
The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies

 ,
,  ,
,
Abstract
1. Cardiac Channelopathies
2. Long QT Syndrome and Short QT Syndrome
3. Brugada Syndrome
3.1. Clinical Background
3.2. Genetic Background
3.3. Pathomechanism
4. Catecholaminergic Polymorphic Ventricular Tachycardia
4.1. Clinical Background
4.2. Pathomechanism
5. Human-Induced Pluripotent Stem Cells (hiPSCs)
6. Human-Induced Pluripotent Stem Cells Derived Cardiomyocytes (hiPSCs-CMs)
7. Human-Induced Pluripotent Stem Cells Derived Cardiomyocytes from Brugada Syndrome Patients
8. Human-Induced Pluripotent Stem-Cells-Derived Cardiomyocytes from Catecholaminergic Polymorphic Ventricular Tachycardia Patients
9. Functional Studies in hiPSC-CMs
10. Patch Clamp
11. Mechanics
12. Multielectrode Array
13. Optical Mapping
14. Impedance
15. Limitations
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yücel, G.; Sattler, K.; Zimmermann, W.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Am. Heart Assoc. 2018, 7, e007394. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, X.; El-Battrawy, I.; Lan, H.; Zhong, R.; Xu, Q.; Huang, M.; Liao, Z.; Lang, S.; Zimmermann, W.-H.; et al. Drug Testing in Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes from a Patient With Short QT Syndrome Type 1. Clin. Pharmacol. Ther. 2019, 106, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Yang, G.; Kowitz, J.; Duru, F.; Saguner, A.M.; Akin, I.; Zhou, X.; El-Battrawy, I. Preclinical Short QT Syndrome Models: Studying the Phenotype and Drug-Screening. EP Eur. 2022, 24, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Doerer, J.J.; Haas, T.S.; Tierney, D.M.; Mueller, F.O. Sudden Deaths in Young Competitive Athletes: Analysis of 1866 Deaths in the United States, 1980–2006. Circulation 2009, 119, 1085–1092. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.-D.; Zimmermann, W.-H.; Cyganek, L.; et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Electrical Dysfunctions in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with an Arrhythmogenic Right Ventricular Cardiomyopathy. Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef] [PubMed]
- Hanses, U.; Kleinsorge, M.; Roos, L.; Yigit, G.; Li, Y.; Barbarics, B.; El-Battrawy, I.; Lan, H.; Tiburcy, M.; Hindmarsh, R.; et al. Intronic CRISPR Repair in a Preclinical Model of Noonan Syndrome-Associated Cardiomyopathy. Circulation 2020, 142, 1059–1076. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Rebs, S.; El-Battrawy, I.; Chasan, S.; Krause, T.; Haas, J.; Zhong, R.; Liao, Z.; Xu, Q.; Zhou, X.; et al. Identification of SCN5a p.C335R Variant in a Large Family with Dilated Cardiomyopathy and Conduction Disease. Int. J. Mol. Sci. 2021, 22, 12990. [Google Scholar] [CrossRef]
- Wang, Y.; Elsherbiny, A.; Kessler, L.; Cordero, J.; Shi, H.; Serke, H.; Lityagina, O.; Trogisch, F.A.; Mohammadi, M.M.; El-Battrawy, I.; et al. Lamin A/C-Dependent Chromatin Architecture Safeguards Naïve Pluripotency to Prevent Aberrant Cardiovascular Cell Fate and Function. Nat. Commun. 2022, 13, 6663. [Google Scholar] [CrossRef]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.-A.; Gummert, J.; Gärtner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. [Google Scholar] [CrossRef]
- Brugada, P.; Brugada, J. Right Bundle Branch Block, Persistent ST Segment Elevation and Sudden Cardiac Death: A Distinct Clinical and Electrocardiographic Syndrome: A Multicenter Report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Vutthikraivit, W.; Rattanawong, P.; Putthapiban, P.; Sukhumthammarat, W.; Vathesatogkit, P.; Ngarmukos, T.; Thakkinstian, A. Worldwide Prevalence of Brugada Syndrome: A Systematic Review and Meta-Analysis. Acta Cardiol. Sin. 2018, 34, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.-X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave Syndromes Expert Consensus Conference Report: Emerging Concepts and Gaps in Knowledge. J. Arrhythmia 2016, 32, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Minier, M.; Probst, V.; Berthome, P.; Tixier, R.; Briand, J.; Geoffroy, O.; Clementy, N.; Mansourati, J.; Jesel, L.; Dupuis, J.-M.; et al. Age at Diagnosis of Brugada Syndrome: Influence on Clinical Characteristics and Risk of Arrhythmia. Heart Rhythm 2020, 17 Pt A, 743–749. [Google Scholar] [CrossRef]
- Probst, V.; Veltmann, C.; Eckardt, L.; Meregalli, P.; Gaita, F.; Tan, H.; Babuty, D.; Sacher, F.; Giustetto, C.; Schulze-Bahr, E.; et al. Long-Term Prognosis of Patients Diagnosed with Brugada Syndrome. Circulation 2010, 121, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Pannone, L.; Osei, R.; Gauthey, A.; Sorgente, A.; Della Rocca, D.G.; Overeinder, I.; Bala, G.; Almorad, A.; Stroker, E.; Sieira, J.; et al. Age and Genetics Are Associated with Ventricular Fibrillation but Not with Monomorphic Ventricular Tachycardia in Brugada Syndrome. Europace 2023, 25 (Suppl. 1), euad122.268. [Google Scholar] [CrossRef]
- Shinohara, T.; Takagi, M.; Kamakura, T.; Sekiguchi, Y.; Yokoyama, Y.; Aihara, N.; Hiraoka, M.; Aonuma, K.; Japan Idiopathic Ventricular Fibrillation Study (J-IVFS) Investigators. Risk Stratification in Asymptomatic Patients with Brugada Syndrome: Utility of Multiple Risk Factor Combination Rather than Programmed Electrical Stimulation. J. Cardiovasc. Electrophysiol. 2021, 32, 507–514. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An International Compendium of Mutations in the SCN5A-Encoded Cardiac Sodium Channel in Patients Referred for Brugada Syndrome Genetic Testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef]
- Nielsen, M.W.; Holst, A.G.; Olesen, S.-P.; Olesen, M.S. The Genetic Component of Brugada Syndrome. Front. Physiol. 2013, 4, 179. [Google Scholar] [CrossRef]
- Smits, J.P.P.; Eckardt, L.; Probst, V.; Bezzina, C.R.; Schott, J.J.; Remme, C.A.; Haverkamp, W.; Breithardt, G.; Escande, D.; Schulze, B.E.; et al. Genotype-Phenotype Relationship in Brugada Syndrome: Electrocardiographic Features Differentiate SCN5A-Related Patients from Non–SCN5A-Related Patients. J. Am. Coll. Cardiol. 2002, 40, 350–356. [Google Scholar] [CrossRef]
- Probst, V.; Wilde, A.A.M.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A Mutations and the Role of Genetic Background in the Pathophysiology of Brugada Syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic Basis and Molecular Mechanism for Idiopathic Ventricular Fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas Martinez, H.; Kahlig, K.; Rajamani, S.; Belardinelli, L.; Pfeiffer, R.; Dezi, F.; Antzelevitch, C. Genetic Variants in SCN10A Associated with Brugada Syndrome, Right Bundle Branch Block and Atrioventricular Block [Abstract]. Heart Rhythm 2012, 9, S395. [Google Scholar]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. Mog1. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.-J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium Channel Β1 Subunit Mutations Associated with Brugada Syndrome and Cardiac Conduction Disease in Humans. J. Clin. Investig. 2008, 118, 2260–2268. [Google Scholar] [CrossRef]
- A Missense Mutation in the Sodium Channel β2 Subunit Reveals SCN2B as a New Candidate Gene for Brugada Syndrome—Riuró—2013—Human Mutation—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/humu.22328 (accessed on 3 October 2024).
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A.; et al. A Mutation in the Β3 Subunit of the Cardiac Sodium Channel Associated with Brugada ECG Phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270–278. [Google Scholar] [CrossRef]
- Molecular and Functional Characterization of Novel Glycerol-3-Phosphate Dehydrogenase 1–Like Gene (GPD1-L) Mutations in Sudden Infant Death Syndrome | Circulation. Available online: https://www.ahajournals.org/doi/full/10.1161/CIRCULATIONAHA.107.704627 (accessed on 3 October 2024).
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.-E.; Nakamura, K.; et al. A Novel Disease Gene for Brugada Syndrome. Circ. Arrhythmia Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; A McDermott, D.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; A MacRae, C.; et al. Mutations in the Desmosomal Protein Plakophilin-2 Are Common in Arrhythmogenic Right Ventricular Cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Buja, G.; Nava, A.; Rossi, L.; Thiene, G. Right Bundle Branch Block, Right Precordial St-Segment Elevation, and Sudden Death in Young People. Circulation 2001, 103, 710–717. [Google Scholar] [CrossRef]
- Epicardial Ventricular Tachycardia Ablation in a Patient with Brugada ECG Pattern and Mutation of PKP2 and DSP Genes | Circulation: Arrhythmia and Electrophysiology. Available online: https://www.ahajournals.org/doi/full/10.1161/CIRCEP.114.002342 (accessed on 30 September 2024).
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-Function Mutations in the Cardiac Calcium Channel Underlie a New Clinical Entity Characterized by ST-Segment Elevation, Short QT Intervals, and Sudden Cardiac Death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular Genetics and Functional Anomalies in a Series of 248 Brugada Cases with 11 Mutations in the TRPM4 Channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Zankov, D.P.; Ding, W.-G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) Variants Are Novel Modulators of Brugada Syndrome and Idiopathic Ventricular Fibrillation. Circ. Arrhythmia Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R.; Schulze-Bahr, E.; Beekman, L.; Bhuiyan, Z.A.; Bertrand, J.; Eckardt, L.; Lin, D.; Borggrefe, M.; Breithardt, G.; et al. Role of Sequence Variations in the Human Ether-a-Go-Go-Related Gene (HERG, KCNH2) in the Brugada Syndrome. Cardiovasc. Res. 2005, 68, 441–453. [Google Scholar] [CrossRef]
- Boczek, N.J.; Ye, D.; Johnson, E.K.; Wang, W.; Crotti, L.; Tester, D.J.; Dagradi, F.; Mizusawa, Y.; Torchio, M.; Alders, M.; et al. Characterization of SEMA3A-Encoded Semaphorin as a Naturally Occurring Kv4.3 Protein Inhibitor and Its Contribution to Brugada Syndrome. Circ. Res. 2014, 115, 460–469. [Google Scholar] [CrossRef]
- Delpón, E.; Cordeiro, J.M.; Núñez, L.; Thomsen, P.E.B.; Guerchicoff, A.; Pollevick, G.D.; Wu, Y.; Kanters, J.K.; Larsen, C.T.; Hofman-Bang, J.; et al. Functional Effects of KCNE3 Mutation and Its Role in the Development of Brugada Syndrome. Circ. Arrhythmia Electrophysiol. 2008, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martínez, H.; Hu, D.; Ferrer, T.; Onetti, C.G.; Wu, Y.; Burashnikov, E.; Boyle, M.; Surman, T.; Urrutia, J.; Veltmann, C.; et al. Molecular Genetic and Functional Association of Brugada and Early Repolarization Syndromes with S422L Missense Mutation in KCNJ8. Heart Rhythm 2012, 9, 548–555. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient Outward Current (Ito) Gain-of-Function Mutations in the KCND3-Encoded Kv4.3 Potassium Channel and Brugada Syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef]
- Biel, S.; Aquila, M.; Hertel, B.; Berthold, A.; Neumann, T.; DiFrancesco, D.; Moroni, A.; Thiel, G.; Kauferstein, S. Mutation in S6 Domain of HCN4 Channel in Patient with Suspected Brugada Syndrome Modifies Channel Function. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 1663–1671. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 Is a Novel Brugada and Early Repolarization Syndrome Susceptibility Gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Aweimer, A.; Mügge, A.; Akin, I.; El-Battrawy, I. [Asymptomatic channelopathies: Risk stratification and primary prophylaxis]. Herzschrittmacherther. Elektrophysiol. 2023, 34, 101–108. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Roterberg, G.; Liebe, V.; Ansari, U.; Lang, S.; Zhou, X.; Borggrefe, M.; Akin, I. Implantable Cardioverter-Defibrillator in Brugada Syndrome: Long-Term Follow-Up. Clin. Cardiol. 2019, 42, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, B. Management of Brugada Syndrome 2016: Should All High Risk Patients Receive an ICD? Circ. Arrhythmia Electrophysiol. 2016, 9, e004185. [Google Scholar] [CrossRef]
- ESC Guidelines on Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Available online: https://www.escardio.org/Guidelines/Clinical-Practice-Guidelines/Ventricular-Arrhythmias-and-the-Prevention-of-Sudden-Cardiac-Death (accessed on 5 September 2023).
- Yan, G.-X.; Antzelevitch, C. Cellular Basis for the Brugada Syndrome and Other Mechanisms of Arrhythmogenesis Associated With ST-Segment Elevation. Circulation 1999, 100, 1660–1666. [Google Scholar] [CrossRef]
- Di Diego, J.M.; Sun, Z.Q.; Antzelevitch, C. I(to) and Action Potential Notch Are Smaller in Left vs. Right Canine Ventricular Epicardium. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H548–H561. [Google Scholar] [CrossRef] [PubMed]
- Transient Outward Current Prominent in Canine Ventricular Epicardium but Not Endocardium | Circulation Research. Available online: https://www.ahajournals.org/doi/abs/10.1161/01.RES.62.1.116 (accessed on 25 September 2023).
- Ionic and Cellular Basis for the Predominance of the Brugada Syndrome Phenotype in Males | Circulation. Available online: https://www.ahajournals.org/doi/full/10.1161/01.CIR.0000032002.22105.7A (accessed on 25 September 2023).
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of Ventricular Fibrillation Episodes in Brugada Syndrome by Catheter Ablation Over the Anterior Right Ventricular Outflow Tract Epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Wilde, A.A.M.; Tan, H.L. Pathophysiological Mechanisms of Brugada Syndrome: Depolarization Disorder, Repolarization Disorder, or More? Cardiovasc. Res. 2005, 67, 367–378. [Google Scholar] [CrossRef]
- Bruns, H.-J.; Eckardt, L.; Vahlhaus, C.; Schulze-Bahr, E.; Haverkamp, W.; Borggrefe, M.; Breithardt, G.; Wichter, T. Body Surface Potential Mapping in Patients with Brugada Syndrome: Right Precordial ST Segment Variations and Reverse Changes in Left Precordial Leads. Cardiovasc. Res. 2002, 54, 58–66. [Google Scholar] [CrossRef]
- Conrath, C.E.; Wilders, R.; Coronel, R.; de Bakker, J.M.T.; Taggart, P.; de Groot, J.R.; Opthof, T. Intercellular Coupling through Gap Junctions Masks M Cells in the Human Heart. Cardiovasc. Res. 2004, 62, 407–414. [Google Scholar] [CrossRef]
- Marx, A.; Lange, B.; Nalenz, C.; Hoffmann, B.; Rostock, T.; Konrad, T. A 35-Year Effective Treatment of Catecholaminergic Polymorphic Ventricular Tachycardia with Propafenone. Heart Rhythm Case Rep. 2018, 5, 74–77. [Google Scholar] [CrossRef]
- Kallas, D.; Lamba, A.; Roston, T.M.; Arslanova, A.; Franciosi, S.; Tibbits, G.F.; Sanatani, S. Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist. Int. J. Mol. Sci. 2021, 22, 9293. [Google Scholar] [CrossRef] [PubMed]
- Olubando, D.; Hopton, C.; Eden, J.; Caswell, R.; Lowri Thomas, N.; Roberts, S.A.; Morris-Rosendahl, D.; Venetucci, L.; Newman, W.G. Classification and Correlation of RYR2 Missense Variants in Individuals with Catecholaminergic Polymorphic Ventricular Tachycardia Reveals Phenotypic Relationships. J. Hum. Genet. 2020, 65, 531–539. [Google Scholar] [CrossRef]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.-M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.A.M.; Guicheney, P.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia: RYR2 Mutations, Bradycardia, and Follow up of the Patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef]
- Abbas, M.; Miles, C.; Behr, E. Catecholaminergic Polymorphic Ventricular Tachycardia. Arrhythmia Electrophysiol. Rev. 2022, 11, e20. [Google Scholar] [CrossRef]
- Singh, M.; Morin, D.P.; Link, M.S. Sudden Cardiac Death in Long QT Syndrome (LQTS), Brugada Syndrome, and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT). Prog. Cardiovasc. Dis. 2019, 62, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bergeman, A.T.; Wilde, A.A.M.; van der Werf, C. Catecholaminergic Polymorphic Ventricular Tachycardia: A Review of Therapeutic Strategies. Card. Electrophysiol. Clin. 2023, 15, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Capes, E.; Loaiza, R.; Valdivia, H.H. Ryanodine Receptors. Skelet. Muscle 2011, 1, 18. [Google Scholar] [CrossRef]
- Priori, S.G.; Mazzanti, A.; Santiago, D.J.; Kukavica, D.; Trancuccio, A.; Kovacic, J.C. Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia: JACC Focus Seminar 5/5. J. Am. Coll. Cardiol. 2021, 77, 2592–2612. [Google Scholar] [CrossRef]
- Hou, C.; Jiang, X.; Zhang, Y.; Xu, M.; Sun, X.; Jia, J.; Li, Y.; Zhao, Y.; Xie, L.; Xiao, T. A de Novo Heterozygous Cardiac Ryanodine Receptor Gene (RYR2) Mutation in a Catecholaminergic Polymorphic Ventricular Tachycardia Patient. Gene Rep. 2019, 16, 100439. [Google Scholar] [CrossRef]
- Terentyev, D.; Hamilton, S. Regulation of Sarcoplasmic Reticulum Ca2+ Release by Serine-Threonine Phosphatases in the Heart. J. Mol. Cell Cardiol. 2016, 101, 156–164. [Google Scholar] [CrossRef]
- Zhao, Y.-T.; Valdivia, C.R.; Gurrola, G.B.; Powers, P.P.; Willis, B.C.; Moss, R.L.; Jalife, J.; Valdivia, H.H. Arrhythmogenesis in a Catecholaminergic Polymorphic Ventricular Tachycardia Mutation That Depresses Ryanodine Receptor Function. Proc. Natl. Acad. Sci. USA 2015, 112, E1669–E1677. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; Amenta, S.; Feilotter, H.; Nannenberg, E.A.; Mazzarotto, F.; et al. Evaluation of Gene Validity for CPVT and Short QT Syndrome in Sudden Arrhythmic Death. Eur. Heart J. 2022, 43, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.-A.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a New Life-Threatening Inherited Arrhythmia Gene Associated with Overlapping Clinical Features of Both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef]
- Deb, A.; Tow, B.D.; Qing, Y.; Walker, M.; Hodges, E.R.; Stewart, J.A.; Knollmann, B.C.; Zheng, Y.; Wang, Y.; Liu, B. Genetic Inhibition of Mitochondrial Permeability Transition Pore Exacerbates Ryanodine Receptor 2 Dysfunction in Arrhythmic Disease. Cells 2023, 12, 204. [Google Scholar] [CrossRef]
- Gonano, L.A.; Jones, P.P. FK506-Binding Proteins 12 and 12.6 (FKBPs) as Regulators of Cardiac Ryanodine Receptors: Insights from New Functional and Structural Knowledge. Channels 2017, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Xiao, B.; Yang, D.; Wang, R.; Choi, P.; Zhang, L.; Cheng, H.; Chen, S.R.W. RyR2 Mutations Linked to Ventricular Tachycardia and Sudden Death Reduce the Threshold for Store-Overload-Induced Ca2+ Release (SOICR). Proc. Natl. Acad. Sci. USA 2004, 101, 13062–13067. [Google Scholar] [CrossRef]
- Sumitomo, N. Current Topics in Catecholaminergic Polymorphic Ventricular Tachycardia. J. Arrhythm. 2016, 32, 344–351. [Google Scholar] [CrossRef]
- Pappone, C.; Negro, G.; Ciconte, G. Ventricular Fibrillation Ablation in Cardiomyopathies and Arrhythmic Storm. Eur. Heart J. Suppl. 2021, 23 (Suppl. E), E112–E117. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Apostolopoulos, E.J.; Apostolaki, N.E.; Melita, H.; Manolis, A.S. The Role of the Autonomic Nervous System in Cardiac Arrhythmias: The Neuro-Cardiac Axis, More Foe than Friend? Trends Cardiovasc. Med. 2021, 31, 290–302. [Google Scholar] [CrossRef]
- Kashimura, T.; Briston, S.J.; Trafford, A.W.; Napolitano, C.; Priori, S.G.; Eisner, D.A.; Venetucci, L.A. In the RyR2(R4496C) Mouse Model of CPVT, β-Adrenergic Stimulation Induces Ca Waves by Increasing SR Ca Content and Not by Decreasing the Threshold for Ca Waves. Circ. Res. 2010, 107, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-Dependent Protein Kinase II: Linking Heart Failure and Arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythmia Electrophysiol. 2016, 9, e004161. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A Cardiac Arrhythmia Syndrome Caused by Loss of Ankyrin-B Function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef]
- Boczek, N.J.; Will, M.L.; Loporcaro, C.G.; Tester, D.J.; Ackerman, M.J. Abstract 14699: Spectrum and Prevalence of CALM1, CALM2, and CALM3 Mutations in Long QT Syndrome, Catecholaminergic Polymorphic Ventricular Tachycardia, Idiopathic Ventricular Fibrillation, and Sudden Unexplained Death in the Young. Circulation 2013, 128 (Suppl. 22), A14699. [Google Scholar]
- Chopra, N.; Yang, T.; Asghari, P.; Moore, E.D.; Huke, S.; Akin, B.; Cattolica, R.A.; Perez, C.F.; Hlaing, T.; Knollmann-Ritschel, B.E.C.; et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation–contraction coupling, and cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 2009, 106, 7636–7641. [Google Scholar] [CrossRef] [PubMed]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A Missense Mutation in a Highly Conserved Region of CASQ2 Is Associated with Autosomal Recessive Catecholamine-Induced Polymorphic Ventricular Tachycardia in Bedouin Families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef]
- Postma, A.V.; Bhuiyan, Z.A.; Shkolnikova, M.A. Involvement of the Kir2 Gene Family in Catecholaminergic Polymorphic Ventricular Tachycardia; Analysis for Mutations and Identification of Numerous Pseudogenes. Eur. Heart J. 2004, 25, 66. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.-H.; Gepstein, L. Cardiomyocyte Differentiation of Human Induced Pluripotent Stem Cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef]
- Generation of Integration-Free Induced Pluripotent Stem Cells from Urine-Derived Cells Isolated from Individuals with Down Syndrome | Stem Cells Translational Medicine | Oxford Academic. Available online: https://academic.oup.com/stcltm/article/6/6/1465/6398814 (accessed on 11 September 2023).
- Ye, L.; Muench, M.O.; Fusaki, N.; Beyer, A.I.; Wang, J.; Qi, Z.; Yu, J.; Kan, Y.W. Blood Cell-Derived Induced Pluripotent Stem Cells Free of Reprogramming Factors Generated by Sendai Viral Vectors. Stem Cells Transl. Med. 2013, 2, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Schimanski, T.; Zhang, F.; Lan, H.; Hohn, A.; Xu, Q.; Huang, M.; Liao, Z.; Qiao, L.; Yang, Z.; et al. A Preclinical Study on Brugada Syndrome with a CACNB2 Variant Using Human Cardiomyocytes from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2022, 23, 8313. [Google Scholar] [CrossRef] [PubMed]
- Larribere, L.; Wu, H.; Novak, D.; Galach, M.; Bernhardt, M.; Orouji, E.; Weina, K.; Knappe, N.; Sachpekidis, C.; Umansky, L.; et al. NF1 Loss Induces Senescence during Human Melanocyte Differentiation in an iPSC-Based Model. Pigment. Cell Melanoma Res. 2015, 28, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Yücel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.-H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides Induced Inflammatory Responses and Electrophysiological Dysfunctions in Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Zhao, Z.; Lan, H.; El-Battrawy, I.; Li, X.; Buljubasic, F.; Sattler, K.; Yücel, G.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Ion Channel Expression and Characterization in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Int. 2018, 2018, 6067096. [Google Scholar] [CrossRef]
- Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. Available online: https://arpi.unipi.it/handle/11568/1029349 (accessed on 11 September 2023).
- Hubbard, J.J.; Sullivan, S.K.; Mills, J.A.; Hayes, B.J.; Torok-Storb, B.J.; Ramakrishnan, A. Efficient iPS Cell Generation from Blood Using Episomes and HDAC Inhibitors. J. Vis. Exp. 2014, 52009. [Google Scholar] [CrossRef]
- Generation of Human Induced Pluripotent Stem Cells (hiPSCs) from Fibroblasts Using Episomal Vectors—DE. Available online: https://www.thermofisher.com/de/de/home/references/protocols/cell-culture/stem-cell-protocols/ipsc-protocols/generation-human-induced-pluripotent-stem-cells-fibroblasts.html (accessed on 30 September 2023).
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient Induction of Transgene-Free Human Pluripotent Stem Cells Using a Vector Based on Sendai Virus, an RNA Virus That Does Not Integrate into the Host Genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. iPSC Reprogramming from Human Peripheral Blood Using Sendai Virus Mediated Gene Transfer. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2014. [Google Scholar] [CrossRef]
- Patel, A.; Garcia Diaz, A.; Moore, J.C.; Sirabella, D.; Corneo, B. Establishment and Characterization of Two iPSC Lines Derived from Healthy Controls. Stem Cell Res. 2020, 47, 101926. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A Cellular Model of Brugada Syndrome with SCN10A Variants Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Europace 2019, 21, 1410–1421. [Google Scholar] [CrossRef]
- Buljubasic, F.; El-Battrawy, I.; Lan, H.; Lomada, S.K.; Chatterjee, A.; Zhao, Z.; Li, X.; Zhong, R.; Xu, Q.; Huang, M.; et al. Nucleoside Diphosphate Kinase B Contributes to Arrhythmogenesis in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Med. 2020, 9, 486. [Google Scholar] [CrossRef]
- Warren, L.; Lin, C. mRNA-Based Genetic Reprogramming. Mol. Ther. 2019, 27, 729–734. [Google Scholar] [CrossRef]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence: Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of Catecholaminergic Polymorphic Ventricular Tachycardia with Patient-Specific Human-Induced Pluripotent Stem Cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Kong, C.-W.; Ren, L.; Karakikes, I.; Geng, L.; He, J.; Chow, M.Z.Y.; Mok, C.F.; Chan, H.Y.S.; Webb, S.E.; et al. A Simple, Cost-Effective but Highly Efficient System for Deriving Ventricular Cardiomyocytes from Human Pluripotent Stem Cells. Stem Cells Dev. 2014, 23, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-Dimensional Control of Wnt Pathway for hiPSC-CMs Differentiation. Sci. Rep. 2019, 9, 19389. [Google Scholar] [CrossRef] [PubMed]
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.-H.; et al. Deep Phenotyping of Human Induced Pluripotent Stem Cell–Derived Atrial and Ventricular Cardiomyocytes. JCI Insight 2018, 3, e99941. [Google Scholar] [CrossRef]
- Ma, D.; Liu, Z.; Loh, L.J.; Zhao, Y.; Li, G.; Liew, R.; Islam, O.; Wu, J.; Chung, Y.Y.; Teo, W.S.; et al. Identification of an INa-Dependent and Ito-Mediated Proarrhythmic Mechanism in Cardiomyocytes Derived from Pluripotent Stem Cells of a Brugada Syndrome Patient. Sci. Rep. 2018, 8, 11246. [Google Scholar] [CrossRef]
- Pang, L.; Sager, P.; Yang, X.; Shi, H.; Sannajust, F.; Brock, M.; Wu, J.C.; Abi-Gerges, N.; Lyn-Cook, B.; Berridge, B.R.; et al. Workshop Report. Circ. Res. 2019, 125, 855–867. [Google Scholar] [CrossRef]
- Lo Sardo, V.; Ferguson, W.; Erikson, G.A.; Topol, E.J.; Baldwin, K.K.; Torkamani, A. Influence of Donor Age on Induced Pluripotent Stem Cells. Nat. Biotechnol. 2017, 35, 69–74. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the Adult Human Heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Gentile, C. Cardiac Spheroids as in Vitro Bioengineered Heart Tissues to Study Human Heart Pathophysiology. J. Vis. Exp. 2021, e61962. [Google Scholar] [CrossRef]
- Ergir, E.; Oliver-De La Cruz, J.; Fernandes, S.; Cassani, M.; Niro, F.; Pereira-Sousa, D.; Vrbský, J.; Vinarský, V.; Perestrelo, A.R.; Debellis, D.; et al. Generation and Maturation of Human iPSC-Derived 3D Organotypic Cardiac Microtissues in Long-Term Culture. Sci. Rep. 2022, 12, 17409. [Google Scholar] [CrossRef]
- Beauchamp, P.; Moritz, W.; Kelm, J.M.; Ullrich, N.D.; Agarkova, I.; Anson, B.D.; Suter, T.M.; Zuppinger, C. Development and Characterization of a Scaffold-Free 3D Spheroid Model of Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes. Tissue Eng. Part C Methods 2015, 21, 852–861. [Google Scholar] [CrossRef]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; Van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-Cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879.e11. [Google Scholar] [CrossRef]
- Fong, A.H.; Romero-López, M.; Heylman, C.M.; Keating, M.; Tran, D.; Sobrino, A.; Tran, A.Q.; Pham, H.H.; Fimbres, C.; Gershon, P.D.; et al. Three-Dimensional Adult Cardiac Extracellular Matrix Promotes Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Tissue Eng. Part A 2016, 22, 1016–1025. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Chen, Y.; Yan, Q.; Li, X.; Ding, L.; Wei, T.; Zeng, D. Three-Dimensional Poly-(ε-Caprolactone) Nanofibrous Scaffolds Promote the Maturation of Human Pluripotent Stem Cells-Induced Cardiomyocytes. Front. Cell Dev. Biol. 2022, 10, 875278. [Google Scholar] [CrossRef]
- Chen, Y.; Chan, J.P.Y.; Wu, J.; Li, R.-K.; Santerre, J.P. Compatibility and Function of Human Induced Pluripotent Stem Cell Derived Cardiomyocytes on an Electrospun Nanofibrous Scaffold, Generated from an Ionomeric Polyurethane Composite. J. Biomed. Mater. Res. Part A 2022, 110, 1932–1943. [Google Scholar] [CrossRef]
- Qin, J.; van Mil, A.; Sluijter, J.P.G. Biophysical Stretch Induced Differentiation and Maturation of Induced Pluripotent Stem Cell-Derived Cardiomyocytes. In Cardiac Mechanobiology in Physiology and Disease; Hecker, M., Duncker, D.J., Eds.; Cardiac and Vascular Biology; Springer International Publishing: Cham, Switzerland, 2023; pp. 141–179. [Google Scholar] [CrossRef]
- Hong, Y.; Zhao, Y.; Li, H.; Yang, Y.; Chen, M.; Wang, X.; Luo, M.; Wang, K. Engineering the Maturation of Stem Cell-Derived Cardiomyocytes. Front. Bioeng. Biotechnol. 2023, 11, 1155052. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Matamoros, M.; Alfayate, S.; Nieto-Marín, P.; Utrilla, R.G.; Tinaquero, D.; de Andrés, R.; Crespo, T.; Ponce-Balbuena, D.; Willis, B.C.; et al. Brugada Syndrome Trafficking–Defective Nav1.5 Channels Can Trap Cardiac Kir2.1/2.2 Channels. JCI Insight 2018, 3, e96291. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Casini, S.; van den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes Derived from Pluripotent Stem Cells Recapitulate Electrophysiological Characteristics of an Overlap Syndrome of Cardiac Sodium Channel Disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, G.; Veerman, C.C.; Casini, S.; Verkerk, A.O.; van de Pas, S.; Bellin, M.; Wilde, A.A.M.; Mummery, C.L.; Bezzina, C.R. Readthrough-Promoting Drugs Gentamicin and PTC124 Fail to Rescue Nav1.5 Function of Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes Carrying Nonsense Mutations in the Sodium Channel Gene SCN5A. Circ. Arrhythmia Electrophysiol. 2016, 9, e004227. [Google Scholar] [CrossRef]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Termglichan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell–Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- Selga, E.; Sendfeld, F.; Martinez-Moreno, R.; Medine, C.N.; Tura-Ceide, O.; Wilmut, S.I.; Pérez, G.J.; Scornik, F.S.; Brugada, R.; Mills, N.L. Sodium Channel Current Loss of Function in Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Brugada Syndrome Patient. J. Mol. Cell. Cardiol. 2018, 114, 10–19. [Google Scholar] [CrossRef]
- de la Roche, J.; Angsutararux, P.; Kempf, H.; Janan, M.; Bolesani, E.; Thiemann, S.; Wojciechowski, D.; Coffee, M.; Franke, A.; Schwanke, K.; et al. Comparing Human iPSC-Cardiomyocytes versus HEK293T Cells Unveils Disease-Causing Effects of Brugada Mutation A735V of NaV1.5 Sodium Channels. Sci. Rep. 2019, 9, 11173. [Google Scholar] [CrossRef] [PubMed]
- Embryonic Type Na+ Channel β-Subunit, SCN3B Masks the Disease Phenotype of BRUGADA Syndrome | Scientific Reports. Available online: https://www.nature.com/articles/srep34198 (accessed on 8 October 2024).
- El-Battrawy, I.; Müller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome with an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Zhang, F.; Yang, Z.; Li, Y.; Xu, Q.; Lan, H.; Cyganek, L.; El-Battrawy, I.; Zhou, X.; Akin, I.; et al. Epigenetic Mechanism of L-Type Calcium Channel β-Subunit Downregulation in Short QT Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes with CACNB2 Mutation. Europace 2022, 24, 2028–2036. [Google Scholar] [CrossRef]
- Belbachir, N.; Portero, V.; Al Sayed, Z.R.; Gourraud, J.-B.; Dilasser, F.; Jesel, L.; Guo, H.; Wu, H.; Gaborit, N.; Guilluy, C.; et al. RRAD Mutation Causes Electrical and Cytoskeletal Defects in Cardiomyocytes Derived from a Familial Case of Brugada Syndrome. Eur. Heart J. 2019, 40, 3081–3094. [Google Scholar] [CrossRef]
- Miller, D.C.; Harmer, S.C.; Poliandri, A.; Nobles, M.; Edwards, E.C.; Ware, J.S.; Sharp, T.V.; McKay, T.R.; Dunkel, L.; Lambiase, P.D.; et al. Ajmaline Blocks INa and IKr without Eliciting Differences between Brugada Syndrome Patient and Control Human Pluripotent Stem Cell-Derived Cardiac Clusters. Stem Cell Res. 2017, 25, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Guan, K.; Stauske, M.; Barc, J.; Tan, H.L.; Wilde, A.A.M.; Verkerk, A.O.; Bezzina, C.R. hiPSC-Derived Cardiomyocytes from Brugada Syndrome Patients without Identified Mutations Do Not Exhibit Clear Cellular Electrophysiological Abnormalities. Sci. Rep. 2016, 6, 30967. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Stauske, M.; Luo, X.; Wagner, S.; Vollrath, M.; Mehnert, C.S.; Schubert, M.; Cyganek, L.; Chen, S.; Hasheminasab, S.-M.; et al. Disease Phenotypes and Mechanisms of iPSC-Derived Cardiomyocytes from Brugada Syndrome Patients with a Loss-of-Function SCN5A Mutation. Front. Cell Dev. Biol. 2020, 8, 592893. [Google Scholar] [CrossRef]
- Simons, E.; Nijak, A.; Loeys, B.; Alaerts, M. Generation of Two Induced Pluripotent Stem Cell (iPSC) Lines (BBANTWi006-A, BBANTWi007-A) from Brugada Syndrome Patients Carrying an SCN5A Mutation. Stem Cell Res. 2022, 60, 102719. [Google Scholar] [CrossRef]
- Kashiwa, A.; Makiyama, T.; Kohjitani, H.; Maurissen, T.L.; Ishikawa, T.; Yamamoto, Y.; Wuriyanghai, Y.; Gao, J.; Huang, H.; Imamura, T.; et al. Disrupted CaV1.2 Selectivity Causes Overlapping Long QT and Brugada Syndrome Phenotypes in the CACNA1C-E1115K iPS Cell Model. Heart Rhythm 2023, 20, 89–99. [Google Scholar] [CrossRef]
- Li, Y.; Dinkel, H.; Pakalniskyte, D.; Busley, A.V.; Cyganek, L.; Zhong, R.; Zhang, F.; Xu, Q.; Maywald, L.; Aweimer, A.; et al. Novel Insights in the Pathomechanism of Brugada Syndrome and Fever-Related Type 1 ECG Changes in a Preclinical Study Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Clin. Transl. Med. 2023, 13, e1130. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Wang, X.; Sun, Y.; Fan, H.; Zhou, J.; Yang, Z.; Qiu, H.; Wang, J.; Su, J.; Gong, T.; et al. Patient-Specific iPSC-Derived Cardiomyocytes Reveal Aberrant Activation of Wnt/β-Catenin Signaling in SCN5A-Related Brugada Syndrome. Stem Cell Res. Ther. 2023, 14, 241. [Google Scholar] [CrossRef]
- Kamga, M.V.K.; Reppel, M.; Hescheler, J.; Nguemo, F. Modeling Genetic Cardiac Channelopathies Using Induced Pluripotent Stem Cells—Status Quo from an Electrophysiological Perspective. Biochem. Pharmacol. 2021, 192, 114746. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martinez, H.; Smith, M.; Hu, D.; Goodrow, R.J.; Puleo, C.; Hasdemir, C.; Antzelevitch, C.; Pfeiffer, R.; Treat, J.A.; Cordeiro, J.M. Susceptibility to Ventricular Arrhythmias Resulting from Mutations in FKBP1B, PXDNL, and SCN9A Evaluated in hiPSC Cardiomyocytes. Stem Cells Int. 2020, 2020, 8842398. [Google Scholar] [CrossRef]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug Screening Using a Library of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Reveals Disease-Specific Patterns of Cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Cui, C.; Qin, H.; Chen, H.; Chen, S.; Lin, Y.; Cheng, H.; Jiang, X.; Chen, M. Pathogenesis and Drug Response of iPSC-Derived Cardiomyocytes from Two Brugada Syndrome Patients with Different Nav1.5-Subunit Mutations. J. Biomed. Res. 2021, 35, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Marín, P.; Tinaquero, D.; Utrilla, R.G.; Cebrián, J.; González-Guerra, A.; Crespo-García, T.; Cámara-Checa, A.; Rubio-Alarcón, M.; Dago, M.; Alfayate, S.; et al. Tbx5 Variants Disrupt Nav1.5 Function Differently in Patients Diagnosed with Brugada or Long QT Syndrome. Cardiovasc. Res. 2022, 118, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Li, W.; Henze, S.; Luo, X.; Ulbricht, Y.; Richter, A.; Di Donato, N.; Wilde, A.A.M.; Guan, K. Generation of iPSC Lines from CPVT Patient Carrying Heterozygous Mutation p.A2254V in the Ryanodine Receptor 2 Gene. Stem Cell Res. 2021, 53, 102259. [Google Scholar] [CrossRef]
- Jung, C.B.; Moretti, A.; Schnitzler, M.M.Y.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene Rescues Arrhythmogenic RYR2 Defect in a Patient-Specific Stem Cell Model of Catecholaminergic Polymorphic Ventricular Tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef]
- Pölönen, R.P.; Swan, H.; Aalto-Setälä, K. Mutation-Specific Differences in Arrhythmias and Drug Responses in CPVT Patients: Simultaneous Patch Clamp and Video Imaging of iPSC Derived Cardiomyocytes. Mol. Biol. Rep. 2020, 47, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Preininger, M.K.; Jha, R.; Maxwell, J.T.; Wu, Q.; Singh, M.; Wang, B.; Dalal, A.; Mceachin, Z.T.; Rossoll, W.; Hales, C.M.; et al. A Human Pluripotent Stem Cell Model of Catecholaminergic Polymorphic Ventricular Tachycardia Recapitulates Patient-Specific Drug Responses. Dis. Models Mech. 2016, 9, 927–939. [Google Scholar] [CrossRef]
- Novak, A.; Barad, L.; Lorber, A.; Gherghiceanu, M.; Reiter, I.; Eisen, B.; Eldor, L.; Itskovitz-Eldor, J.; Eldar, M.; Arad, M.; et al. Functional Abnormalities in iPSC-Derived Cardiomyocytes Generated from CPVT1 and CPVT2 Patients Carrying Ryanodine or Calsequestrin Mutations. J. Cell. Mol. Med. 2015, 19, 2006–2018. [Google Scholar] [CrossRef]
- Gao, J.; Makiyama, T.; Yamamoto, Y.; Kobayashi, T.; Aoki, H.; Maurissen, T.L.; Wuriyanghai, Y.; Kashiwa, A.; Imamura, T.; Aizawa, T.; et al. Novel Calmodulin Variant p.E46K Associated with Severe Catecholaminergic Polymorphic Ventricular Tachycardia Produces Robust Arrhythmogenicity in Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Arrhythmia Electrophysiol. 2023, 16, e011387. [Google Scholar] [CrossRef]
- Maizels, L.; Huber, I.; Arbel, G.; Tijsen, A.J.; Gepstein, A.; Khoury, A.; Gepstein, L. Patient-Specific Drug Screening Using a Human Induced Pluripotent Stem Cell Model of Catecholaminergic Polymorphic Ventricular Tachycardia Type 2. Circ. Arrhythmia Electrophysiol. 2017, 10, e004725. [Google Scholar] [CrossRef]
- Nijak, A.; Saenen, J.; Labro, A.J.; Schepers, D.; Loeys, B.L.; Alaerts, M. iPSC-Cardiomyocyte Models of Brugada Syndrome—Achievements, Challenges and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 2825. [Google Scholar] [CrossRef] [PubMed]
- Mansor, M.A.; Ahmad, M.R. Single Cell Electrical Characterization Techniques. Int. J. Mol. Sci. 2015, 16, 12686–12712. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Luo, X.; Ulbricht, Y.; Wagner, M.; Piorkowski, C.; El-Armouche, A.; Guan, K. Establishment of an Automated Patch-Clamp Platform for Electrophysiological and Pharmacological Evaluation of hiPSC-CMs. Stem Cell Res. 2019, 41, 101662. [Google Scholar] [CrossRef]
- Forced Aggregation and Defined Factors Allow Highly Uniform-Sized Embryoid Bodies and Functional Cardiomyocytes from Human Embryonic and Induced Pluripotent Stem Cells | Heart and Vessels. Available online: https://link.springer.com/article/10.1007/s00380-013-0436-9 (accessed on 30 September 2024).
- Pesl, M.; Pribyl, J.; Acimovic, I.; Vilotic, A.; Jelinkova, S.; Salykin, A.; Lacampagne, A.; Dvorak, P.; Meli, A.C.; Skladal, P.; et al. Atomic Force Microscopy Combined with Human Pluripotent Stem Cell Derived Cardiomyocytes for Biomechanical Sensing. Biosens. Bioelectron. 2016, 85, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Pardon, G.; Vander Roest, A.S.; Chirikian, O.; Birnbaum, F.; Lewis, H.; Castillo, E.A.; Wilson, R.; Denisin, A.K.; Blair, C.A.; Holbrook, C.; et al. Tracking Single hiPSC-Derived Cardiomyocyte Contractile Function Using CONTRAX an Efficient Pipeline for Traction Force Measurement. Nat. Commun. 2024, 15, 5427. [Google Scholar] [CrossRef]
- Miccoli, B.; Lopez, C.M.; Goikoetxea, E.; Putzeys, J.; Sekeri, M.; Krylychkina, O.; Chang, S.-W.; Firrincieli, A.; Andrei, A.; Reumers, V.; et al. High-Density Electrical Recording and Impedance Imaging with a Multi-Modal CMOS Multi-Electrode Array Chip. Front. Neurosci. 2019, 13, 641. [Google Scholar] [CrossRef]
- Kussauer, S.; David, R.; Lemcke, H. hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells 2019, 8, 1331. [Google Scholar] [CrossRef]
- Natarajan, A.; Stancescu, M.; Dhir, V.; Armstrong, C.; Sommerhage, F.; Hickman, J.J.; Molnar, P. Patterned Cardiomyocytes on Microelectrode Arrays as a Functional, High Information Content Drug Screening Platform. Biomaterials 2011, 32, 4267–4274. [Google Scholar] [CrossRef]
- Zwartsen, A.; de Korte, T.; Nacken, P.; de Lange, D.W.; Westerink, R.H.S.; Hondebrink, L. Cardiotoxicity Screening of Illicit Drugs and New Psychoactive Substances (NPS) in Human iPSC-Derived Cardiomyocytes Using Microelectrode Array (MEA) Recordings. J. Mol. Cell. Cardiol. 2019, 136, 102–112. [Google Scholar] [CrossRef]
- Detection of Drug-Induced Torsades de Pointes Arrhythmia Mechanisms Using hiPSC-CM Syncytial Monolayers in a High-Throughput Screening Voltage Sensitive Dye Assay | Toxicological Sciences | Oxford Academic. Available online: https://academic.oup.com/toxsci/article/173/2/402/5640507?login=false (accessed on 11 September 2023).
- Slotvitsky, M.; Tsvelaya, V.; Frolova, S.; Dementyeva, E.; Agladze, K. Arrhythmogenicity Test Based on a Human-Induced Pluripotent Stem Cell (iPSC)-Derived Cardiomyocyte Layer. Toxicol. Sci. 2019, 168, 70–77. [Google Scholar] [CrossRef]
- Juang, J.-M.J.; Binda, A.; Lee, S.-J.; Hwang, J.-J.; Chen, W.-J.; Liu, Y.-B.; Lin, L.-Y.; Yu, C.-C.; Ho, L.-T.; Huang, H.-C.; et al. GSTM3 Variant Is a Novel Genetic Modifier in Brugada Syndrome, a Disease with Risk of Sudden Cardiac Death. eBioMedicine 2020, 57, 102843. [Google Scholar] [CrossRef]
- Shiti, A.; Arbil, G.; Shaheen, N.; Huber, I.; Setter, N.; Gepstein, L. Utilizing Human Induced Pluripotent Stem Cells to Study Atrial Arrhythmias in the Short QT Syndrome. J. Mol. Cell. Cardiol. 2023, 183, 42–53. [Google Scholar] [CrossRef]
- Lamore, S.D.; Scott, C.W.; Peters, M.F. Cardiomyocyte Impedance Assay; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Dias, T.P.; Pinto, S.N.; Santos, J.I.; Fernandes, T.G.; Fernandes, F.; Diogo, M.M.; Prieto, M.; Cabral, J.M.S. Biophysical Study of Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Structural Maturation during Long-Term Culture. Biochem. Biophys. Res. Commun. 2018, 499, 611–617. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Liu, Z.; Alimohamadi, S.; Yin, C.; Liu, J.; Qian, L. Comparative Gene Expression Analyses Reveal Distinct Molecular Signatures between Differentially Reprogrammed Cardiomyocytes. Cell Rep. 2017, 20, 3014–3024. [Google Scholar] [CrossRef]
- Mummery, C.L. Perspectives on the Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Biomedical Research. Stem Cell Rep. 2018, 11, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic Memory in Induced Pluripotent Stem Cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Kosmidis, G.; Mummery, C.L.; Casini, S.; Verkerk, A.O.; Bellin, M. Immaturity of Human Stem-Cell-Derived Cardiomyocytes in Culture: Fatal Flaw or Soluble Problem? Stem Cells Dev. 2015, 24, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.C.; Karakikes, I.; Serrao, G.W.; Backeris, P.; Lee, J.-J.; Xie, C.; Senyei, G.; Gordon, R.E.; Li, R.A.; Akar, F.G.; et al. Advancing Functional Engineered Cardiac Tissues toward a Preclinical Model of Human Myocardium. FASEB J. 2014, 28, 644–654. [Google Scholar] [CrossRef]
- Shaheen: Human Induced Pluripotent Stem Cell-Derived...—Google Scholar. Available online: https://scholar.google.com/scholar_lookup?title=human%20induced%20pluripotent%20stem%20cell-derived%20cardiac%20cell%20sheets%20expressing%20genetically%20encoded%20voltage%20indicator%20for%20pharmacological%20and%20arrhythmia%20studies&author=N%20Shaheen&author=A%20Shiti&author=I%20Huber&author=R%20Shinnawi&author=G%20Arbel&author=A%20Gepstein&author=N%20Setter&author=I%20Goldfracht&author=A%20Gruber&author=SV%20Chorna&author=L.%20Gepstein&publication_year=2018&journal=Stem%20Cell%20Rep&volume=10&pages=1879-1894 (accessed on 10 September 2023).
- Personalised Organs-on-Chips: Functional Testing for Precision Medicine—Lab on a Chip (RSC Publishing). Available online: https://pubs.rsc.org/en/content/articlehtml/2019/lc/c8lc00827b (accessed on 10 September 2023). [CrossRef]
- Eder, A.; Vollert, I.; Hansen, A.; Eschenhagen, T. Human Engineered Heart Tissue as a Model System for Drug Testing. Adv. Drug Deliv. Rev. 2016, 96, 214–224. [Google Scholar] [CrossRef]
- Perbellini, F.; Thum, T. Living Myocardial Slices: A Novel Multicellular Model for Cardiac Translational Research. Eur. Heart J. 2020, 41, 2405–2408. [Google Scholar] [CrossRef]
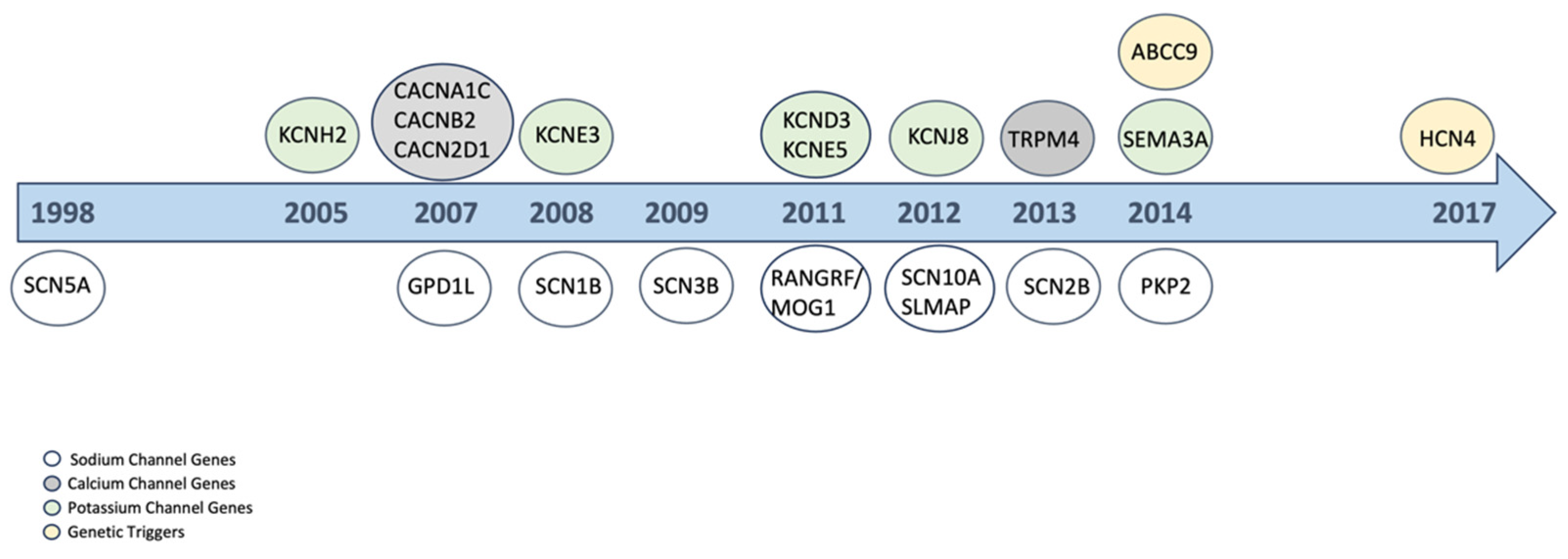
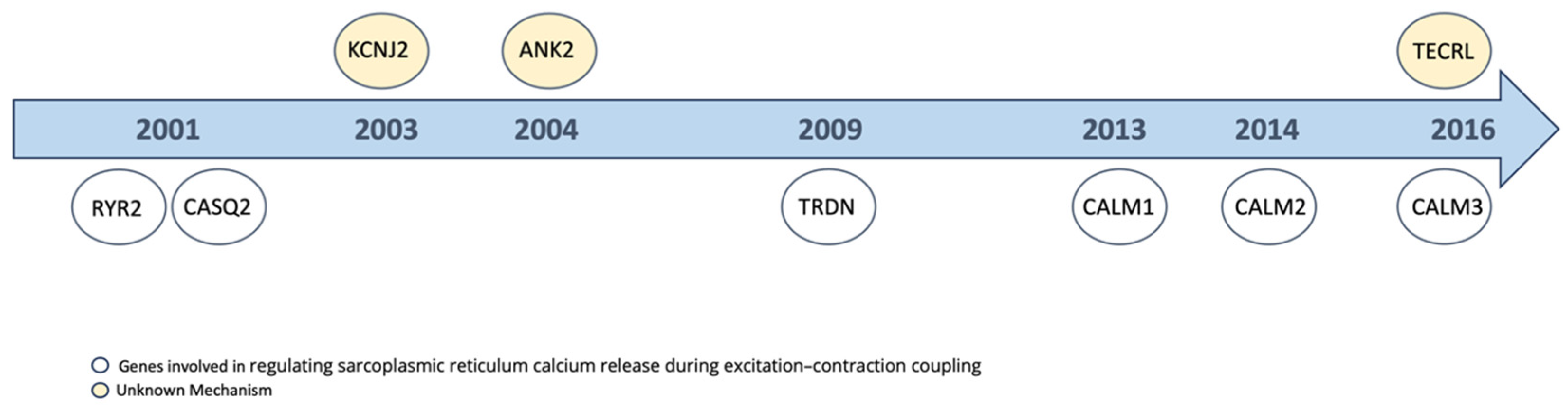
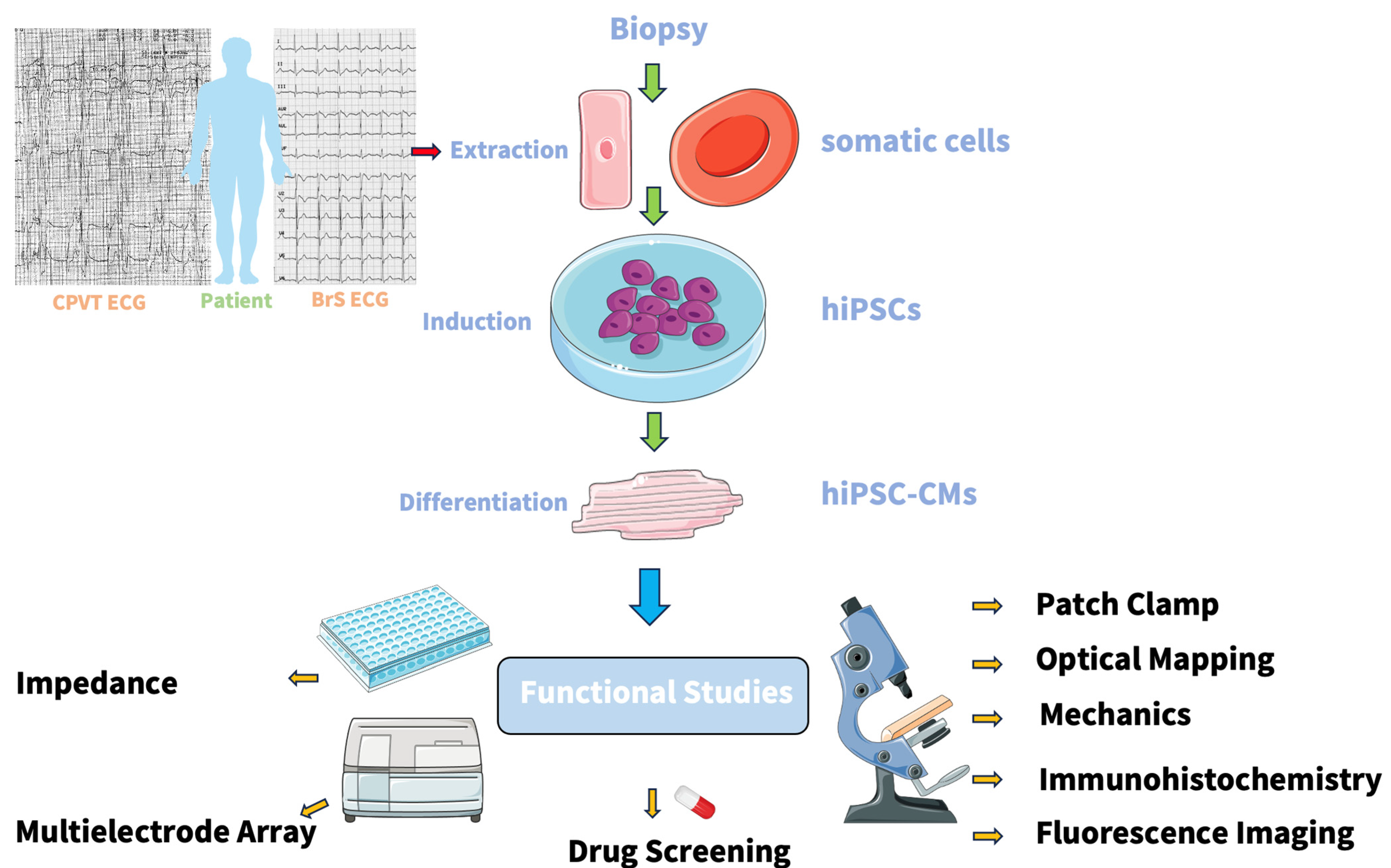

{kind=link}
{kind=link}
{kind=link}
| Reference | Gene | OMIM | Variant | Phenotype |
|---|---|---|---|---|
| Davis et al., 2012 [124] | SCN5A | 601144/ 603830 | c.5537insTGA (p.1795insD) | Reduced INa; reduced Vmax. |
| Kosmidis et al., 2016 [125] | SCN5A | 600163 | c.4912C> T (p.R1638X) c.468G> A (p.W156X) | Reduced INa; reduced Vmax. |
| Liang et al., 2016 [126] | SCN5A | 600163.006 | c.2053G> A (p.R620H) and c.2626G> A (p.R811H) c.4190delA (p.K1397Gfs*5) | Reduced INa; reduced Vmax; abnormal Ca2+ transients. |
| Ma et al., 2018 [108] | SCN5A | 600163.004 | c.677> T (p.A226V) and c.4885C> T (p.R1629X) c.4859C> T p.T1620M | Reduced INa; reduced Vmax. |
| Selga et al., 2018 [127] | SCN5A | 601144 | c.1100G> A (p.367H) | Reduced INa density. |
| De la Roche et al., 2019 [128] | SCN5A | 601144 | c.2204C> T (p.A735V) | Reduced INa; reduced Vmax; rightward shift of the steady-state activation curve. |
| Okata et al., 2016 [129] | SCN5A | 600163 | c.5349G> A (p.E1784K) | Reduced INa. |
| El-Battrawy, Albers et al., 2019 [100] | SCN10A | 604427 | c.3749G> A (p.R1250Q) and c.3808G> A (p.R1268Q) | Reduced INa density; reduced Vmax. |
| El-Battrawy, Müller et al., 2019 [130] | SCN1B | 600235 | c.629T> C (p.L210P) and c.637C> A (p.P213T) | Reduced INa density; reduced Vmax; rightward shift of the steady-state activation curve. |
| El-Battrawy et al., 2021 [131] | CACNB2 | 600003 | c.1439C> T/p.S480L | Reduced APD; reduced ICaL. |
| Belbachir et al., 2019 [132] | RRAD | 179503 | p.R211H | Reduced INa density; reduced Vmax; prolonged AP; increased EAD; reduced ICaL. |
| Cerrone et al., 2014 [22] | PKP2 | 602861 | c.1904G> A (R635Q) | Reduced INa. |
| Miller et al., 2017 [133] | Undefined | Undefined | Reduced INa density. | |
| Undefined | Undefined | |||
| PKP2 | 602861 | c.302G> A (p.R101H) | ||
| Veerman et al., 2016 [134] | Undefined | Undefined | Reduced INa; reduced Vmax. | |
| Undefined | Undefined | |||
| CACNA1C | 114205 | Int19 position -7 (benign) | ||
| Li et al., 2020 [135] | SCN5A | 600163 | p.S1812X11 | Reduced INa density; reduced Vmax; increased ICaL. |
| Simons et al., 2022 [136] | SCN5A | 600163 | c.4813+3_4813+6dupGGGT | - |
| Kashiwa et al., 2023 [137] | CACNA1C | 114205.0019 | E1115K | Longer APD; increased EAD; reduced ICaL. |
| Li et al., 2023 [138] | SCN5A | 600163 | c.3148G> A/p.Ala1050Thr | Reduced INa; reduced Vmax. |
| Zhong et al., 2022 [90] | CACNB2 | 600003 | c.425C> T/p.S142F | Reduced ICaL. |
| Cai et al., 2023 [139] | SCN5A | 600163 | T1788fs | Reduced INa; reduced Vmax. |
| Kamga et al., 2021 [140] | SCN5A | 600163 | Undefined | Reduced INa; reduced Vmax. |
| Reference | Gene | OMIM | Variant | Phenotype |
|---|---|---|---|---|
| Jung et al., 2012 [147] | RYR2 | 180902.0007 | p.(Ser406Leu) p.(Pro2328Ser) | Increased diastolic [Ca2+]; reduced SR Ca2+; increased DAD. |
| Itzhaki et al., 2012 [104] | RYR2 | 180902 | p.(Met4109Arg) | Increased DAD; store-overload-induced Ca2+ release. |
| Preiniger et al., 2016 [149] | RYR2 | 180902 | p.(Leu3741Pro) | Increased diastolic [Ca2+]; reduced SR Ca2+. |
| Novak et al., 2015 [150] | RYR2 | 180902 | p.(Ile4587Val) | Increased diastolic [Ca2+]; reduced SR Ca2+. |
| Novak et al., 2015 & Nijak et al., 2021 [150,153] | CASQ2 | 114251 | p.(Asp307His) | Increased diastolic [Ca2+]; reduced SR Ca2+; increased DAD. |
| Pölönen et al., 2020 [148] | RYR2 | 180902.0011 | Exon 3 Deletion (E3D) L4115F | Reduced APD; increased Vmax; increased diastolic [Ca2+]. |
| Maizels et al., 2017 [152] | CASQ2 | 114251 | p.(Asp307His) | Increased EAD; reduced threshold for store-overload-induced Ca2+ release. |
| Gao et al., 2023 [151] | CALM2 | 114182 | p.E46K | Reduced SR Ca2+. |
| Kamga et al., 2021 [140] | RYR2 | 180902 | Undefined | Increased diastolic [Ca2+]; reduced SR Ca2+. |
| Li et al., 2021 [146] | RYR2 CASQ2 | 180902 114251.0001 | p.A2254V D307H | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begovic, M.; Schneider, L.; Zhou, X.; Hamdani, N.; Akin, I.; El-Battrawy, I. The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies. Int. J. Mol. Sci. 2024, 25, 12034. https://doi.org/10.3390/ijms252212034
Begovic M, Schneider L, Zhou X, Hamdani N, Akin I, El-Battrawy I. The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies. International Journal of Molecular Sciences. 2024; 25(22):12034. https://doi.org/10.3390/ijms252212034
Chicago/Turabian StyleBegovic, Merima, Luca Schneider, Xiaobo Zhou, Nazha Hamdani, Ibrahim Akin, and Ibrahim El-Battrawy. 2024. "The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies" International Journal of Molecular Sciences 25, no. 22: 12034. https://doi.org/10.3390/ijms252212034
APA StyleBegovic, M., Schneider, L., Zhou, X., Hamdani, N., Akin, I., & El-Battrawy, I. (2024). The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies. International Journal of Molecular Sciences, 25(22), 12034. https://doi.org/10.3390/ijms252212034