
Broad Neutralization Capacity of an Engineered Thermostable Three-Helix Angiotensin-Converting Enzyme 2 Polypeptide Targeting the Receptor-Binding Domain of SARS-CoV-2

 , , ,
, , ,  , , ,
, , ,
Abstract
1. Introduction
2. Results
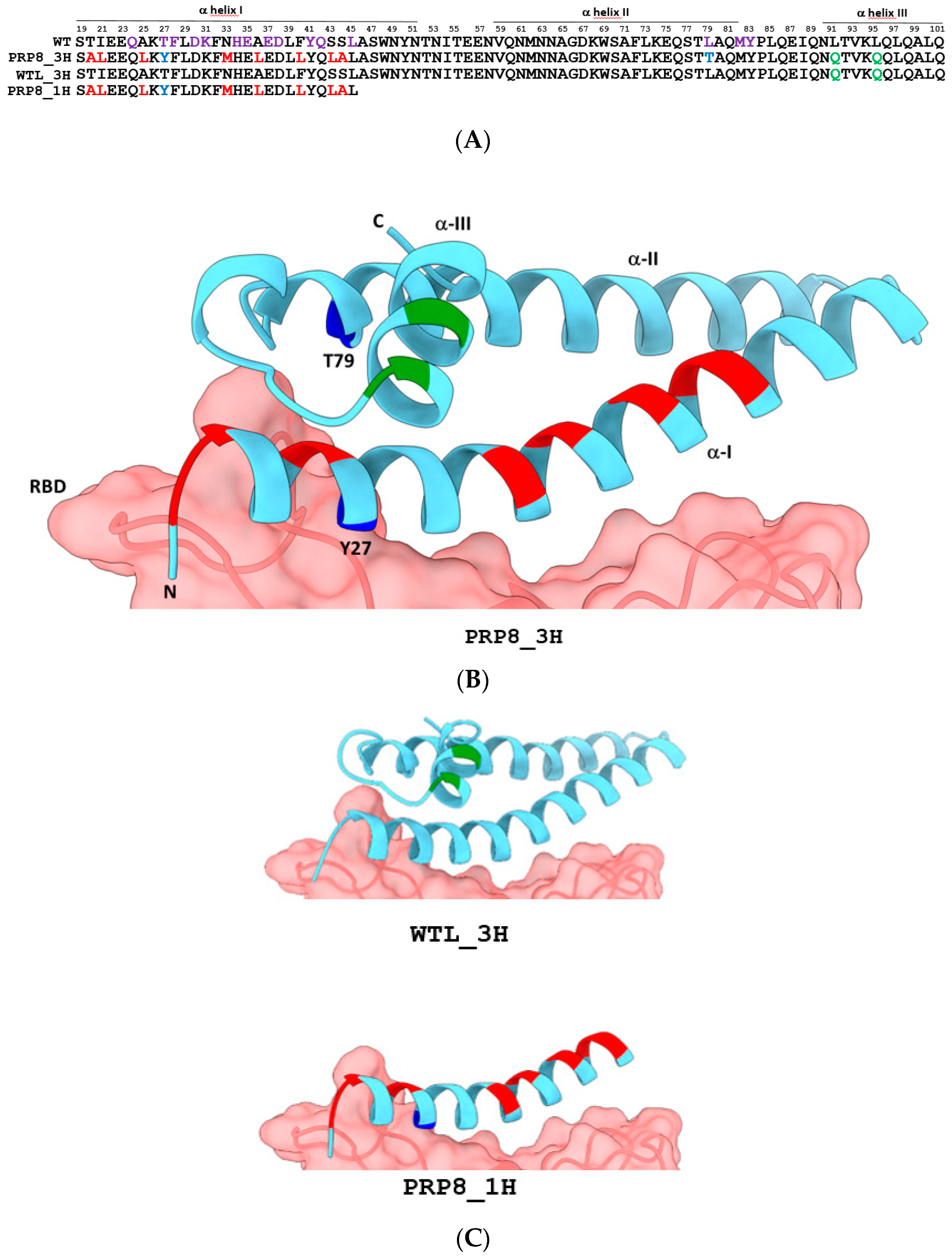
2.1. Design of an Engineered Tri-Helical ACE2 Polypeptide Fragment
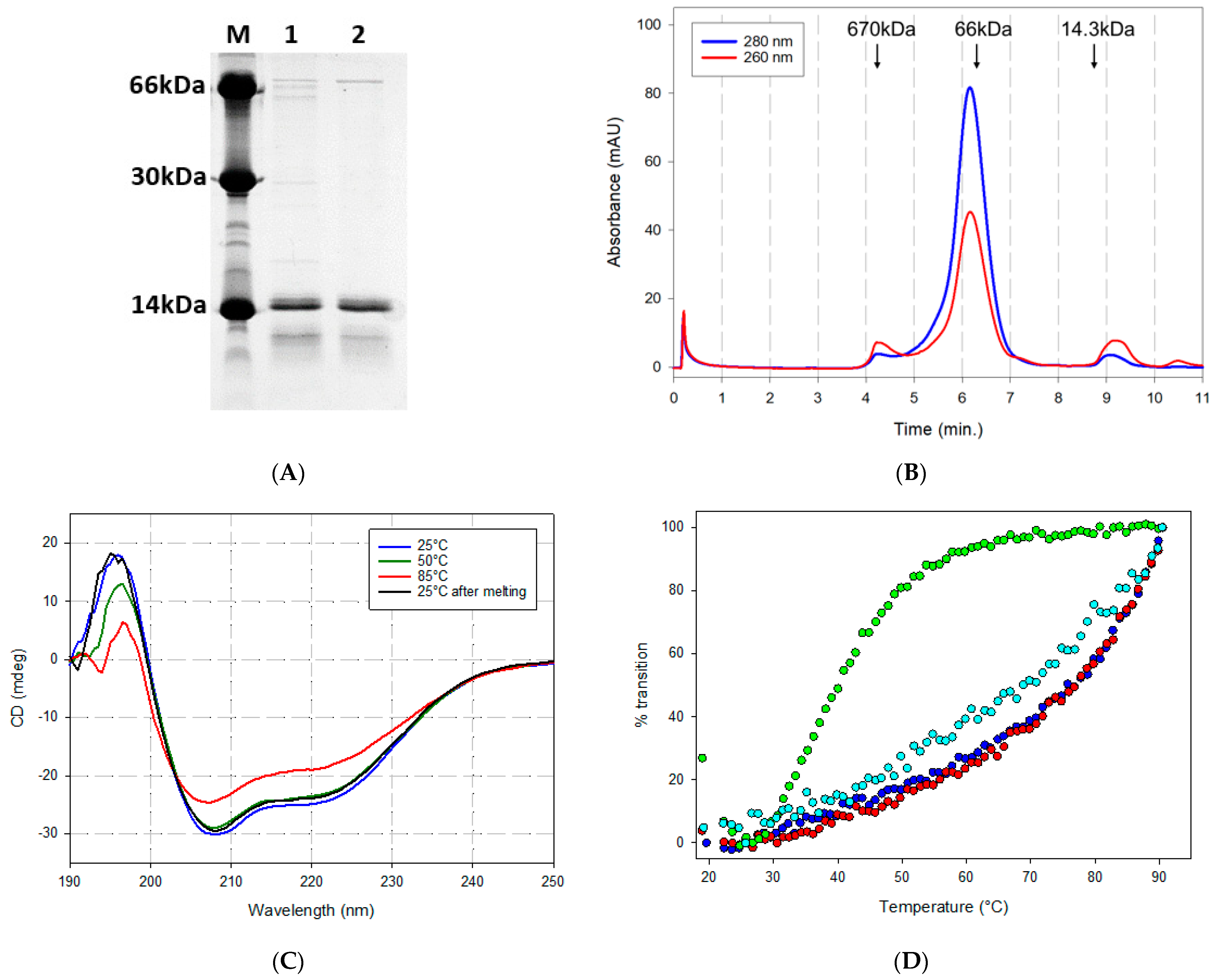
2.2. Biochemical Properties of Recombinantly Expressed ACE2 Fragment Polypeptides
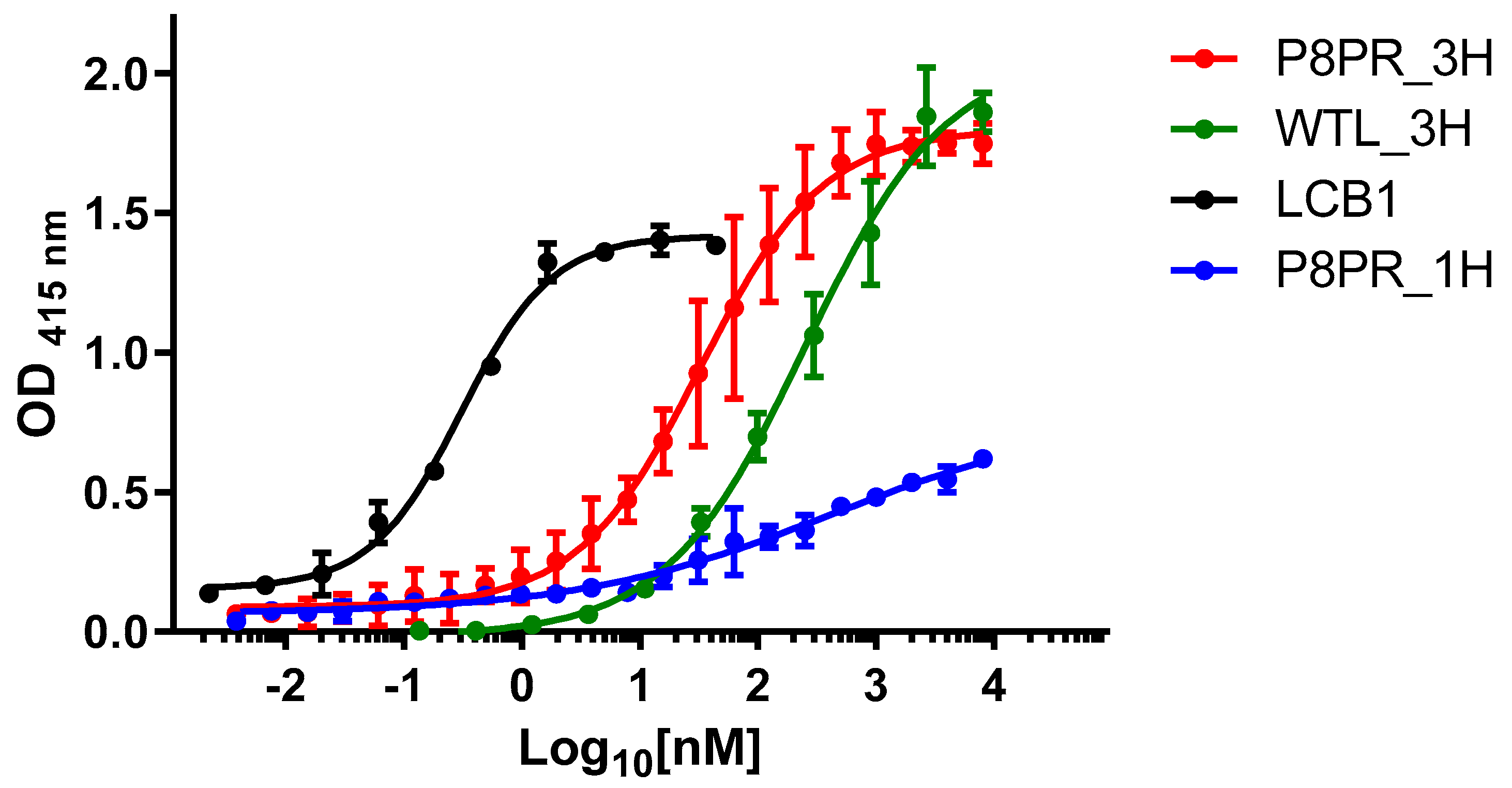
2.3. Spike RBD Binding by PRP8_3H and Other ACE2 Fragments
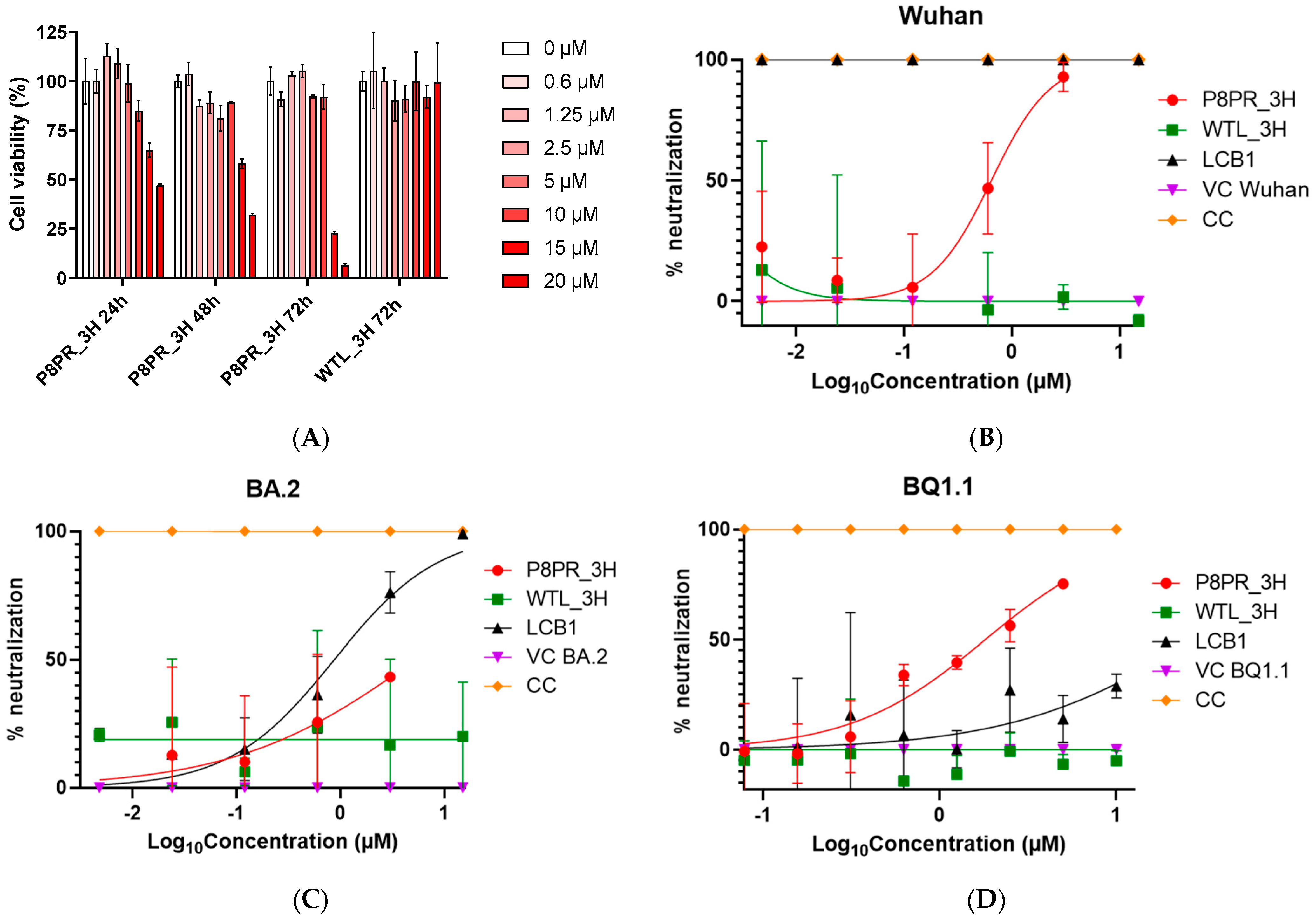
2.4. Multivalent SARS-CoV 2 Pseudovirus Neutralization by the PRP8_3H Decoy
3. Discussion
4. Materials and Methods
4.1. Structure Predictions
4.2. Expression and Purification of ACE2-Derived Polypeptide Fragments
4.3. Analytical Size Exclusion Chromatography
4.4. Far-UV Circular Dichroism
4.5. ELISA
4.6. Cell Viability Assays
4.7. Neutralization Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Koelle, K.; Martin, M.A.; Antia, R.; Lopman, B.; Dean, N.E. The changing epidemiology of SARS-CoV-2. Science 2022, 375, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Lu, R.M.; Li, M.C.; Huang, J.-L.; Hsu, F.-F.; Ko, S.-H.; Ke, F.-Y.; Su, S.-C.; Liang, K.-H.; Yuan, J.P.-Y.; et al. A critical overview of current progress for COVID-19: Development of vaccines, antiviral drugs, and therapeutic antibodies. J. Biomed. Sci. 2022, 29, 68. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Mutair, A.A.; Hajissa, K.; Alfaraj, A.H.; Al-Jishi, J.M.; Alhajri, M.; Alwarthan, S.; Alsuliman, S.A.; Al-Najjar, A.H.; Al Zaydani, I.A.; et al. A Comprehensive Review on the Current Vaccines and Their Efficacies to Combat SARS-CoV-2 Variants. Vaccines 2022, 10, 1655. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Flores, D.; Zepeda-Cervantes, J.; Cruz-Reséndiz, A.; Aguirre-Sampieri, S.; Sampieri, A.; Vaca, L. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol. 2021, 12, 701501. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Lu, R.M.; Su, S.C.; Chiang, P.Y.; Ko, S.H.; Ke, F.Y.; Liang, K.H.; Hsieh, T.Y.; Wu, H.C. Monoclonal antibodies for COVID-19 therapy and SARS-CoV-2 detection. J. Biomed. Sci. 2022, 29, 1. [Google Scholar] [CrossRef]
- Zhou, H.; Møhlenberg, M.; Thakor, J.C.; Tuli, H.S.; Wang, P.; Assaraf, Y.G.; Dhama, K.; Jiang, S. Sensitivity to Vaccines, Therapeutic Antibodies, and Viral Entry Inhibitors and Advances To Counter the SARS-CoV-2 Omicron Variant. Clin. Microbiol. Rev. 2022, 35, e0001422. [Google Scholar] [CrossRef]
- Tada, T.; Zhou, H.; Dcosta, B.M.; Samanovic, M.I.; Chivukula, V.; Herati, R.S.; Hubbard, S.R.; Mulligan, M.J.; Landau, N.R. Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine 2022, 78, 103944. [Google Scholar] [CrossRef]
- Iketani, S.; Ho, D.D. SARS-CoV-2 resistance to monoclonal antibodies and small-molecule drugs. Cell Chem. Biol. 2024, 31, 632–657. [Google Scholar] [CrossRef]
- Zhao, F.; Zai, X.; Zhang, Z.; Xu, J.; Chen, W. Challenges and developments in universal vaccine design against SARS-CoV-2 variants. NPJ Vaccines 2022, 7, 167. [Google Scholar] [CrossRef]
- Chi, W.Y.; Li, Y.D.; Huang, H.C.; Chan, T.E.H.; Chow, S.-Y.; Su, J.-H.; Ferrall, L.; Hung, C.-F.; Wu, T.-C. COVID-19 vaccine update: Vaccine effectiveness, SARS-CoV-2 variants, boosters, adverse effects, and immune correlates of protection. J. Biomed. Sci. 2022, 29, 82. [Google Scholar] [CrossRef]
- Miller, J.; Hachmann, N.P.; Collier, A.Y.; Lasrado, N.; Mazurek, C.R.; Patio, R.C.; Powers, O.; Surve, N.; Theiler, J.; Korber, B.; et al. Substantial Neutralization Escape by SARS-CoV-2 Omicron Variants BQ.1.1 and XBB.1. N. Engl. J. Med. 2023, 388, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.-J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J.; et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Addetia, A.; Piccoli, L.; Case, J.B.; Park, Y.-J.; Beltramello, M.; Guarino, B.; Dang, H.; Pinto, D.; Scheaffer, S.; Sprouse, K.; et al. Therapeutic and vaccine-induced cross-reactive antibodies with effector function against emerging Omicron variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Khattri, A.; Verma, V. Structural and antigenic variations in the spike protein of emerging SARS-CoV-2 variants. PLoS Pathog. 2022, 18, e1010260. [Google Scholar] [CrossRef]
- Higuchi, Y.; Suzuki, T.; Arimori, T.; Ikemura, N.; Mihara, E.; Kirita, Y.; Ohgitani, E.; Mazda, O.; Motooka, D.; Nakamura, S.; et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat. Commun. 2021, 12, 3802. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor-binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Costa, C.F.S.; Barbosa, A.J.M.; Dias, A.M.G.C.; Roque, A.C.A. Native, engineered and de novo designed ligands targeting the SARS-CoV-2 spike protein. Biotechnol. Adv. 2022, 59, 107986. [Google Scholar] [CrossRef]
- Chitsike, L.; Duerksen-Hughes, P. Keep out! SARS-CoV-2 entry inhibitors: Their role and utility as COVID-19 therapeutics. Virol. J. 2021, 18, 154. [Google Scholar] [CrossRef]
- Arimori, T.; Ikemura, N.; Okamoto, T.; Takagi, J.; Standley, D.M.; Hoshino, A. Engineering ACE2 decoy receptors to combat viral escapability. Trends Pharmacol. Sci. 2022, 43, 838–851. [Google Scholar] [CrossRef]
- Zhang, H.; Lv, P.; Jiang, J.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Advances in developing ACE2 derivatives against SARS-CoV-2. Lancet Microbe 2023, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Lu, J.; Zhang, J.; Johnson, R.I.; McKay, L.G.A.; Storm, N.; Lavine, C.L.; Peng, H.; Cai, Y.; Rits-Volloch, S.; et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat. Struct. Mol. Biol. 2021, 28, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, E.; Zhang, L.; Wang, W.; Jin, Y.; Sun, J.; Huang, S.; Yin, W.; Dai, J.; Zhuang, Z.; et al. Potent prophylactic and therapeutic efficacy of recombinant human ACE2-Fc against SARS-CoV-2 infection in vivo. Cell Discov. 2021, 7, 65. [Google Scholar] [CrossRef]
- Torchia, J.A.; Tavares, A.H.; Carstensen, L.S.; Chen, D.-Y.; Huang, J.; Xiao, T.; Mukherjee, S.; Reeves, P.M.; Tu, H.; Sluder, A.E.; et al. Optimized ACE2 decoys neutralize antibody-resistant SARS-CoV-2 variants through functional receptor mimicry and treat infection in vivo. Sci. Adv. 2022, 8, eabq6527. [Google Scholar] [CrossRef]
- Svilenov, H.L.; Bester, R.; Sacherl, J.; Absmeier, R.; Peters, C.; Protzer, U.; Brockmeyer, C.; Buchner, J. Multimeric ACE2-IgM fusions as broadly active antivirals that potently neutralize SARS-CoV-2 variants. Commun. Biol. 2022, 5, 1237. [Google Scholar] [CrossRef]
- Sims, J.J.; Greig, J.A.; Michalson, K.T.; Lian, S.; Martino, R.A.; Meggersee, R.; Turner, K.B.; Nambiar, K.; Dyer, C.; Hinderer, C.; et al. Intranasal gene therapy to prevent infection by SARS-CoV-2 variants. PLoS Pathog. 2021, 17, e1009544. [Google Scholar] [CrossRef]
- Ikemura, N.; Taminishi, S.; Inaba, T.; Arimori, T.; Motooka, D.; Katoh, K.; Kirita, Y.; Higuchi, Y.; Li, S.; Suzuki, T.; et al. An engineered ACE2 decoy neutralizes the SARS-CoV-2 Omicron variant and confers protection against infection in vivo. Sci. Transl. Med. 2022, 14, eabn7737. [Google Scholar] [CrossRef]
- Zhang, L.; Dutta, S.; Xiong, S.; Chan, M.; Chan, K.K.; Fan, T.M.; Bailey, K.L.; Lindeblad, M.; Cooper, L.M.; Rong, L.; et al. Engineered ACE2 decoy mitigates lung injury and death induced by SARS-CoV-2 variants. Nat. Chem. Biol. 2022, 18, 342–351. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, L.; Ullah, I.; Beaudoin-Bussières, G.; Anand, S.P.; Hederman, A.P.; Tolbert, W.D.; Sherburn, R.; Nguyen, D.N.; Marchitto, L.; et al. Engineered ACE2-Fc counters murine lethal SARS-CoV-2 infection through direct neutralization and Fc-effector activities. Sci. Adv. 2022, 8, eabn4188. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Vishweshwaraiah, Y.L.; Hnath, B.; Wang, J.; Chandler, M.; Mukherjee, A.; Yennawar, N.H.; Booker, S.J.; Afonin, K.A.; Dokholyan, N.V. A Piecewise Design Approach to Engineering a Miniature ACE2 Mimic to Bind SARS-CoV-2. ACS Appl. Bio Mater. 2024, 7, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Renzi, F.; Seamann, A.; Ganguly, K.; Pandey, K.; Byrareddy, S.N.; Batra, S.; Kumar, S.; Ghersi, D. Engineering an ACE2-Derived Fragment as a Decoy for Novel SARS-CoV-2 Virus. ACS Pharmacol. Transl. Sci. 2023, 6, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Vishvakarma, V.; Dey, A.; Dey, S.; Gupta, A.; Das, M.; Vishwakarma, K.K.; Roy, D.S.; Yadav, S.; Kesarwani, S.; et al. Biophysical properties of the isolated spike protein binding helix of human ACE2. Biophys. J. 2021, 120, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Tan, X.; Loas, A.; Pentelute, B.L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. bioRxiv 2020. [Google Scholar] [CrossRef]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Berisio, R. An engineered stable mini-protein to plug SARS-Cov-2 Spikes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.-T.K.; Hillyer, C.D.; Debnath, A.K. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Karoyan, P.; Vieillard, V.; Gómez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.-E.; Denis, A.; Grondin, P.; Lequin, O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.-Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.C.; Case, J.B.; Park, Y.J.; Cao, L.; Wu, K.; Walls, A.C.; Liu, Z.; Bowen, J.E.; Yeh, H.-W.; Saini, S.; et al. Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice. Sci. Transl. Med. 2022, 14, eabn1252. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Chen, R.E.; Cao, L.; Ying, B.; Winkler, E.S.; Johnson, M.; Goreshnik, I.; Pham, M.N.; Shrihari, S.; Kafai, N.M.; et al. Ultrapotent miniproteins targeting the SARS-CoV-2 receptor-binding domain protect against infection and disease. Cell Host Microbe 2021, 29, 1151–1161.e5. [Google Scholar] [CrossRef] [PubMed]
- Henchey, L.K.; Jochim, A.L.; Arora, P.S. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr. Opin. Chem. Biol. 2008, 12, 692–697. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2--a server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 2012, 80, 374–381. [Google Scholar] [CrossRef]
- Fernandes, L.A.; Gomes, A.A.; Guimarães, B.G.; Magalhães, M.d.L.B.; Ray, P.; da Silva, G.F. Engineering defensin α-helix to produce high-affinity SARS-CoV-2 spike protein binding ligands. Protein Sci. 2022, 31, e4355. [Google Scholar] [CrossRef]
- Beatty, J.D.; Beatty, B.G.; Vlahos, W.G. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. J. Immunol. Methods 1987, 100, 173–179. [Google Scholar] [CrossRef]
- Guo, H.; Ha, S.; Botten, J.W.; Xu, K.; Zhang, N.; An, Z.; Strohl, W.R.; Shiver, J.W.; Fu, T.-M. SARS-CoV-2 Omicron: Viral Evolution, Immune Evasion, and Alternative Durable Therapeutic Strategies. Viruses 2024, 16, 697. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zou, F.; Yu, F.; Li, R.; Yuan, Y.; Zhang, Y.; Zhang, X.; Deng, J.; Chen, T.; Song, Z.; et al. Nanoparticle Vaccines Based on the Receptor Binding Domain (RBD) and Heptad Repeat (HR) of SARS-CoV-2 Elicit Robust Protective Immune Responses. Immunity 2020, 53, 1315–1330.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, L.; Liu, Z.; Xing, L.; Zhu, Y.; Xu, W.; Xia, S.; Lu, L.; Jiang, S. An engineered recombinant protein containing three structural domains in SARS-CoV-2 S2 protein has potential to act as a pan-human coronavirus entry inhibitor or vaccine antigen. Emerg. Microbes Infect. 2023, 12, 2244084. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.V.; Wall, S.C.; Kramer, K.J.; Holt, C.M.; Periasamy, S.; Richardson, S.I.; Manamela, N.P.; Suryadevara, N.; Andreano, E.; Paciello, I.; et al. Discovery and Characterization of a Pan-betacoronavirus S2-binding antibody. bioRxiv 2024. [Google Scholar] [CrossRef]
- Liang, L.; Wang, B.; Zhang, Q.; Zhang, S.; Zhang, S. Antibody drugs targeting SARS-CoV-2: Time for a rethink? Biomed. Pharmacother. 2024, 176, 116900. [Google Scholar] [CrossRef]
- Li, M.; Ye, Z.W.; Tang, K.; Guo, L.; Bi, W.; Zhang, Y.; Tang, Y.-D.; Rong, G.; Sawan, M.; Yin, X.; et al. Enhanced trimeric ACE2 exhibits potent prophylactic and therapeutic efficacy against the SARS-CoV-2 Delta and Omicron variants in vivo. Cell Res. 2022, 32, 589–592. [Google Scholar] [CrossRef]
- Urano, E.; Itoh, Y.; Suzuki, T.; Sasaki, T.; Kishikawa, J.-I.; Akamatsu, K.; Higuchi, Y.; Sakai, Y.; Okamura, T.; Mitoma, S.; et al. An inhaled ACE2 decoy confers protection against SARS-CoV-2 infection in preclinical models. Sci. Transl. Med. 2023, 15, eadi2623. [Google Scholar] [CrossRef]
- Havranek, B.; Lindsey, G.W.; Higuchi, Y.; Itoh, Y.; Suzuki, T.; Okamoto, T.; Hoshino, A.; Procko, E.; Islam, S.M. A computationally designed ACE2 decoy has broad efficacy against SARS-CoV-2 omicron variants and related viruses in vitro and in vivo. Commun. Biol. 2023, 6, 513. [Google Scholar] [CrossRef]
- Glieca, S.; Cavazzini, D.; Levati, E.; Garrapa, V.; Bolchi, A.; Franceschi, V.; Odau, S.; Ottonello, S.; Donofrio, G.; Füner, J.; et al. A dry powder formulation for peripheral lung delivery and absorption of an anti-SARS-CoV-2 ACE2 decoy polypeptide. Eur. J. Pharm. Sci. 2023, 191, 106609. [Google Scholar] [CrossRef]
- Zhou, Z.; Liao, B.; Wang, S.; Tang, J.; Zhao, H.; Tong, M.; Li, K.; Xiong, S. Improved Production of Anti-FGF-2 Nanobody Using Pichia pastoris and Its Effect on Antiproliferation of Keratinocytes and Alleviation of Psoriasis. Arch. Immunol. Ther. Exp. 2023, 71, 20. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polypeptides | Kd ± SD (nM) * |
|---|---|
| P8PR_3H | 24.3 ± 2.6 |
| WTL_3H | 191.8 + 21.5 |
| LCB1 | 0.21 + 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavazzini, D.; Levati, E.; Germani, S.; Ta, B.L.; Monica, L.; Bolchi, A.; Donofrio, G.; Garrapa, V.; Ottonello, S.; Montanini, B. Broad Neutralization Capacity of an Engineered Thermostable Three-Helix Angiotensin-Converting Enzyme 2 Polypeptide Targeting the Receptor-Binding Domain of SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 12319. https://doi.org/10.3390/ijms252212319
Cavazzini D, Levati E, Germani S, Ta BL, Monica L, Bolchi A, Donofrio G, Garrapa V, Ottonello S, Montanini B. Broad Neutralization Capacity of an Engineered Thermostable Three-Helix Angiotensin-Converting Enzyme 2 Polypeptide Targeting the Receptor-Binding Domain of SARS-CoV-2. International Journal of Molecular Sciences. 2024; 25(22):12319. https://doi.org/10.3390/ijms252212319
Chicago/Turabian StyleCavazzini, Davide, Elisabetta Levati, Saveria Germani, Bao Loc Ta, Lara Monica, Angelo Bolchi, Gaetano Donofrio, Valentina Garrapa, Simone Ottonello, and Barbara Montanini. 2024. "Broad Neutralization Capacity of an Engineered Thermostable Three-Helix Angiotensin-Converting Enzyme 2 Polypeptide Targeting the Receptor-Binding Domain of SARS-CoV-2" International Journal of Molecular Sciences 25, no. 22: 12319. https://doi.org/10.3390/ijms252212319
APA StyleCavazzini, D., Levati, E., Germani, S., Ta, B. L., Monica, L., Bolchi, A., Donofrio, G., Garrapa, V., Ottonello, S., & Montanini, B. (2024). Broad Neutralization Capacity of an Engineered Thermostable Three-Helix Angiotensin-Converting Enzyme 2 Polypeptide Targeting the Receptor-Binding Domain of SARS-CoV-2. International Journal of Molecular Sciences, 25(22), 12319. https://doi.org/10.3390/ijms252212319