
Epigenetic Regulation of Anthocyanin Biosynthesis in Betula pendula ‘Purple Rain’

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
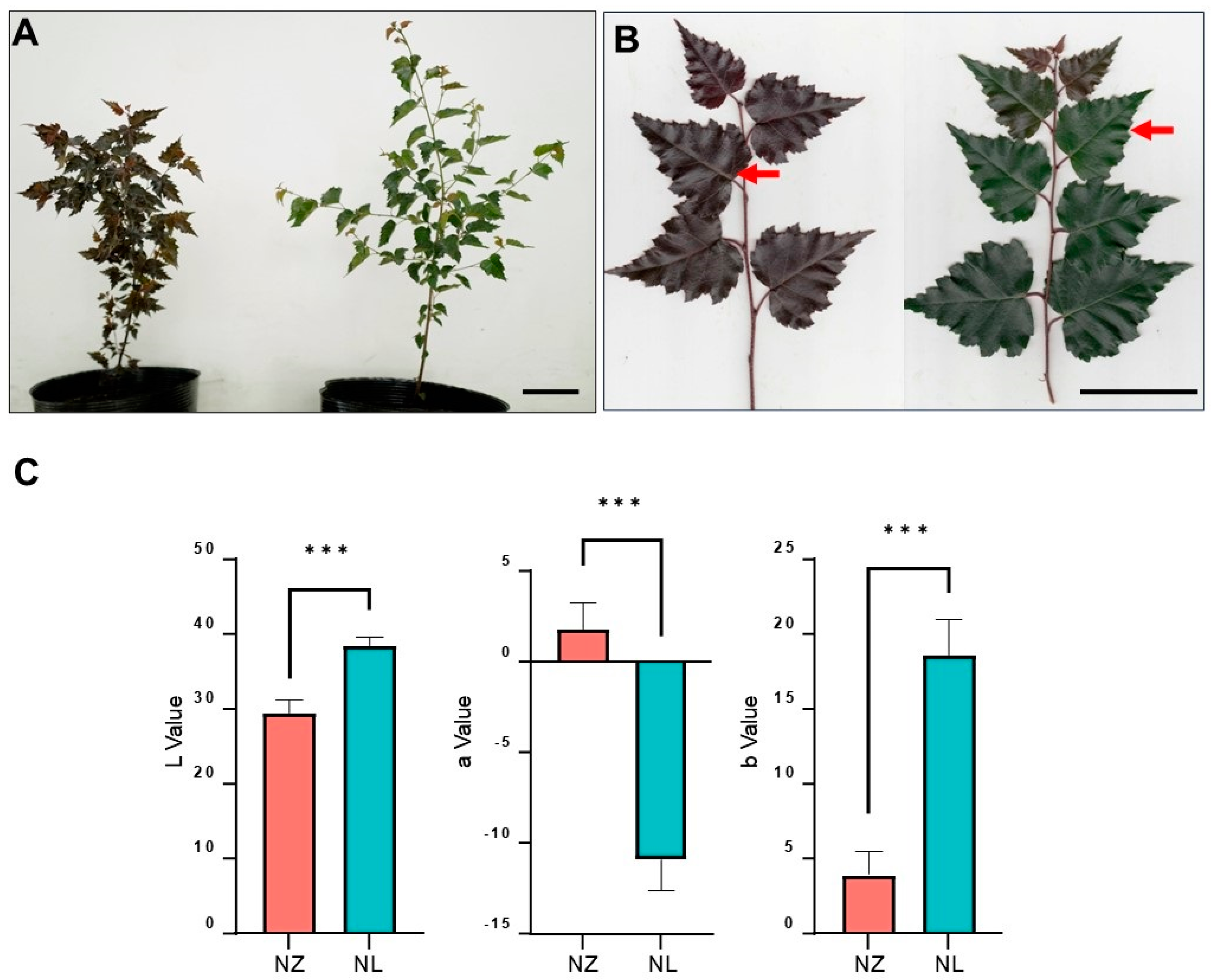
2.1. Leaf Color Phenotype Difference
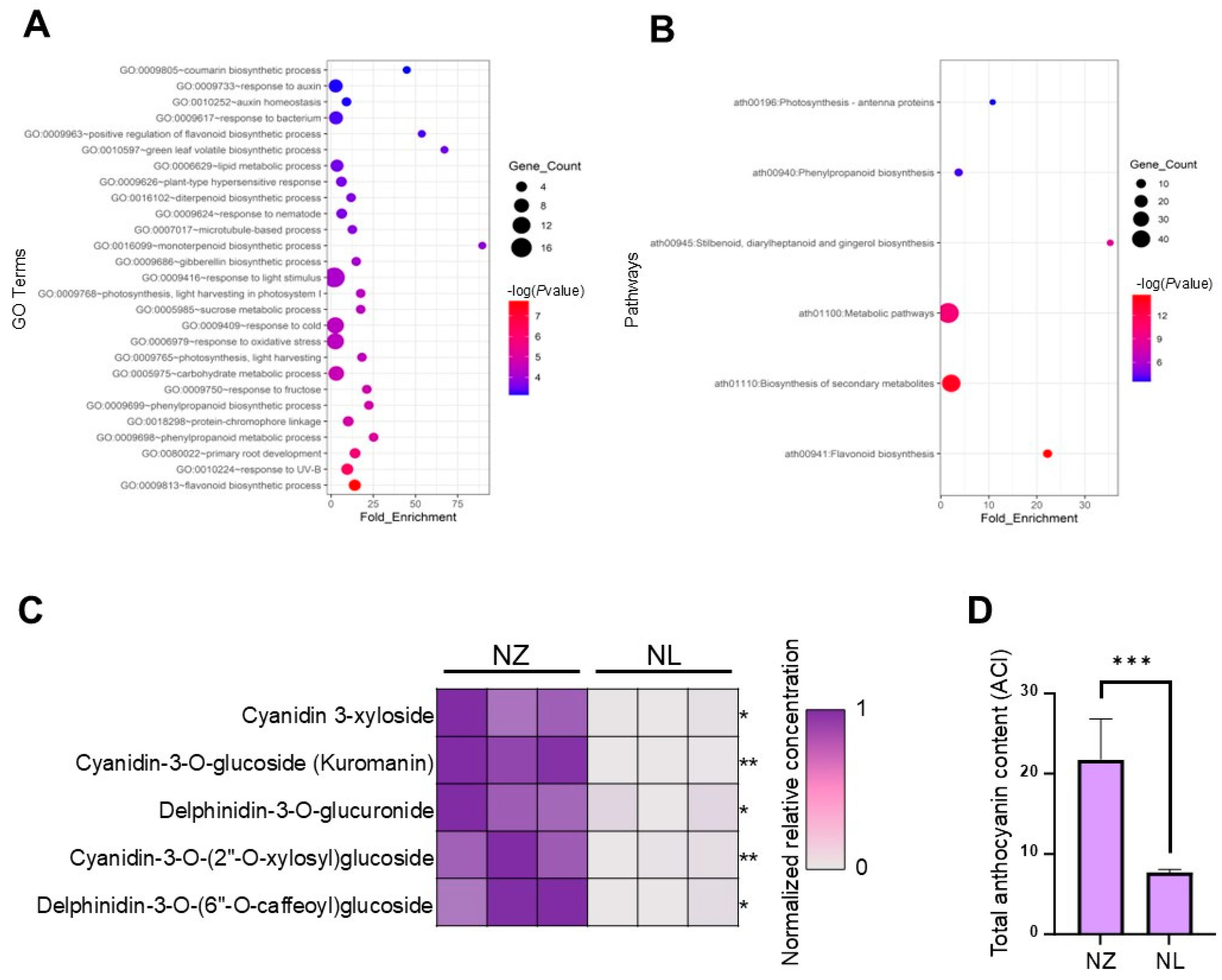
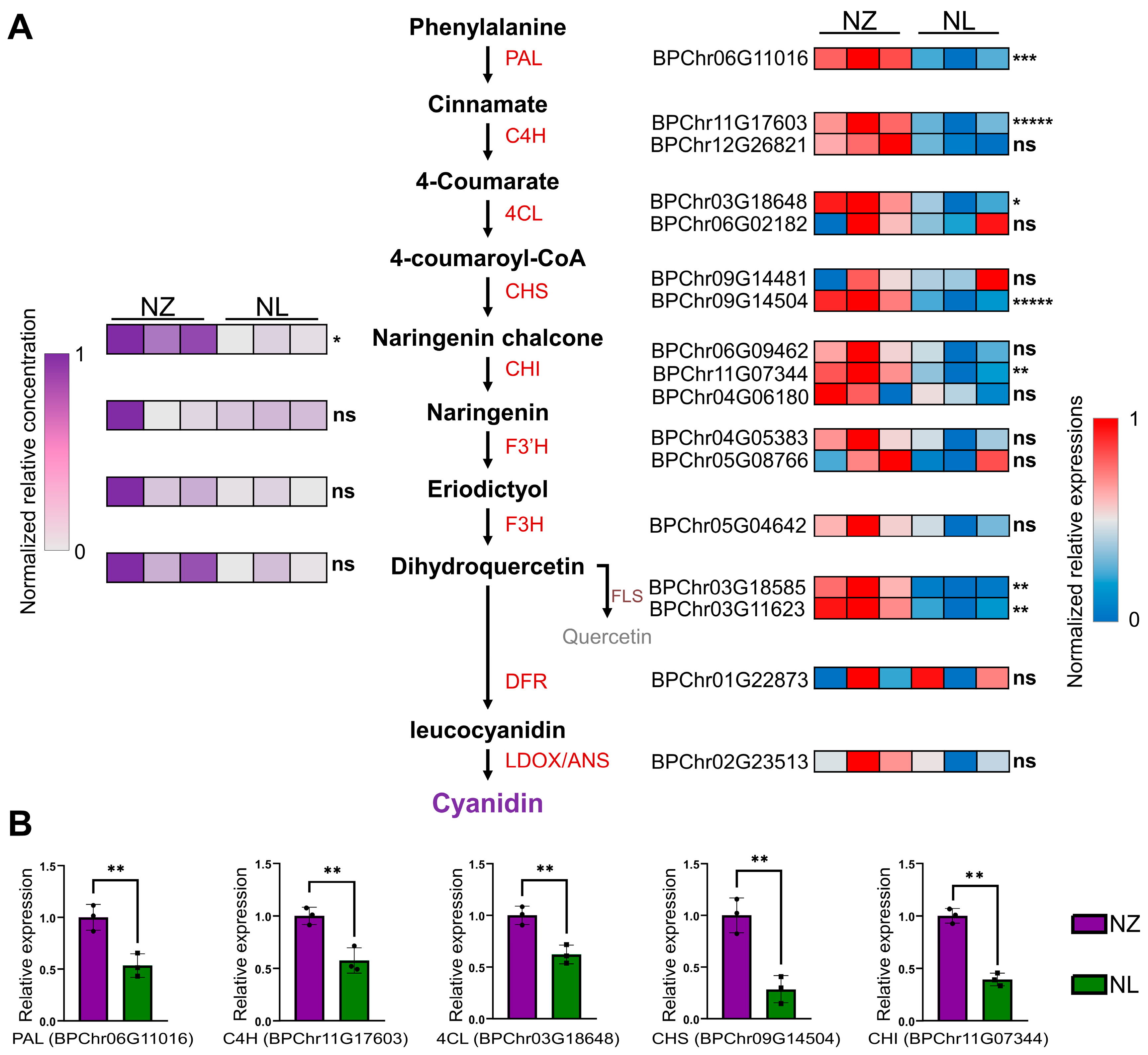
2.2. RNA-Seq and Metabolomic Analysis Showed That the Anthocyanin Biosynthesis Pathway Was Down-Regulated by Changes in Some TFs
2.3. SNPs/InDels Mutations Are Not Supported as a Cause of Phenotypic Variation
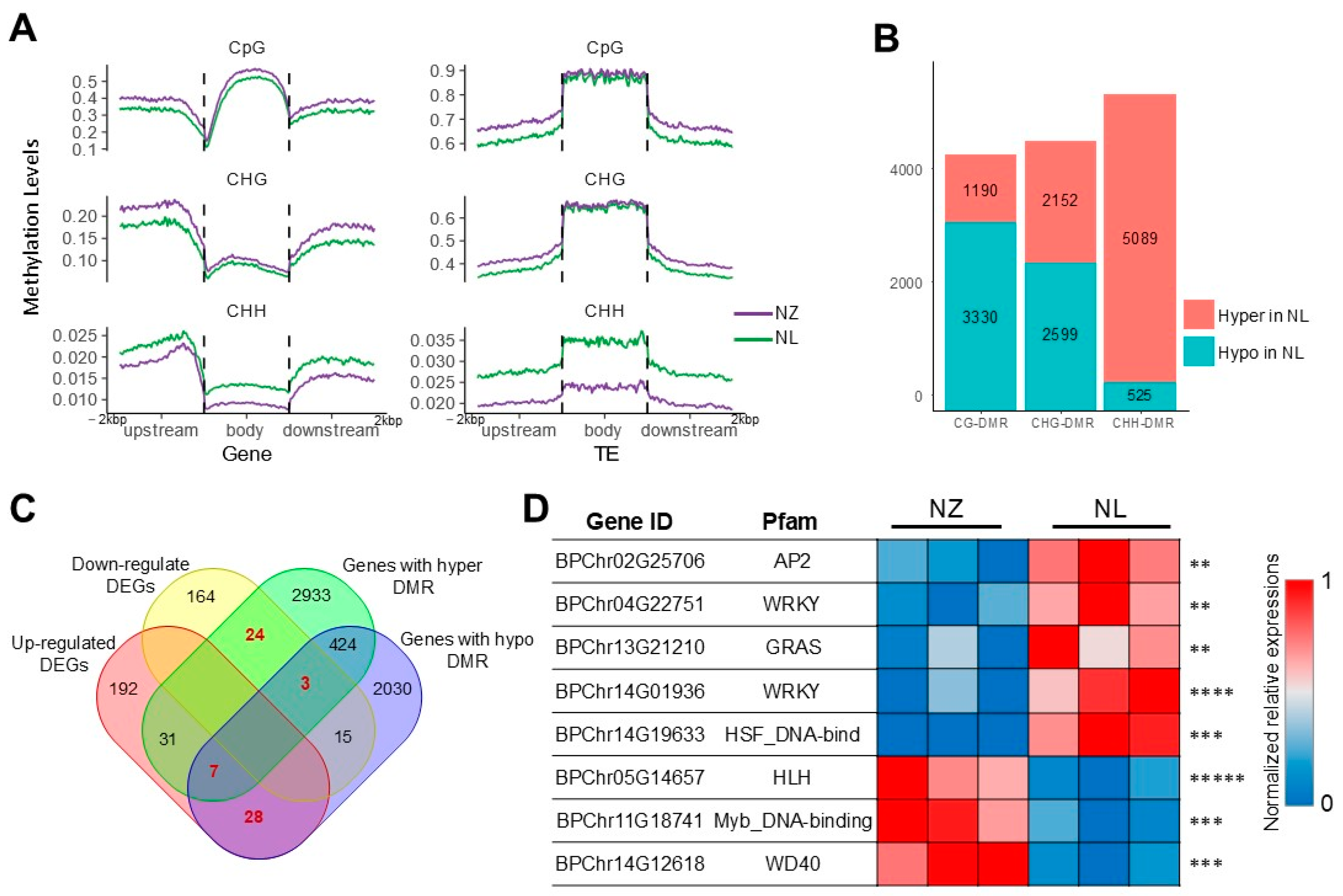
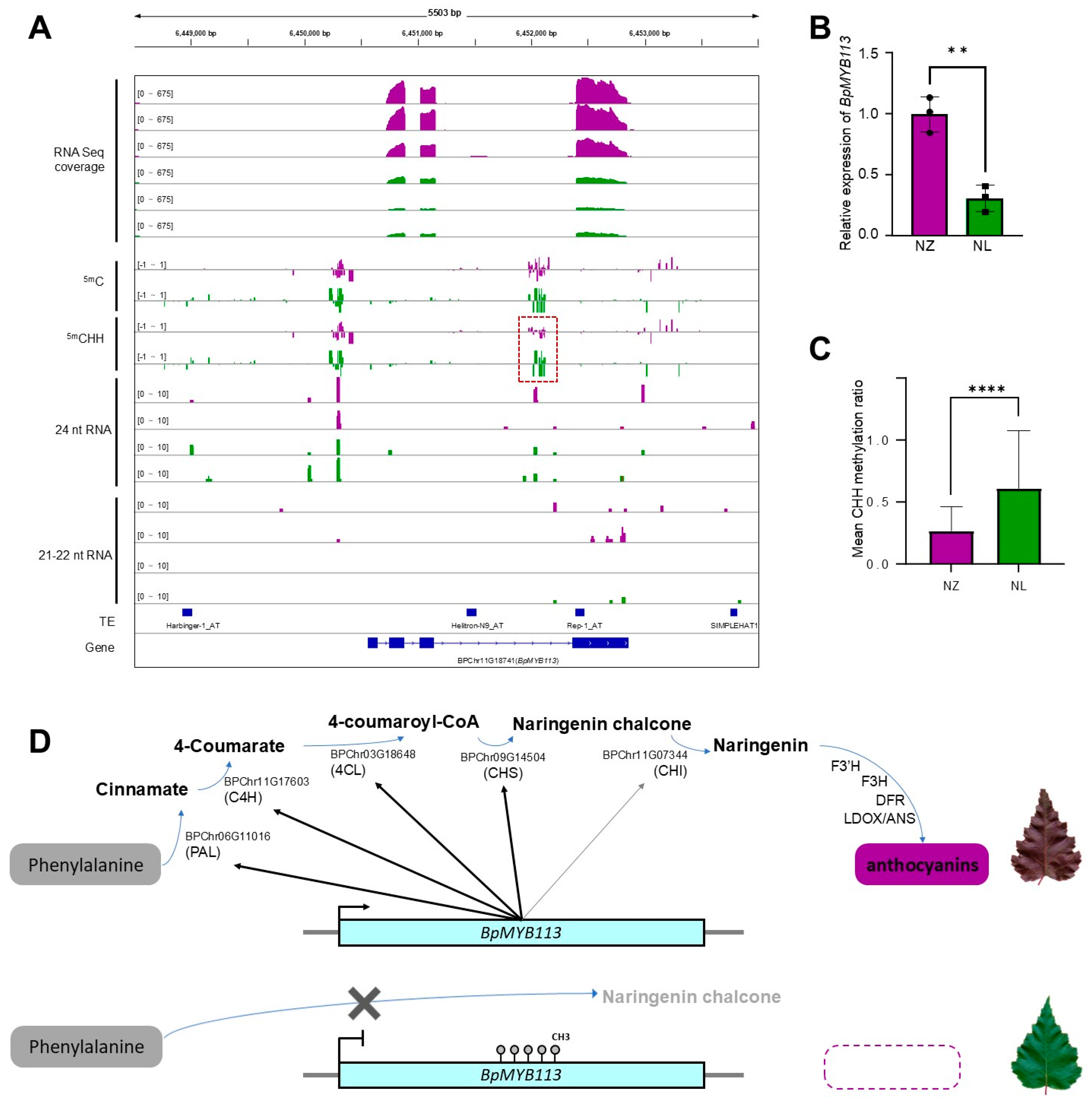
2.4. Epigenetic Analysis Reveals Regulation of Anthocyanin Biosynthesis by MYB113
3. Materials and Methods
3.1. Plant Materials
3.2. Leaf Color Measurement
3.3. Flavonoids-Targeted Metabolomics Determination
3.4. Whole Genome Re-Sequencing (WGS)
3.5. RNA Sequencing
3.6. Small RNA Sequencing
3.7. Whole Genome Bisulfite Sequencing (WGBS)
3.8. qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Qian, J.; Li, J.; Xing, M.; Grierson, D.; Sun, C.; Xu, C.; Li, X.; Chen, K. Hydroxylation decoration patterns of flavonoids in horticultural crops: Chemistry, bioactivity, and biosynthesis. Hortic. Res. 2022, 9, uhab068. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Gao, L.; Fang, Z.; Lei, T.; Xing, D.; Ding, Y.; Rashid, A.; Zhuang, J.; Zhang, Q.; Gu, C.; et al. A flavonoid metabolon: Cytochrome b 5 enhances B-ring trihydroxylated flavan-3-ols synthesis in tea plants. Plant J. 2024, 118, 1793–1814. [Google Scholar] [CrossRef] [PubMed]
- Liga, S.; Paul, C.; Péter, F. Flavonoids: Overview of Biosynthesis, Biological Activity, and Current Extraction Techniques. Plants 2023, 12, 2732. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Gentile, C.; Ertani, A.; Serio, G.; Bertea, C.M. Anthocyanins: Biosynthesis, Distribution, Ecological Role, and Use of Biostimulants to Increase Their Content in Plant Foods—A Review. Agriculture 2021, 11, 212. [Google Scholar] [CrossRef]
- Mekapogu, M.; Vasamsetti, B.M.K.; Kwon, O.-K.; Ahn, M.-S.; Lim, S.-H.; Jung, J.-A. Anthocyanins in Floral Colors: Biosynthesis and Regulation in Chrysanthemum Flowers. Int. J. Mol. Sci. 2020, 21, 6537. [Google Scholar] [CrossRef]
- Tang, M.; Xue, W.; Li, X.; Wang, L.; Wang, M.; Wang, W.; Yin, X.; Chen, B.; Qu, X.; Li, J.; et al. Mitotically heritable epigenetic modifications of CmMYB6 control anthocyanin biosynthesis in chrysanthemum. New Phytol. 2022, 236, 1075–1088. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Liu, Z.; Wang, L.; Bi, Z.; Sun, C.; Yao, P.; Zhang, J.; Bai, J.; Zeng, Y. Integrated transcriptomic and metabolomic analysis revealed altitude-related regulatory mechanisms on flavonoid accumulation in potato tubers. Food Res. Int. 2023, 170, 112997. [Google Scholar] [CrossRef]
- Song, M.; Wang, L.; Zhang, Y.; Wang, Q.; Han, X.; Yang, Q.; Zhang, J.; Tong, Z. Temporospatial pattern of flavonoid metabolites and potential regulatory pathway of PbMYB211-coordinated kaempferol-3-O-rhamnoside biosynthesis in Phoebe bournei. Plant Physiol. Biochem. 2023, 202, 107913. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Long, T.; Wang, S.; Yang, J. Regulation Mechanism of Plant Pigments Biosynthesis: Anthocyanins, Carotenoids, and Betalains. Metabolites 2022, 12, 871. [Google Scholar] [CrossRef]
- Fu, Z.; Jiang, H.; Chao, Y.; Dong, X.; Yuan, X.; Wang, L.; Zhang, J.; Xu, M.; Wang, H.; Li, Y.; et al. Three Paralogous R2R3-MYB Genes Contribute to Delphinidin-Related Anthocyanins Synthesis in Petunia hybrida. J. Plant Growth Regul. 2021, 40, 1687–1700. [Google Scholar] [CrossRef]
- He, G.; Zhang, R.; Jiang, S.; Wang, H.; Ming, F. The MYB transcription factor RcMYB1 plays a central role in rose anthocyanin biosynthesis. Hortic. Res. 2023, 10, uhad080. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yue, M.; Liu, Y.; Zhang, N.; Lin, Y.; Zhang, Y.; Wang, Y.; Li, M.; Luo, Y.; Zhang, Y.; et al. A novel R2R3-MYB transcription factor FaMYB5 positively regulates anthocyanin and proanthocyanidin biosynthesis in cultivated strawberries (Fragaria x ananassa). Plant Biotechnol. J. 2023, 21, 1140–1158. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.; Wu, J.; Cui, M.-L. The combination of R2R3-MYB gene AmRosea1 and hairy root culture is a useful tool for rapidly induction and production of anthocyanins in Antirrhinum majus L. AMB Express 2021, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gómez, S.; Suárez-Baron, H.; Alzate, J.F.; González, F.; Pabón-Mora, N. Evolution of the Subgroup 6 R2R3-MYB Genes and Their Contribution to Floral Color in the Perianth-Bearing Piperales. Front. Plant Sci. 2021, 12, 633227. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Zhu, L.; Liao, Y.; Lin, K.; Wu, W.; Duan, L.; Wang, P.; Xiao, X.; Zhang, T.; Chen, X.; Wang, J.; et al. Cytokinin promotes anthocyanin biosynthesis via regulating sugar accumulation and MYB113 expression in Eucalyptus. Tree Physiol. 2024, 44, tpad154. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, H.; Liu, Z.; Zhang, T.; Li, Z.; Cao, L.; Wu, S.; Liu, Y.; Yu, S.; Zhang, Q.; et al. A naturally-occurring phenomenon of flower color change during flower development in Xanthoceras sorbifolium. Front. Plant Sci. 2022, 13, 1072185. [Google Scholar] [CrossRef]
- Abid, M.A.; Wei, Y.; Meng, Z.; Wang, Y.; Ye, Y.; Wang, Y.; He, H.; Zhou, Q.; Li, Y.; Wang, P.; et al. Increasing floral visitation and hybrid seed production mediated by beauty mark in Gossypium hirsutum. Plant Biotechnol. J. 2022, 20, 1274–1284. [Google Scholar] [CrossRef]
- Jiang, S.; Sun, Q.; Zhang, T.; Liu, W.; Wang, N.; Chen, X. MdMYB114 regulates anthocyanin biosynthesis and functions downstream of MdbZIP4-like in apple fruit. J. Plant Physiol. 2021, 257, 153353. [Google Scholar] [CrossRef]
- He, K.; Du, J.; Han, X.; Li, H.; Kui, M.; Zhang, J.; Huang, Z.; Fu, Q.; Jiang, Y.; Hu, Y. PHOSPHATE STARVATION RESPONSE1 (PHR1) interacts with JASMONATE ZIM-DOMAIN (JAZ) and MYC2 to modulate phosphate deficiency-induced jasmonate signaling in Arabidopsis. Plant Cell 2023, 35, 2132–2156. [Google Scholar] [CrossRef]
- Li, Y.; Xing, M.; Yang, Q.; Wang, Y.; Jiang, J.; Zhao, Y.; Zhao, X.; Shen, A.; Feng, Y.; Zhao, X.; et al. SmCIP7, a COP1 interactive protein, positively regulates anthocyanin accumulation and fruit size in eggplant. Int. J. Biol. Macromol. 2023, 234, 123729. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Martín-Pizarro, C.; Zhou, L.; Hou, B.; Wang, Y.; Shen, Y.; Li, B.; Posé, D.; Qin, G. Deciphering the regulatory network of the NAC transcription factor FvRIF, a key regulator of strawberry (Fragaria vesca) fruit ripening. Plant Cell 2023, 35, 4020–4045. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Wang, X.; Yang, R.; Wu, Z.; Wang, H.; Wang, L.; Hu, Z.; Guo, S.; Zhang, H.; et al. MiR156 regulates anthocyanin biosynthesis through SPL targets and other microRNAs in poplar. Hortic. Res. 2020, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, D.; Li, Y.; Hu, P.; Xu, R.; Wang, X. Genome-wide identification and bioinformatics analysis of the WRKY transcription factors and screening of candidate genes for anthocyanin biosynthesis in azalea (Rhododendron simsii). Front. Genet. 2023, 14, 1172321. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA Methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J. RNA-directed DNA methylation. Curr. Opin. Plant Biol. 2011, 14, 142–147. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, Y.; Su, X.; Liu, W.; Guo, Y.; Jiang, J.; Ma, W. Characterization of the complete chloroplast genome of Betula pendula purple rain (betulaceae). Mitochondrial DNA Part B 2023, 8, 281–284. [Google Scholar] [CrossRef]
- Lyu, D.; Lin, L.; Guo, Y.; Han, R.; Jiang, J. Characterization of gene expression in anthocyanin synthesis and salt tolerance of Betula pendula ‘Purple Rain’ (In Chinese). J. Nanjing For. Univ. 2018, 42, 25–32. [Google Scholar]
- Lin, L.; Mu, H.; Jiang, J.; Liu, G. Transcriptomic analysis of purple leaf determination in birch. Gene 2013, 526, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Han, R.; Liu, C.; Fang, G.; Yuan, Q.; Zheng, Z.; Yu, Q.; Jiang, J.; Liu, S.; Xie, L.; et al. Heritable epigenetic modification of BpPIN1 is associated with leaf shapes in Betula pendula. Tree Physiol. 2023, 43, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gu, C.; Jiang, J.; Guifeng, L.; Huiyu, L. Heritable Epigenetic Modification of BpIAA9 Causes the Reversion Mutation of Leaf Shapes in Betula pendula ‘Dalecarlica’. Forests 2024, 15, 95. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Yu, L.; Zheng, T.; Wang, S.; Yue, Z.; Jiang, J.; Kumari, S.; Zheng, C.; Tang, H.; et al. Genome sequence and evolution ofBetula platyphylla. Hortic. Res. 2021, 8, 1–12. [Google Scholar] [CrossRef]
- Jung, Y.; Han, D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 2022, 38, 2404–2413. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X. miRDeep-P2: Accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics. 2019, 35, 2521–2522. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.C.; Ghosh, A. Phenotypic variation and epigenetic insight into tissue culture berry crops. Front. Plant Sci. 2022, 13, 1042726. [Google Scholar] [CrossRef] [PubMed]
- Müller-Xing, R.; Xing, Q. The plant stem-cell niche and pluripotency: 15 years of an epigenetic perspective. Front. Plant Sci. 2022, 13, 1018559. [Google Scholar] [CrossRef] [PubMed]
- Mateo-Bonmatí, E.; Casanova-Sáez, R.; Ljung, K. Epigenetic Regulation of Auxin Homeostasis. Biomolecules 2019, 9, 623. [Google Scholar] [CrossRef]
- Singroha, G.; Kumar, S.; Gupta, O.P.; Singh, G.P.; Sharma, P. Uncovering the Epigenetic Marks Involved in Mediating Salt Stress Tolerance in Plants. Front. Genet. 2022, 13, 811732. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Sun, H.; Sun, L.; Zhang, L. Transposon-induced methylation of the RsMYB1 promoter disturbs anthocyanin accumulation in red-fleshed radish. J. Exp. Bot. 2020, 71, 2537–2550. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Y.; Wang, Q.; Li, B.; Wang, X.; Zhou, X.; Zhang, H.; Xu, W.; Li, S.; Wang, L. The combination of DNA methylation and positive regulation of anthocyanin biosynthesis by MYB and bHLH transcription factors contributes to the petal blotch formation in Xibei tree peony. Hortic. Res. 2023, 10, uhad100. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Horvath, R.; Laenen, B.; Takuno, S.; Slotte, T. Single-cell expression noise and gene-body methylation in Arabidopsis thaliana. Heredity 2019, 123, 81–91. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, C.; Xu, H.; Yuan, Q.; Huang, J.; Yuan, K.; Zhao, Y.; Liu, G.; Zhang, Q.; Jiang, J. Epigenetic Regulation of Anthocyanin Biosynthesis in Betula pendula ‘Purple Rain’. Int. J. Mol. Sci. 2024, 25, 12030. https://doi.org/10.3390/ijms252212030
Gu C, Xu H, Yuan Q, Huang J, Yuan K, Zhao Y, Liu G, Zhang Q, Jiang J. Epigenetic Regulation of Anthocyanin Biosynthesis in Betula pendula ‘Purple Rain’. International Journal of Molecular Sciences. 2024; 25(22):12030. https://doi.org/10.3390/ijms252212030
Chicago/Turabian StyleGu, Chenrui, Huan Xu, Qihang Yuan, Jinbo Huang, Kunying Yuan, Yihan Zhao, Guifeng Liu, Qingzhu Zhang, and Jing Jiang. 2024. "Epigenetic Regulation of Anthocyanin Biosynthesis in Betula pendula ‘Purple Rain’" International Journal of Molecular Sciences 25, no. 22: 12030. https://doi.org/10.3390/ijms252212030
APA StyleGu, C., Xu, H., Yuan, Q., Huang, J., Yuan, K., Zhao, Y., Liu, G., Zhang, Q., & Jiang, J. (2024). Epigenetic Regulation of Anthocyanin Biosynthesis in Betula pendula ‘Purple Rain’. International Journal of Molecular Sciences, 25(22), 12030. https://doi.org/10.3390/ijms252212030