

Gut Microbiota and Immune System Dynamics in Parkinson’s and Alzheimer’s Diseases




Abstract
1. Introduction
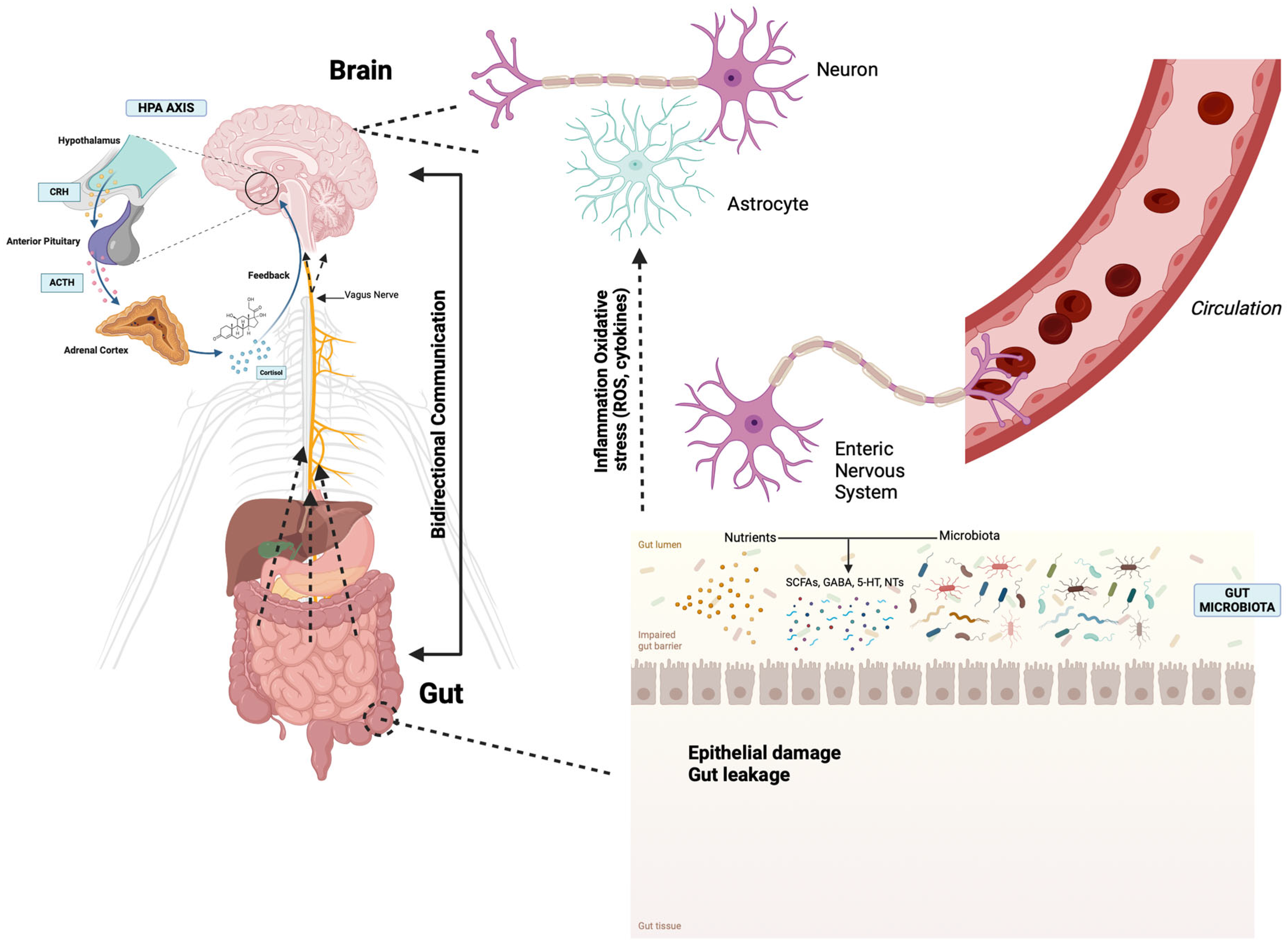
2. The Microbiota–Gut–Brain Axis
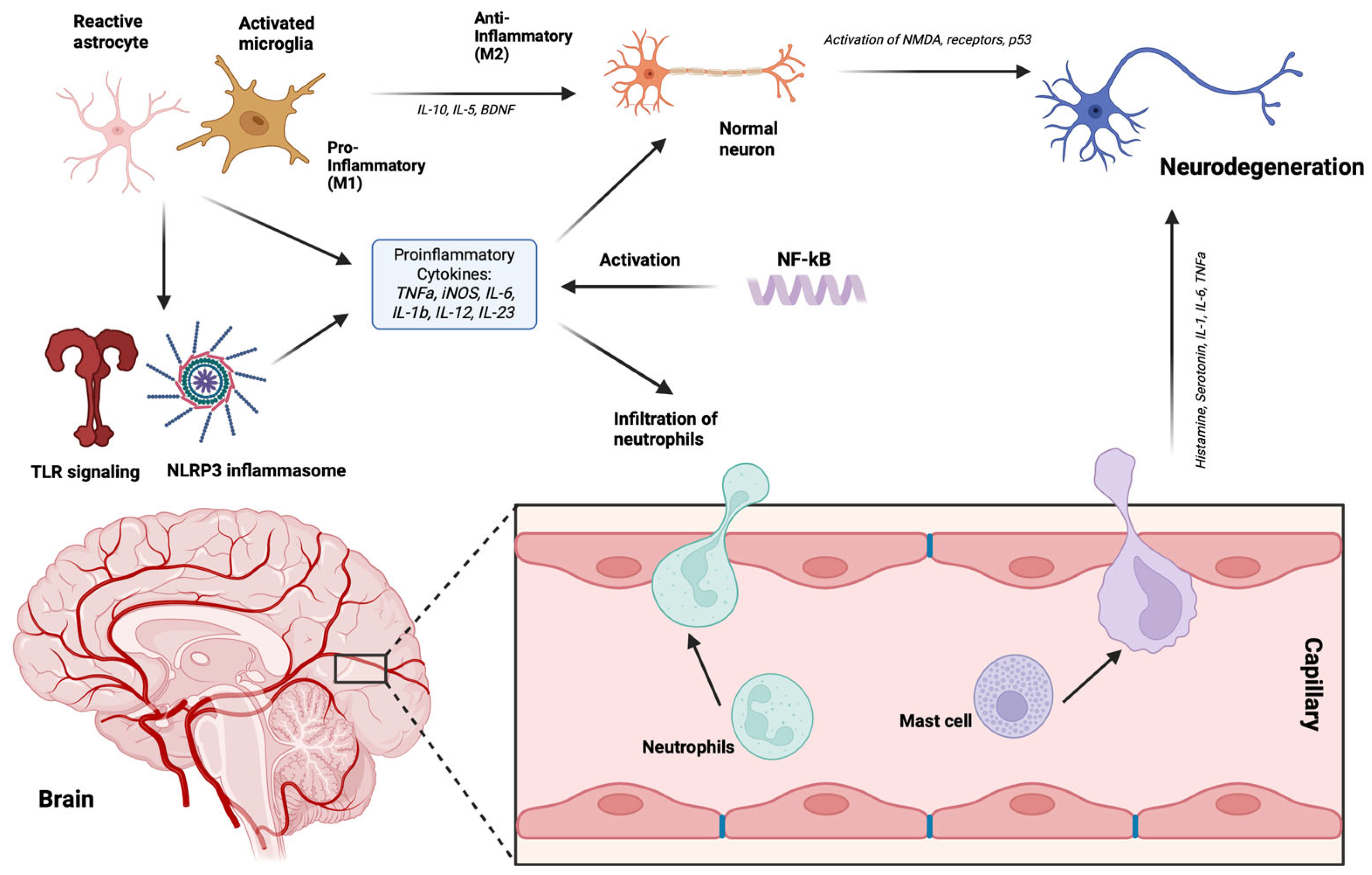
3. Gut Microbiota and Immune System
4. Neurodegenerative Disorders
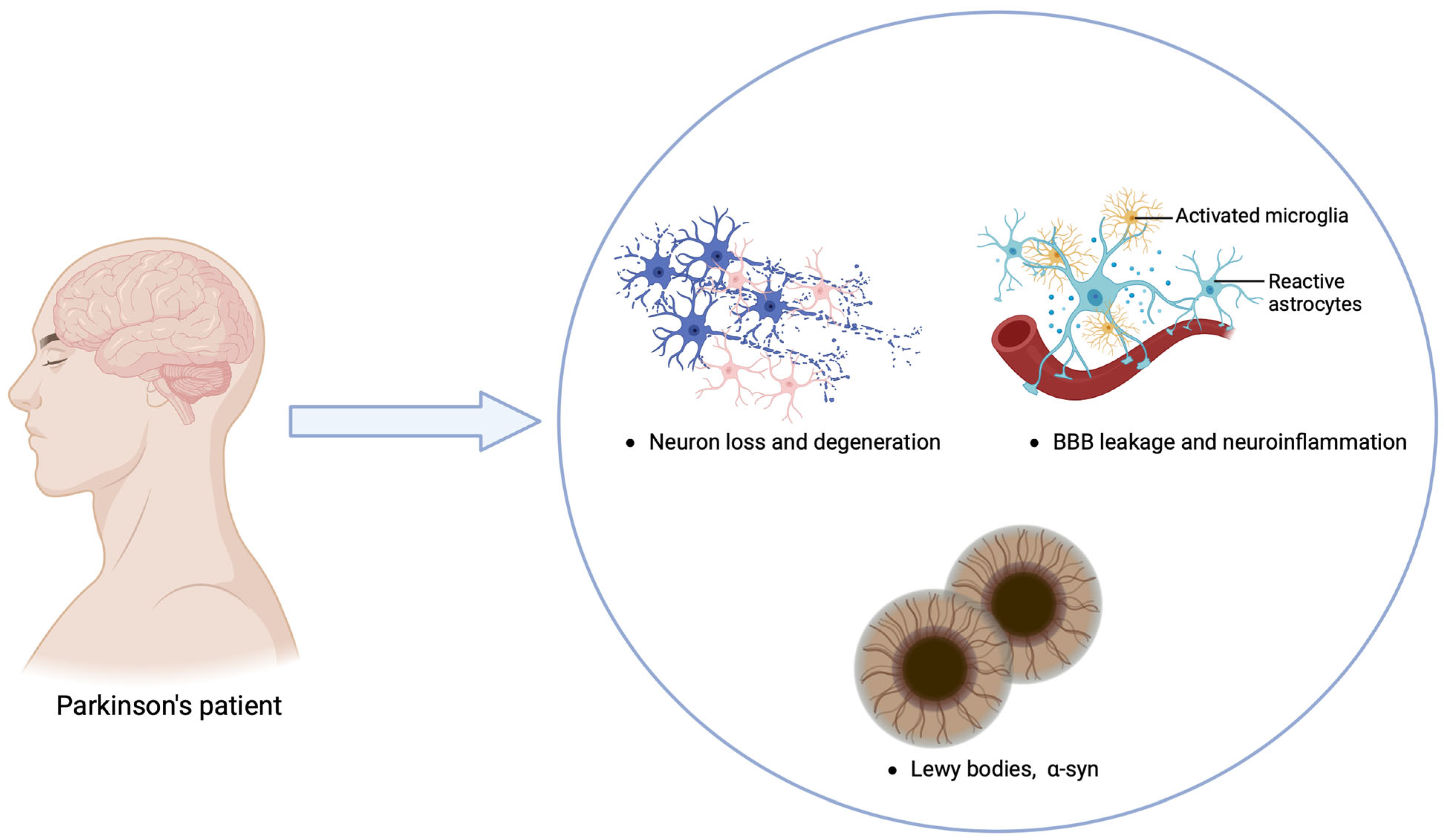
5. Parkinson’s Disease
5.1. Gut Microbiota and α-Synuclein and Immune System Interaction in PD
5.2. Changes in Gut Microbiota in PD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Increased Bacterial Abundance | Decreased Bacterial Abundance |
|---|---|
| Enterobacteriaceae [109,126,133,135,146] | Lachnospiraceae [138] |
| Akkermansia [130] | Faecalibacterium [127,130,131,134] |
| Bifidobacterium [130,136,147] | Blautia [127,128,134] |
| Christensenella [128] | Dorea [128] |
| Catabacter [128] | Bacteroides [128,135] |
| Oscillospiraceae [128] | Prevotellaceae [126,128,131,132,134,135,137] |
| Ruminococcus bromii [128] | Coprococcus [127,129] |
| Papilibacter cinnamivorans [128] | Faecalibacterium [127,134] |
| Proteus [148] | Roseburia [129] |
| Lactobacillaceae [126,128,136,149] | Ralstonia [129] |
| Clostridium Coccoides [147] | B. Fragilis [147] |
| Christinesella [141] | Ruminococcus [134] |
| Oscillospiraceae [134] |
5.3. Microbiota–Gut–Brain Axis for Parkinson’s Disease Treatment
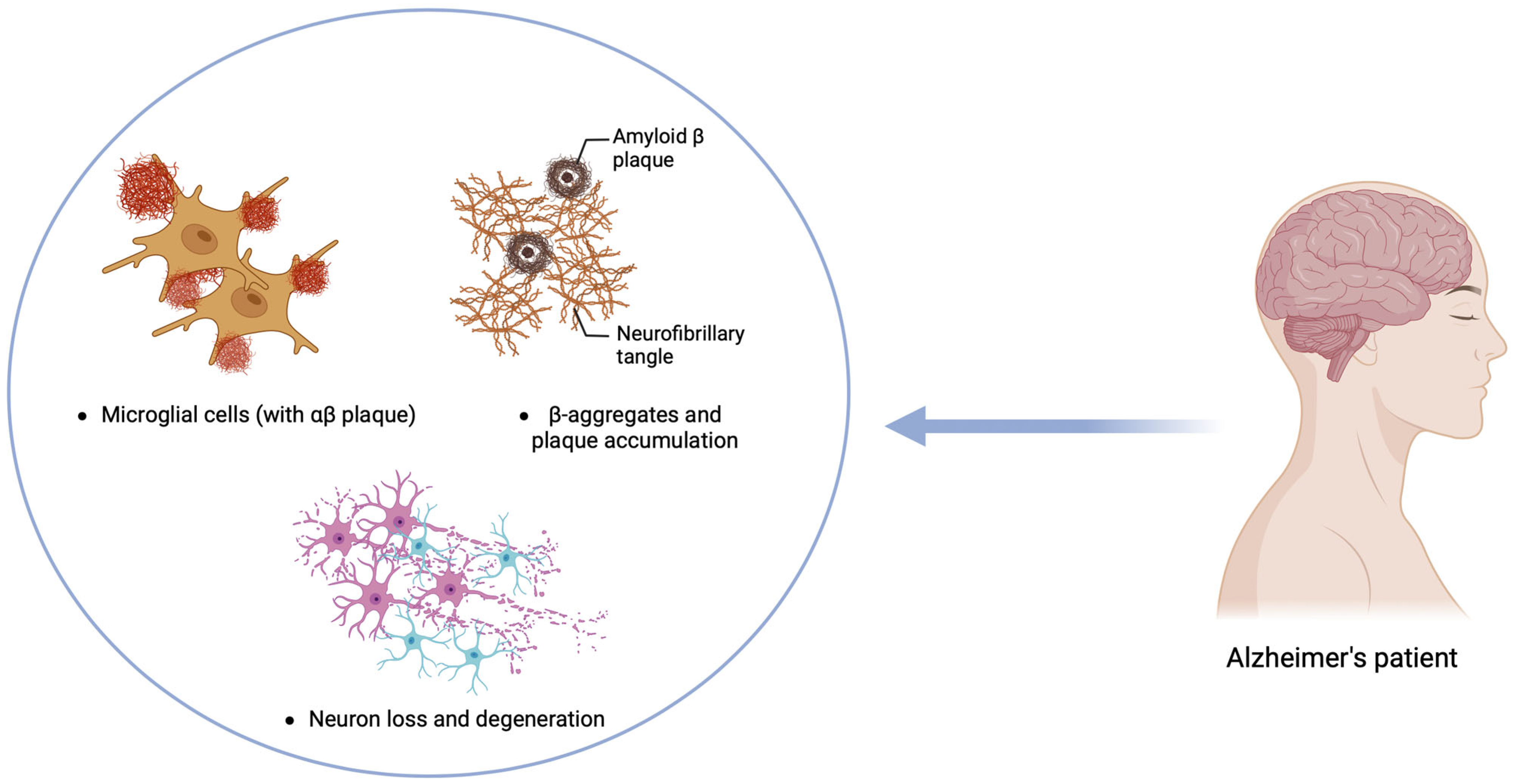
6. Alzheimer’s Disease
6.1. Interaction Between Gut Microbiota and Immune Cells in AD
6.2. Microbiota–Gut–Brain Axis for Alzheimer’s Disease Treatment
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gori, S.; Inno, A.; Belluomini, L.; Bocus, P.; Bisoffi, Z.; Russo, A.; Arcaro, G. Gut microbiota and cancer: How gut microbiota modulates activity, efficacy and toxicity of antitumoral therapy. Crit. Rev. Oncol. Hematol. 2019, 143, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Mousavinasab, F.; Karimi, R.; Taheri, S.; Ahmadvand, F.; Sanaaee, S.; Najafi, S.; Halvaii, M.S.; Haghgoo, A.; Zamany, M.; Majidpoor, J.; et al. Microbiome modulation in inflammatory diseases: Progress to microbiome genetic engineering. Cancer Cell Int. 2023, 23, 271. [Google Scholar] [CrossRef]
- Zhang, W.; Ye, Y.; Song, J.; Sang, T.; Xia, T.; Xie, L.; Qiu, X.; Zeng, Q.; Luo, X. Research Progress of Microbiota-Gut-Brain Axis in Parkinson’s Disease. J. Integr. Neurosci. 2023, 22, 157. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 2022, 13, 6958. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Littman, D.R.; Pamer, E.G. Role of the Commensal Microbiota in Normal and Pathogenic Host Immune Responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef]
- Kelly, D.; King, T.; Aminov, R. Importance of microbial colonization of the gut in early life to the development of immunity. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2007, 622, 58–69. [Google Scholar] [CrossRef]
- Walker, A.W.; Lawley, T.D. Therapeutic modulation of intestinal dysbiosis. Pharmacol. Res. 2013, 69, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome–gut–brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016, 158, 52–62. [Google Scholar] [CrossRef]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef]
- Hattori, N.; Yamashiro, Y. The Gut-Brain Axis. Ann. Nutr. Metab. 2021, 77 (Suppl. S2), 1–3. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Lane, R.D.; Oshinsky, M.L.; Kenny, P.J.; Sinha, R.; Mayer, E.A.; Critchley, H.D. Diseases, disorders, and comorbidities of interoception. Trends Neurosci. 2021, 44, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Nance, K.; Chen, S. The Gut–Brain Axis. Annu. Rev. Med. 2022, 73, 439–453. [Google Scholar] [CrossRef]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef]
- Mayer, E.A. Gut feelings: The emerging biology of gut–brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef]
- Freestone, P. Communication between Bacteria and Their Hosts. Scientifica 2013, 2013, 361073. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef]
- Pascale, A.; Marchesi, N.; Govoni, S.; Barbieri, A. Targeting the microbiota in pharmacology of psychiatric disorders. Pharmacol. Res. 2020, 157, 104856. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Schneeberger, M.; Matheis, F.; Wang, P.; Kerner, Z.; Ilanges, A.; Pellegrino, K.; Del Mármol, J.; Castro, T.B.; Furuichi, M.; et al. Microbiota modulate sympathetic neurons via a gut–brain circuit. Nature 2020, 583, 441–446. [Google Scholar] [CrossRef]
- Gubert, C.; Gasparotto, J.; Morais, L.H. Convergent pathways of the gut microbiota–brain axis and neurodegenerative disorders. Gastroenterol. Rep. 2022, 10, goac017. [Google Scholar] [CrossRef]
- Udit, S.; Gautron, L. Molecular anatomy of the gut-brain axis revealed with transgenic technologies: Implications in metabolic research. Front. Neurosci. 2013, 7, 134. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Bohórquez, D.V. Where the gut meets the brain. Brain Res. 1693, 127, 2018. [Google Scholar] [CrossRef]
- Collins, S.M.; Bercik, P. The Relationship Between Intestinal Microbiota and the Central Nervous System in Normal Gastrointestinal Function and Disease. Gastroenterology 2009, 136, 2003–2014. [Google Scholar] [CrossRef]
- Forsythe, P.; Bienenstock, J.; Kunze, W.A. Vagal Pathways for Microbiome-Brain-Gut Axis Communication. Adv. Exp. Med. Biol. 2014, 817, 115–133. [Google Scholar] [CrossRef]
- Latorre, R.; Sternini, C.; De Giorgio, R.; Meerveld, B.G.-V. Enteroendocrine cells: A review of their role in brain–gut communication. Neurogastroenterol. Motil. 2016, 28, 620–630. [Google Scholar] [CrossRef]
- Cameron, J.; Doucet, É. Getting to the bottom of feeding behaviour: Who’s on top? Appl. Physiol. Nutr. Metab. 2007, 32, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Umesaki, Y.; Setoyama, H.; Matsumoto, S.; Okada, Y. Expansion of alpha beta T-cell receptor-bearing intestinal intraepithelial lymphocytes after microbial colonization in germ-free mice and its independence from thymus. Immunology 1993, 79, 32–37. [Google Scholar] [PubMed]
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.; et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Wesemann, D.R.; Portuguese, A.J.; Meyers, R.M.; Gallagher, M.P.; Cluff-Jones, K.; Magee, J.M.; Panchakshari, R.A.; Rodig, S.J.; Kepler, T.B.; Alt, F.W. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 2013, 501, 112–115. [Google Scholar] [CrossRef]
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007, 2, 328–339. [Google Scholar] [CrossRef]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Wingender, G.; Hiss, M.; Engel, I.; Peukert, K.; Ley, K.; Haller, H.; Kronenberg, M.; von Vietinghoff, S. Neutrophilic granulocytes modulate invariant NKT cell function in mice and humans. J. Immunol. 2012, 188, 3000–3008. [Google Scholar] [CrossRef]
- Martínez-López, M.; Iborra, S.; Conde-Garrosa, R.; Mastrangelo, A.; Danne, C.; Mann, E.R.; Reid, D.M.; Gaboriau-Routhiau, V.; Chaparro, M.; Lorenzo, M.P.; et al. Microbiota Sensing by Mincle-Syk Axis in Dendritic Cells Regulates Interleukin-17 and -22 Production and Promotes Intestinal Barrier Integrity. Immunity 2019, 50, 446–461.e9. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Spires, T.L.; Hannan, A.J. Nature, nurture and neurology: Gene–environment interactions in neurodegenerative disease. FEBS J. 2005, 272, 2347–2361. [Google Scholar] [CrossRef]
- Wang, H.; Yang, F.; Zhang, S.; Xin, R.; Sun, Y. Genetic and environmental factors in Alzheimer’s and Parkinson’s diseases and promising therapeutic intervention via fecal microbiota transplantation. NPJ Parkinsons Dis. 2021, 7, 70. [Google Scholar] [CrossRef]
- Knobel, P.; Litke, R.; Mobbs, C.V. Biological age and environmental risk factors for dementia and stroke: Molecular mechanisms. Front. Aging Neurosci. 2022, 14, 1042488. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Innate immunity, local inflammation, and degenerative disease. Sci. Aging Knowl. Environ. 2002, 2002, re3. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Marino, F.; Cosentino, M. Peripheral Immunity, Immunoaging and Neuroinflammation in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3719–3753. [Google Scholar] [CrossRef]
- Behl, T.; Makkar, R.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Bungau, S.; Andronie-Cioara, F.L.; Munteanu, M.A.; Brisc, M.C.; et al. Current Trends in Neurodegeneration: Cross Talks between Oxidative Stress, Cell Death, and Inflammation. Int. J. Mol. Sci. 2021, 22, 7432. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-κB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, C.M.; Annese, V.; Gómez, A.; Ros-Bernal, F.; Aguado-Llera, D.; Martínez-Pagán, M.E.; De Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. Erratum: IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2012, 3, e379. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G.; Yan, Y.-Q.; Ma, C.-G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1702. [Google Scholar] [CrossRef]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic Glutamatergic Transmission and Its Implications in Neurodegenerative Disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef]
- Sharma, R.; Zamani, A.; Dill, L.K.; Sun, M.; Chu, E.; Robinson, M.J.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. A systemic immune challenge to model hospital-acquired infections independently regulates immune responses after pediatric traumatic brain injury. J. Neuroinflamm. 2021, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, e5665–e5674. [Google Scholar] [CrossRef]
- Malm, T.; Koistinaho, M.; Muona, A.; Magga, J.; Koistinaho, J. The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia 2010, 58, 889–900. [Google Scholar] [CrossRef]
- Funk, N.; Wieghofer, P.; Grimm, S.; Schaefer, R.; Bühring, H.-J.; Gasser, T.; Biskup, S. Characterization of peripheral hematopoietic stem cells and monocytes in Parkinson’s disease. Mov. Disord. 2013, 28, 392–395. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood–brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow. Metab. 2020, 40 (Suppl. S1), S6–S24. [Google Scholar] [CrossRef]
- Zang, X.; Chen, S.; Zhu, J.; Ma, J.; Zhai, Y. The Emerging Role of Central and Peripheral Immune Systems in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 872134. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A. On The Role of Natural Killer Cells in Neurodegenerative Diseases. Toxins 2013, 5, 363–375. [Google Scholar] [CrossRef]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Gut Microbiota: The Conductor in the Orchestra of Immune–Neuroendocrine Communication. Clin. Ther. 2015, 37, 954–967. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; et al. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–949. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathology of Parkinson’s disease. Mol. Chem. Neuropathol. 1991, 14, 153–197. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic Criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 33. [Google Scholar] [CrossRef]
- Roller, W.C. Management of Motor Fluctuations in Parkinson’s Disease. Eur. Neurol. 1996, 36, 43–48. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J. The early diagnosis of parkinson’s disease. Ann. Neurol. 1998, 44, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- de Rijk, M.C.; Tzourio, C.; Breteler, M.M.; Dartigues, J.F.; Amaducci, L.; López-Pousa, S.; Manubens-Bertran, J.M.; Alpérovitch, A.; Rocca, W.A. Prevalence of parkinsonism and Parkinson’s disease in Europe: The EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 10–15. [Google Scholar] [CrossRef]
- Wirdefeldt, K.; Adami, H.-O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, 1–58. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological Treatment of Parkinson Disease. JAMA 2014, 311, 1670. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar]
- Fox, S.H. Non-dopaminergic Treatments for Motor Control in Parkinson’s Disease. Drugs 2013, 73, 1405–1415. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Bieri, G.; Gitler, A.D.; Brahic, M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 2018, 109, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s Disease? Evidence from 3 cases. Mov. Disord. 2012, 27, 716–719. [Google Scholar] [CrossRef]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef]
- Hasuike, Y.; Endo, T.; Koroyasu, M.; Matsui, M.; Chiaki Mori, C.; Misaki Yamadera, M.; Harutoshi Fujimura, H.; Sakoda, S. Bile acid abnormality induced by intestinal dysbiosis might explain lipid metabolism in Parkinson’s disease. Med. Hypotheses 2020, 134, 109436. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Bürmann, J.; Faßbender KSchäfer, K.-H.; Unger, M.M. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Park. Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2018, 68, 829–843. [Google Scholar] [CrossRef]
- Clairembault, T.; Leclair-Visonneau, L.; Coron, E.; Bourreille, A.; Le Dily, S.; Vavasseur, F.; Heymann, M.-F.; Neunlist, M.; Derkinderen, P. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, 12. [Google Scholar] [CrossRef]
- Kelly, L.P.; Carvey, P.M.; Keshavarzian, A.; Shannon, K.M.; Shaikh, M.; Bakay, R.A.E.; Kordower, J.H. Progression of intestinal permeability changes and α-synuclein expression in a mouse model of Parkinson’s disease: GI Dysfunction in a Premotor Model of PD. Mov. Disord. 2014, 29, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of Toxic α-Synuclein Oligomer within Endoplasmic Reticulum Occurs in α-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Comi, C.; Mauri, M.; Minafra, B.; Riboldazzi, G.; et al. Dopaminergic Receptors on CD4+ T Naive and Memory Lymphocytes Correlate with Motor Impairment in Patients with Parkinson’s Disease. Sci. Rep. 2016, 6, 33738. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg, E.; Berry, D.; Yang, D.; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A Role for Neuronal α-Synuclein in Gastrointestinal Immunity. J. Innate Immun. 2017, 9, 456–463. [Google Scholar] [CrossRef]
- Werner, T.; Horvath, I.; Wittung-Stafshede, P. Crosstalk Between Alpha-Synuclein and Other Human and Non-Human Amyloidogenic Proteins: Consequences for Amyloid Formation in Parkinson’s Disease. J. Park. Dis. 2020, 10, 819–830. [Google Scholar] [CrossRef]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances α-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.N.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Miraglia, F.; Colla, E. Microbiome, Parkinson’s Disease and Molecular Mimicry. Cells 2019, 8, 222. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.-H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Tükel, Ç.; Nishimori, J.H.; Wilson, R.P.; Winter, M.G.; Keestra, A.M.; VanPutten, J.P.M.; Bäumler, A.J. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms: TLR2 interacts with TLR1 to recognize curli. Cell. Microbiol. 2010, 12, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Tükel, Ç.; Wilson, R.P.; Nishimori, J.H.; Pezeshki, M.; Chromy, B.A.; Bäumler, A.J. ResponsestoAmyloids of Microbial and Host Origin Are Mediated through Toll-like Receptor 2. Cell Host Microbe 2010, 6, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, J.H.; Newman, T.N.; Oppong, G.O.; Rapsinski, G.J.; Yen, J.-H.; Biesecker, S.G.; Wilson, R.P.; Butler, B.P.; Winter, M.G.; Tsolis, R.M.; et al. Microbial Amyloids Induce Interleukin 17A (IL-17A) and IL-22 Responses via Toll-Like Receptor 2 Activation in the Intestinal Mucosa. Infect. Immun. 2012, 80, 4398–4408. [Google Scholar] [CrossRef]
- Rapsinski, G.J.; Newman, T.N.; Oppong, G.O.; van Putten, J.P.M.; Tükel, Ç. CD14ProteinActsasanAdaptor Molecule for the Immune Recognition of Salmonella Curli Fibers. J. Biol. Chem. 2013, 288, 14178–14188. [Google Scholar] [CrossRef]
- Rapsinski, G.J.; Wynosky-Dolfi, M.A.; Oppong, G.O.; Tursi, S.A.; Wilson, R.P.; Brodsky, I.E.; Tükel, Ç. Toll-Like Receptor 2 and NLRP3 Cooperate to Recognize a Functional Bacterial Amyloid, Curli. Infect. Immun. 2015, 83, 693–701. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Leclair-Visonneau, L.; Bourreille, A.; Coron, E.; Neunlist, M.; Derkinderen, P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J. Neurol. 2020, 267, 2207–2213. [Google Scholar] [CrossRef]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.-C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123. [Google Scholar] [CrossRef]
- Yanagi, H.; Tsuda, A.; Matsushima, M.; Takahashi, S.; Ozawa, G.; Koga, Y.; Takagi, A. Changes in the gut microbiota composition and the plasma ghrelin level in patients with Helicobacter pylori- infected patients with eradication therapy. BMJ Open Gastroenterol. 2017, 4, e000182. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wang, W.; Jia, F.; Du, X.; Xie, A.; He, Q.; Shen, X.; Zhang, J.; Rogers, J.T.; Xie, J.; et al. Assessments of plasma ghrelin levels in the early stages of parkinson’s disease. Mov. Disord. 2017, 32, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.V.D.; Rodrigues, A.L.S.; De Lima, T.C.; de Barioglio, S.R.; Raisman-Vozari, R.; Prediger, R.D. Ghrelin as a Neuroprotective and Palliative Agent in Alzheimer’s and Parkinson’s Disease. Curr. Pharm. Des. 2013, 19, 6773–6790. [Google Scholar] [CrossRef] [PubMed]
- Parashar, A.; Udayabanu, M. Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 1–7. [Google Scholar] [CrossRef]
- Mulak, A. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.O.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.H.; Mackenzie, M.; Huan, T.; Finlay, B.B.; et al. Microbiota Composition and Metabolism Are Associated With Gut Function in Parkinson’s Disease. Mov. Disord. 2020, 35, 1208–1217. [Google Scholar] [CrossRef]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, D.; Teofani, A.; Unida, V.; Cerroni, R.; Biocca, S.; Stefani, A.; Desideri, A. Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach. Brain Sci. 2020, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, D.; Cerroni, R.; Unida, V.; Farcomeni, A.; Pierantozzi, M.; Mercuri, N.B.; Biocca, S.; Stefani, A.; Desideri, A. Dysbiosis of gut microbiota in a selected population of Parkinson’s patients. Park. Relat. Disord. 2019, 65, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.-H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164. [Google Scholar] [CrossRef]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Erratum to: Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 61. [Google Scholar] [CrossRef]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Saanta Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2019, 34, 396–405. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef]
- Dutta, G.; Zhang, P.; Liu, B. The lipopolysaccharide Parkinson’s disease animal model: Mechanistic studies and drug discovery. Fundam. Clin. Pharmacol. 2008, 22, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Genc, S.; Genc, K. The Endotoxin-Induced Neuroinflammation Model of Parkinson’s Disease. Park. Dis. 2011, 2011, 1–25. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. In Advances in Research on Neurodegeneration; Springer: Vienna, Austria, 2000; pp. 277–290. [Google Scholar]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Elfil, M.; Kamel, S.; Kandil, M.; Koo, B.B.; Schaefer, S.M. Implications of the Gut Microbiome in Parkinson’s Disease. Mov. Disord. 2020, 35, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhao, D.; Shah, S.Z.A.; Wu, W.; Lai, M.; Zhang, X.; Li, J.; Guan, Z.; Zhao, H.; Li, W.; et al. The Role of the Gut Microbiota in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2019, 10, 1155. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Hopfner, F.; Künstner, A.; Müller, S.H.; Künzel, S.; Zeuner, K.E.; Margraf, N.G.; Deuschl, G.; Baines, J.F.; Kuhlenbäumer, G. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017, 1667, 41–45. [Google Scholar] [CrossRef]
- Goya, M.E.; Xue, F.; Sampedro-Torres-Quevedo, C.; Arnaouteli, S.; Riquelme-Dominguez, L.; Romanowski, A.; Brydon, J.; Ball, K.L.; Stanley-Wall, N.R.; Doitsidou, M. Probiotic Bacillus subtilis Protects against α-Synuclein Aggregation in C. elegans. Cell Rep. 2020, 30, 367–380.e7. [Google Scholar] [CrossRef]
- Sun, J.; Li, H.; Jin, Y.; Yu, J.; Mao, S.; Su, K.-P.; Ling, Z.; Liu, J. Probiotic Clostridium butyricum ameliorated motor deficits in a mouse model of Parkinson’s disease via gut microbiota-GLP-1 pathway. Brain Behav. Immun. 2021, 91, 703–715. [Google Scholar] [CrossRef]
- Liao, J.-F.; Cheng, Y.-F.; You, S.-T.; Kuo, W.-C.; Huang, C.-W.; Chiou, J.-J.; Hsu, C.-C.; Hsieh-Li, H.-M.; Wang, S.; Tsai, Y.-C. Lactobacillus plantarum PS128 alleviates neurodegenerative progression in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse models of Parkinson’s disease. Brain Behav. Immun. 2020, 90, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Taghizadeh, M.; Kakhaki, R.D.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.S.; Nicholson, M.R.; Khoruts, A.; Kahn, S.A. Fecal Microbiota Transplantation Across the Lifespan: Balancing Efficacy, Safety, and Innovation. Am. J. Gastroenterol. 2023, 118, 435–439. [Google Scholar] [CrossRef]
- Więckowska-Gacek, A.; Mietelska-Porowska, M.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef]
- Hegelmaier, T.; Lebbing, M.; Duscha, A.; Tomaske, L.; Tönges, L.; Holm, J.B.; Nielsen, H.B.; Gatermann, S.G.; Przuntek, H.; Haghikia, A. Interventional Influence of the Intestinal Microbiome Through Dietary Intervention and Bowel Cleansing Might Improve Motor Symptoms in Parkinson’s Disease. Cells 2020, 9, 376. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Jiang, X.-J.; Lu, B.; Gao, Q.; Chen, Y.-F.; Wu, D.-B.; Zeng, W.-Y.; Yang, L.; Li, H.-H.; Yu, B. Roles of Gut Microbiota in Pathogenesis of Alzheimer’s Disease and Therapeutic Effects of Chinese Medicine. Chin. J. Integr. Med. 2022, 28, 1048–1056. [Google Scholar] [CrossRef]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Botchway, B.O.; Okoye, F.C.; Chen, Y.; Arthur, W.E.; Fang, M. Alzheimer Disease: Recent Updates on Apolipoprotein E and Gut Microbiome Mediation of Oxidative Stress, and Prospective Interventional Agents. Aging Dis. 2022, 13, 87–102. [Google Scholar] [CrossRef]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, 71, 2233–2252. [Google Scholar] [CrossRef]
- Askarova, S.; Umbayev, B.; Masoud, A.-R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The Links Between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell. Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Sittipo, P.; Choi, J.; Lee, S.; Lee, Y.K. The function of gut microbiota in immune-related neurological disorders: A review. J. Neuroinflamm. 2022, 19, 154. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-X.; Zeng, M.-X.; Cai, D.; Zhou, H.-H.; Wang, Y.-J.; Liu, Z. Correlation between APOE4 gene and gut microbiota in Alzheimer’s disease. Benef. Microbes 2023, 14, 349–360. [Google Scholar] [CrossRef]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A.; et al. GABA-Modulating Bacteria of the Human Gut Microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef]
- Aaldijk, E.; Vermeiren, Y. The Role of Serotonin within the Microbiota-Gut-Brain Axis in the Development of Alzheimer’s Disease: A Narrative Review. Ageing Res. Rev. 2022, 75, 101556. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, L.; Yang, X. Interaction between Autophagy and the NLRP3 Inflammasome in Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2022, 14, 1018848. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Z.; Fan, B.; Chen, Y.; Zhou, L.; Jiang, B.; Long, H.; Zhong, W.; Li, X.; Li, Y. A Selective NLRP3 Inflammasome Inhibitor Attenuates Behavioral Deficits and Neuroinflammation in a Mouse Model of Parkinson’s Disease. J. Neuroimmunol. 2021, 354, 577543. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef] [PubMed]
- Dhami, M.; Raj, K.; Singh, S. Relevance of gut microbiota to Alzheimer’s Disease (AD): Potential effects of probiotic in management of AD. Aging Health Res. 2023, 3, 100128. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Erratum: Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 46856. [Google Scholar] [CrossRef]
- Kountouras, J.; Doulberis, M.; Papaefthymiou, A.; Polyzos, S.A.; Zavos, C.; Kazakos, E.; Arapoglou, S.; Kyrailidi, F.; Mouratidou, M.C.; Boziki, M.; et al. Controlling the Impact of Helicobacter pylori-Related Hyperhomocysteinemia on Neurodegeneration. Medicina 2023, 59, 504. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- McShane, R.; Westby, M.J.; Roberts, E.; Minakaran, N.; Schneider, L.; Farrimond, L.E.; Maayan, N.; Ware, J.; Debarros, J. Memantine for dementia. Cochrane Database Syst. Rev. 2019, 3, CD003154. [Google Scholar] [CrossRef]
- Berti, V.; Walters, M.; Sterling, J.; Quinn, C.G.; Logue, M.; Andrews, R.; Matthews, D.C.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; et al. Mediterranean diet and 3-year Alzheimer brain biomarker changes in middle-aged adults. Neurology 2018, 90, e1789–e1798. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, S.; Ling, Z.; Wang, F.; Ling, Y.; Gong, T.; Fang, N.; Ye, S.; Si, J.; Liu, J. Fructooligosaccharides Ameliorating Cognitive Deficits and Neurodegeneration in APP/PS1 Transgenic Mice through Modulating Gut Microbiota. J. Agric. Food Chem. 2019, 67, 3006–3017. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Chen, C.; Lin, H.; Cheng, I.H. The beneficial effect of synbiotics consumption on Alzheimer’s disease mouse model via reducing local and systemic inflammation. IUBMB Life 2022, 74, 748–753. [Google Scholar] [CrossRef]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med. Res. 2020, 48, 030006052092593. [Google Scholar] [CrossRef]
- Park, S.-Y.; Seo, G.S. Fecal Microbiota Transplantation: Is It Safe? Clin. Endosc. 2021, 54, 157–160. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kustrimovic, N.; Balkhi, S.; Bilato, G.; Mortara, L. Gut Microbiota and Immune System Dynamics in Parkinson’s and Alzheimer’s Diseases. Int. J. Mol. Sci. 2024, 25, 12164. https://doi.org/10.3390/ijms252212164
Kustrimovic N, Balkhi S, Bilato G, Mortara L. Gut Microbiota and Immune System Dynamics in Parkinson’s and Alzheimer’s Diseases. International Journal of Molecular Sciences. 2024; 25(22):12164. https://doi.org/10.3390/ijms252212164
Chicago/Turabian StyleKustrimovic, Natasa, Sahar Balkhi, Giorgia Bilato, and Lorenzo Mortara. 2024. "Gut Microbiota and Immune System Dynamics in Parkinson’s and Alzheimer’s Diseases" International Journal of Molecular Sciences 25, no. 22: 12164. https://doi.org/10.3390/ijms252212164
APA StyleKustrimovic, N., Balkhi, S., Bilato, G., & Mortara, L. (2024). Gut Microbiota and Immune System Dynamics in Parkinson’s and Alzheimer’s Diseases. International Journal of Molecular Sciences, 25(22), 12164. https://doi.org/10.3390/ijms252212164