predictive biomarkers for subclinical mastitis in dairy cows and new insights into the pathobiology of the disease. J. Agric. Food Chem. 2022, 70, 1724–1746. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Human Serum Metabolome (HUSERMET) Consortium; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, T.; Shen, X.; Liu, J.; Zhao, D.; Sun, Y.; Wang, L.; Liu, Y.; Gong, X.; Xue, F.; et al. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Garcia, A.; Barbas, C. Gas chromatography-mass spectrometry (GC-MS)-based metabolomics. Metab. Profiling Methods Protoc. 2011, 708, 191–204. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.; Li, S.; Yao, S.; Qi, P.; Yang, Z.; Feng, Z.; Hou, J.; Cai, L.; Guo, D.A.; et al. An intelligentized strategy for endogenous small molecules characterization and quality evaluation of earthworm from two geographic origins by ultra-high performance HILIC/QTOF MS E and Progenesis QI. Anal. Bioanal. Chem. 2016, 408, 3881–3890. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, D.; Lei, C.; Li, Y.; Sun, X.; Chen, M. Identification of crucial genes in abdominal aortic aneurysm by WGCNA. PeerJ 2019, 7, e7873. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wang, H.; Sun, L.; Liu, S.; Du, S.; Qiao, W.; Wang, W.; Hou, G.; Zhang, K.; Teng, Q.; et al. Identification of key gene modules and hub genes of human mantle cell lymphoma by coexpression network analysis. PeerJ 2020, 8, e8843. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Gearing, M.; Wang, P.G.; Chin, L.S.; Li, L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 19. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 8, 3322–3335. [Google Scholar] [CrossRef]
- Pang, Z.; Lu, Y.; Zhou, G.; Hui, F.; Xu, L.; Viau, C.; Spigelman, A.F.; MacDonald, P.E.; Wishart, D.S.; Xia, J.; et al. MetaboAnalyst 6.0: Towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 2024, 52, gkae253. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | SCC | Milk Fat | Milk Protein | Lactose | |
|---|---|---|---|---|---|
| XJBC | SCC | 1 | 0.204 ** | 0.238 ** | −0.349 ** |
| Milk Fat | 0.204 ** | 1 | 0.115 * | −0.183 ** | |
| Milk Protein | 0.238 ** | 0.115 * | 1 | −0.433 ** | |
| Lactose | −0.349 ** | −0.183 ** | −0.433 ** | 1 | |
| CSC | SCC | 1 | −0.035 | 0.266 ** | −0.391 ** |
| Milk Fat | −0.035 | 1 | 0.147 | −0.138 | |
| Milk Protein | 0.266 ** | 0.147 | 1 | −0.0457 ** | |
| Lactose | −0.39 1 ** | −0.138 | −0.457 ** | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Wang, D.; Zhang, M.; Xu, L.; Fu, X.; Zhang, T.; Yan, M.; Huang, X. Transcriptomic and Metabolomics Joint Analyses Reveal the Influence of Gene and Metabolite Expression in Blood on the Lactation Performance of Dual-Purpose Cattle (Bos taurus). Int. J. Mol. Sci. 2024, 25, 12375. https://doi.org/10.3390/ijms252212375
Ma S, Wang D, Zhang M, Xu L, Fu X, Zhang T, Yan M, Huang X. Transcriptomic and Metabolomics Joint Analyses Reveal the Influence of Gene and Metabolite Expression in Blood on the Lactation Performance of Dual-Purpose Cattle (Bos taurus). International Journal of Molecular Sciences. 2024; 25(22):12375. https://doi.org/10.3390/ijms252212375
Chicago/Turabian StyleMa, Shengchao, Dan Wang, Menghua Zhang, Lei Xu, Xuefeng Fu, Tao Zhang, Mengjie Yan, and Xixia Huang. 2024. "Transcriptomic and Metabolomics Joint Analyses Reveal the Influence of Gene and Metabolite Expression in Blood on the Lactation Performance of Dual-Purpose Cattle (Bos taurus)" International Journal of Molecular Sciences 25, no. 22: 12375. https://doi.org/10.3390/ijms252212375
APA StyleMa, S., Wang, D., Zhang, M., Xu, L., Fu, X., Zhang, T., Yan, M., & Huang, X. (2024). Transcriptomic and Metabolomics Joint Analyses Reveal the Influence of Gene and Metabolite Expression in Blood on the Lactation Performance of Dual-Purpose Cattle (Bos taurus). International Journal of Molecular Sciences, 25(22), 12375. https://doi.org/10.3390/ijms252212375