


Genome-Wide Identification of the bHLH Gene Family in Callerya speciosa Reveals Its Potential Role in the Regulation of Isoflavonoid Biosynthesis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
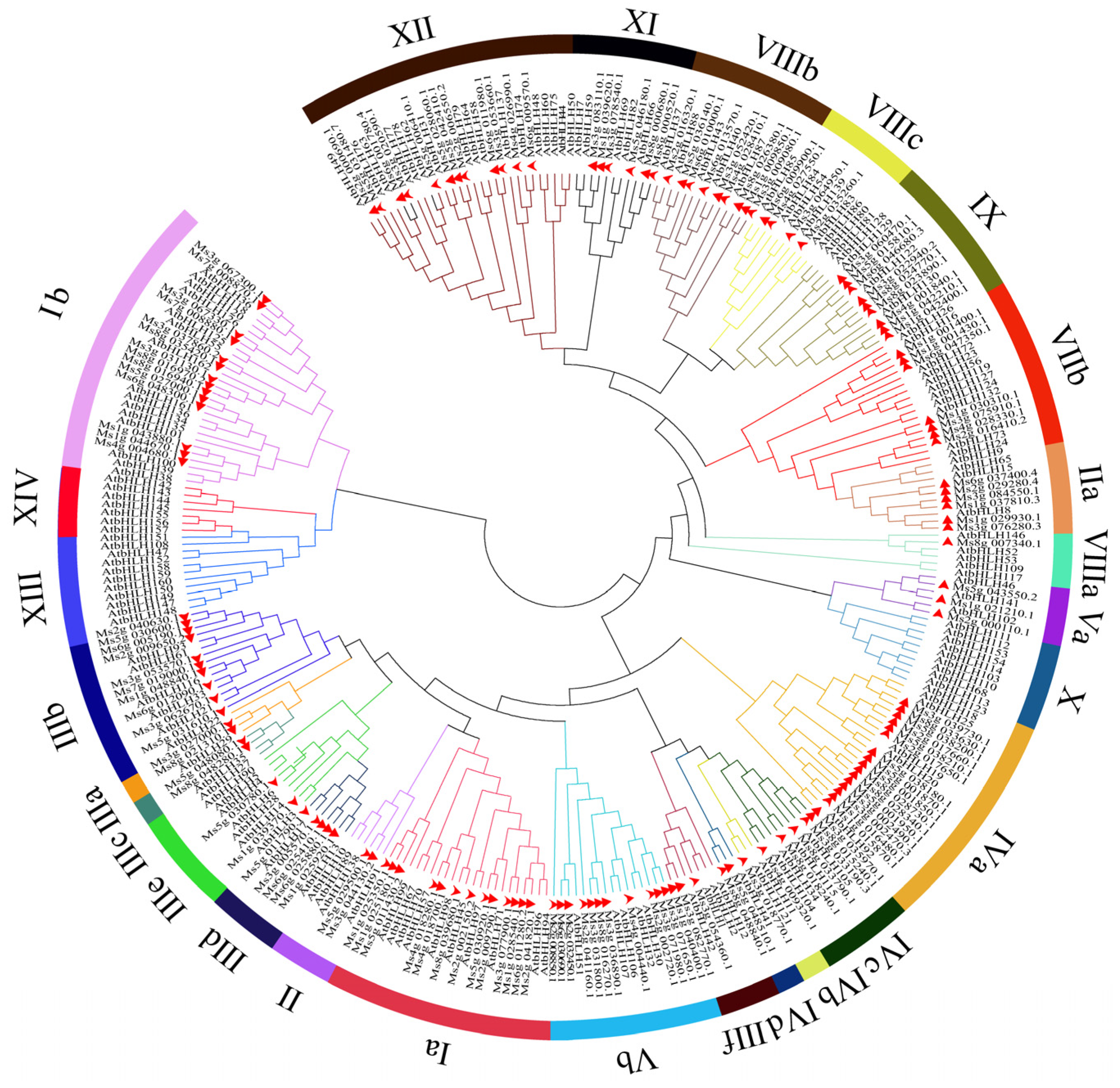
2.1. Genome-Wide Identification of CsbHLH Genes and Their Phylogenetic Analysis
2.2. Conserved Domains, Motif Composition, and Gene Structural Characteristics of the CsbHLH Proteins
2.3. Chromosomal Localization, Gene Duplication, and Synteny Analysis of the CsbHLH Proteins
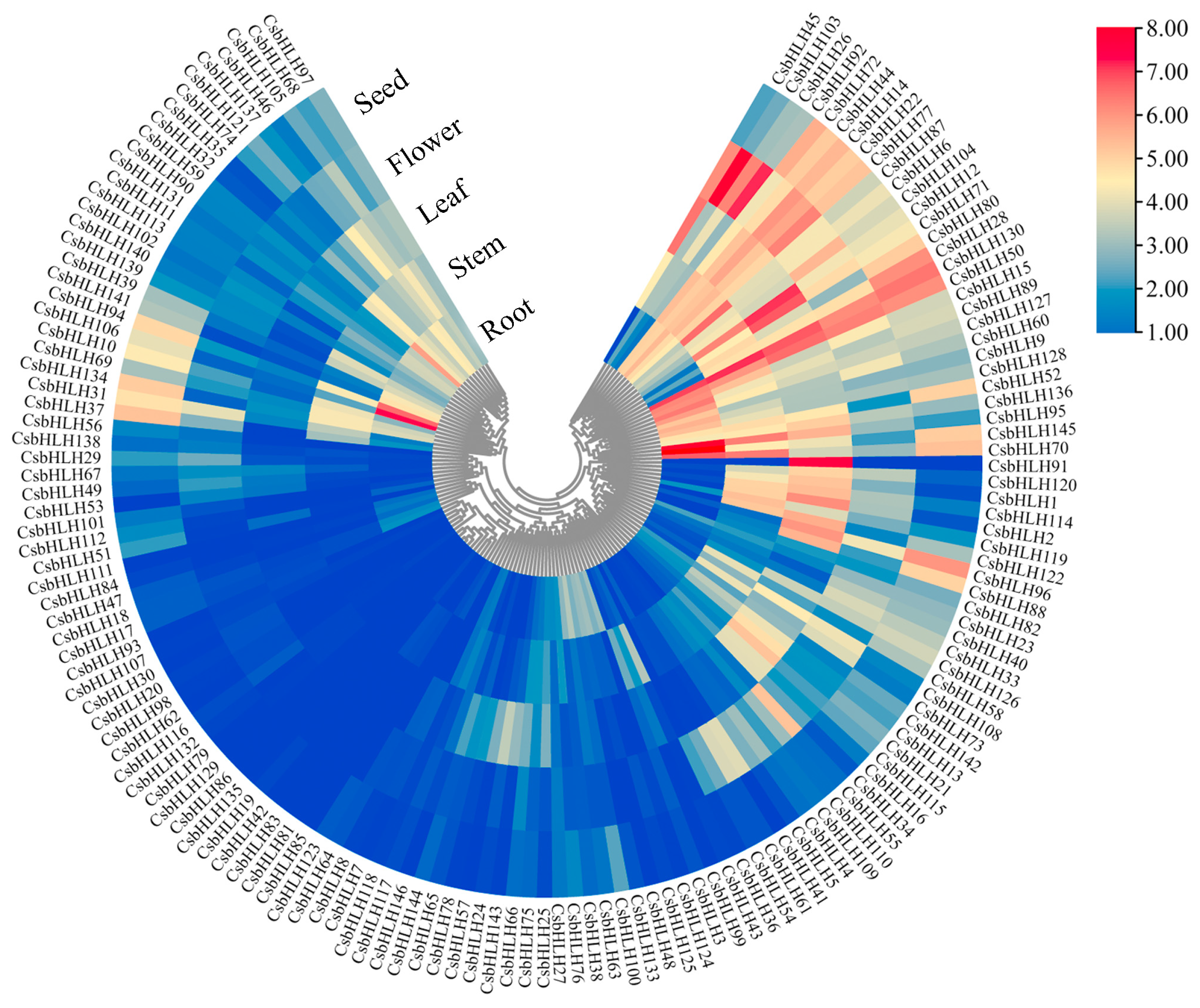
2.4. Expression Pattern of CsbHLH Genes in Different Tissues
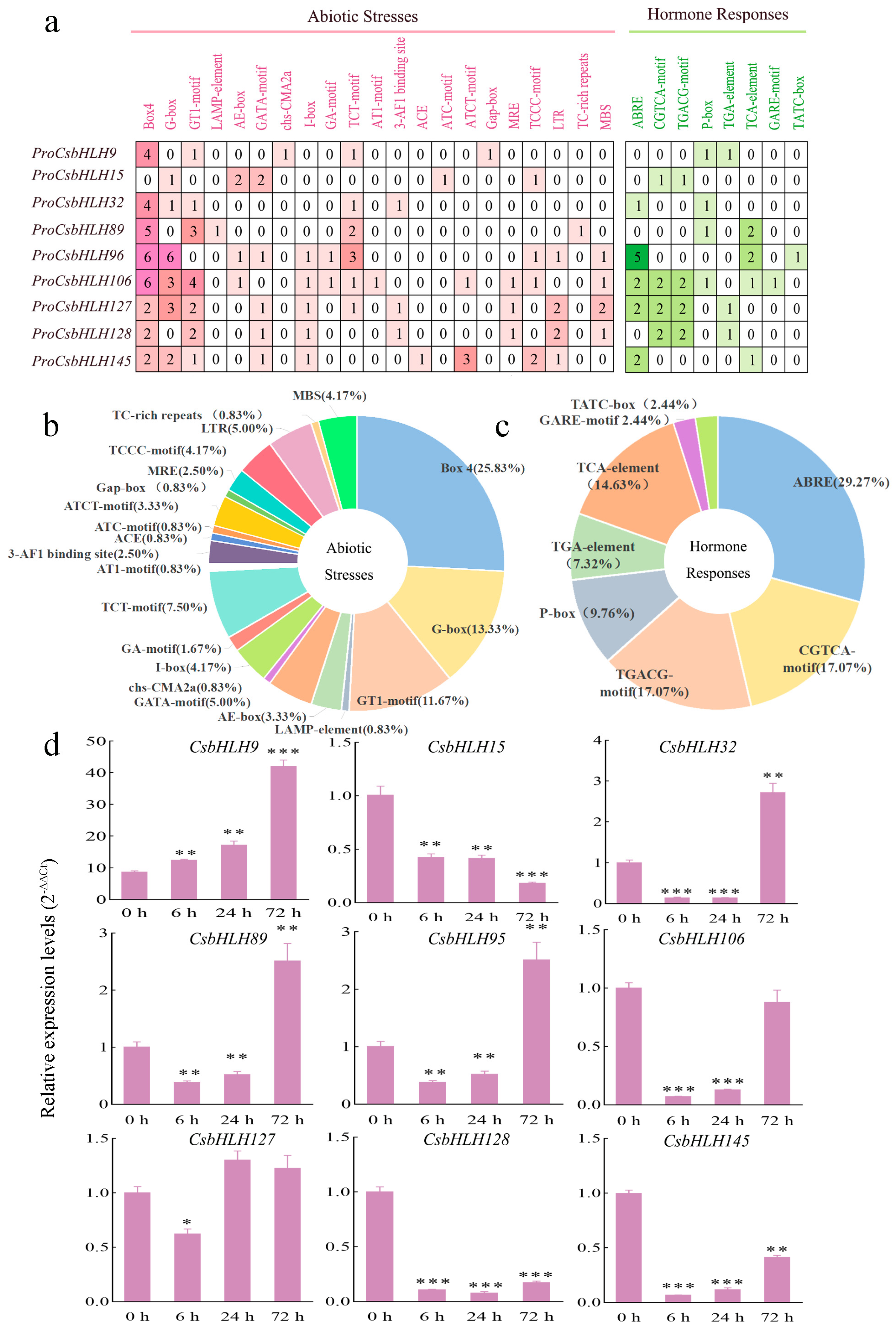
2.5. The Isoflavonoid-Related CsbHLH Genes and Their Cis-Element Analysis
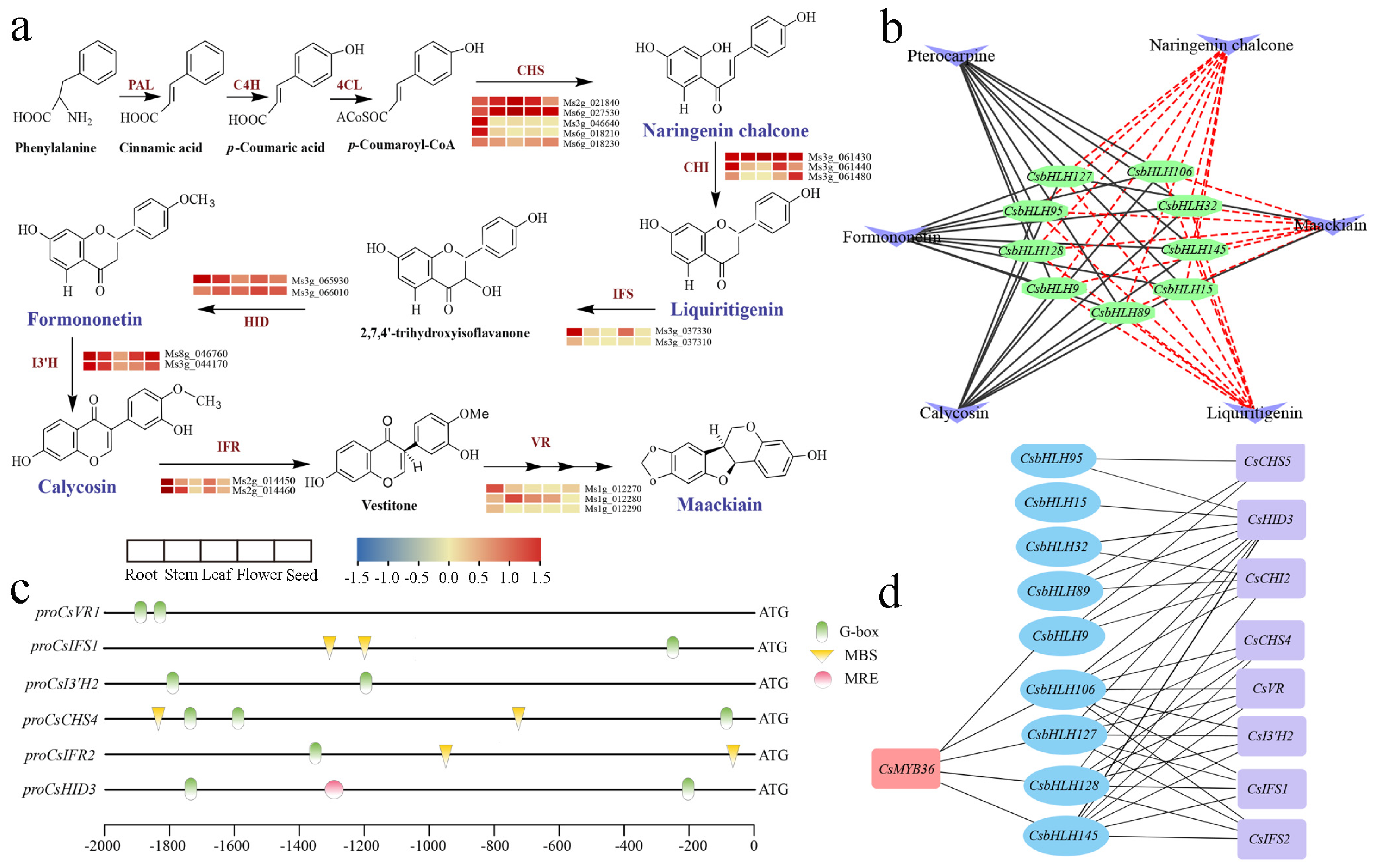
2.6. Prediction of CsbHLHs Involved in the Regulation of (Iso)Flavonoid Biosynthesis
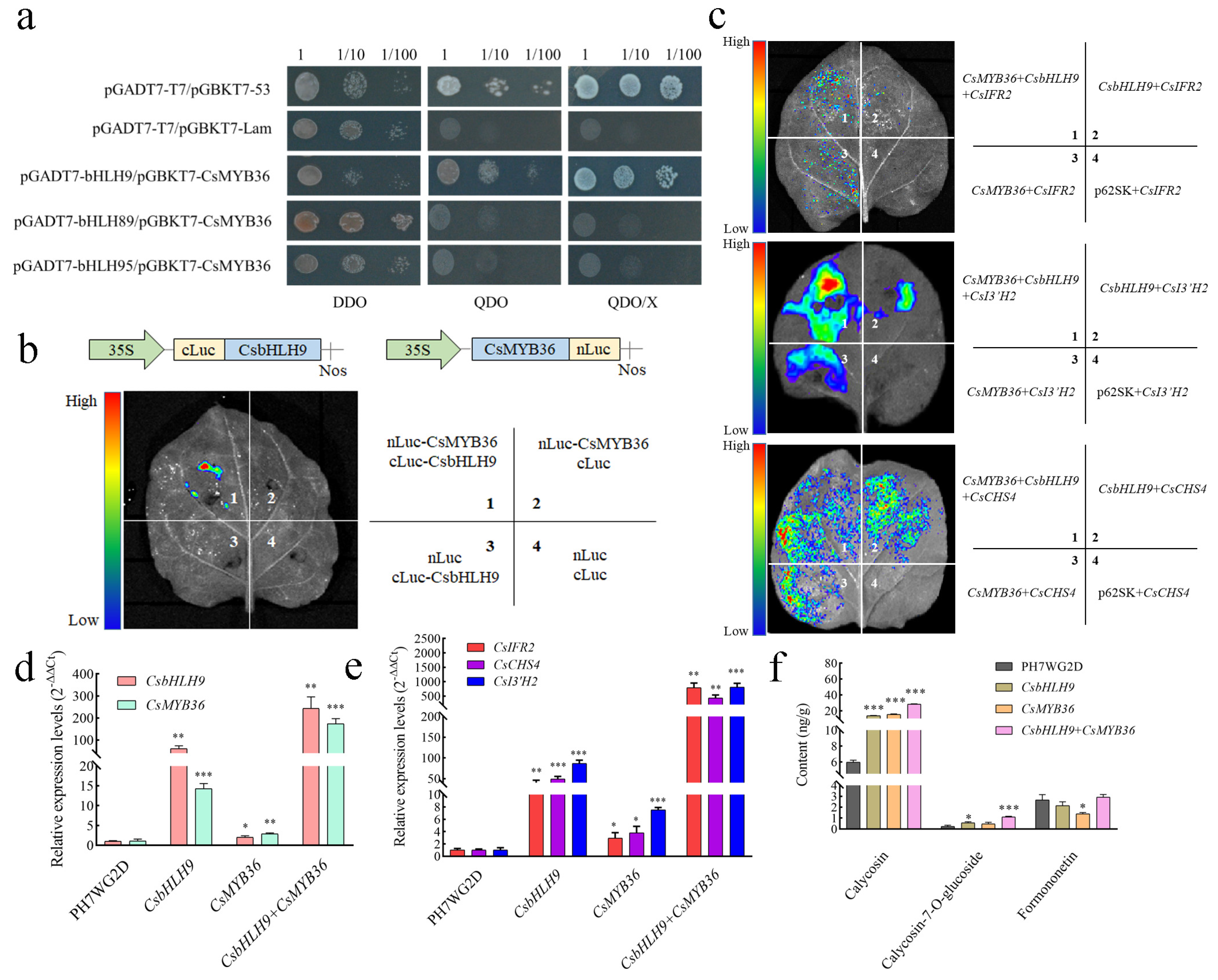
2.7. Verification of the Interaction Between CsMYB36 and CsbHLH9
2.8. Functional Validation of CsMYB36 and CsbHLH9
3. Discussion
4. Materials and Methods
4.1. Plant Materials and MeJA Treatment
4.2. Identification and Annotation of CsbHLH Proteins
4.3. Subcellular Localization and Properties Prediction
4.4. Phylogenetic Analysis and Multiple Sequence Alignment
4.5. Conserved Motif and Gene Structure Analysis
4.6. Chromosomal Locations and Collinearity
4.7. Metabolomics and the Indicative Compound Contents Measurement
4.8. Identification of Co-Expression Modules
4.9. Expression Confirmation by qRT-PCR
4.10. Dual Luciferase (LUC) Assays
4.11. Bimolecular Fluorescence Complementation (BiFC) Assays
4.12. Yeast Two-Hybrid (Y2H) Assays
4.13. Transient Transformation of CsMYB36 and CsbHLH9 in C. Speciosa Leaves
4.14. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, M.; Wang, H.; Wang, Z.; Huang, H.; Chen, S.; Ma, H. Genome-wide characterization and analysis of bHLH transcription factors related to anthocyanin biosynthesis in fig (Ficus carica L.). Front. Plant Sci. 2021, 12, 730692. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Jiang, H.; Sun, Y.; Tang, Y.; Yuan, Z.; Guo, J.; Liang, W.; Chen, L.; Yin, J.; et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006, 141, 1167–1184. [Google Scholar] [CrossRef]
- Qin, Y.; Li, J.; Chen, J.; Yao, S.; Li, L.; Huang, R.; Tan, Y.; Ming, R.; Huang, D. Genome-wide characterization of the bHLH gene family in Gynostemma pentaphyllum reveals its potential role in the regulation of gypenoside biosynthesis. BMC Plant Biol. 2024, 24, 205. [Google Scholar] [CrossRef] [PubMed]
- Grotewold, E.; Sainz, M.B.; Tagliani, L.; Hernandez, J.M.; Bowen, B.; Chandler, V.L. Identification of the residues in the Myb domain of maize C1 that specify the interaction with the bHLH cofactor R. Proc. Natl. Acad. Sci. USA 2000, 97, 13579–13584. [Google Scholar] [CrossRef]
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 2000, 2, 429–440. [Google Scholar] [CrossRef]
- Pires, N.; Dolan, L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 2010, 27, 862–874. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Galstyan, A.; Roig-Villanova, I.; Martínez-García, J.F.; Bilbao-Castro, J.R.; Robertson, D.L. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010, 153, 1398–1412. [Google Scholar] [CrossRef]
- Wang, J.; Hu, Z.; Zhao, T.; Yang, Y.; Chen, T.; Yang, M.; Yu, W.; Zhang, B. Genome-wide analysis of bHLH transcription factor and involvement in the infection by yellow leaf curl virus in tomato (Solanum lycopersicum). BMC Genom. 2015, 16, 39. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, Y.; Vainstein, A.; Chen, S.; Ma, H. Regulation of fig (Ficus carica L.) fruit color: Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front. Plant Sci. 2017, 8, 1990. [Google Scholar] [CrossRef] [PubMed]
- Strygina, K.V.; Khlestkina, E.K. Myc-like transcriptional factors in wheat: Structural and functional organization of the subfamily I members. BMC Plant Biol. 2019, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Tian, T.; Ding, Y.; Yan, N.; Wang, C.; Fang, N.; Liu, Y.; Zhang, Z.; Zhang, H. bHLH transcription factor NtMYC2a regulates carbohydrate metabolism during the pollen development of tobacco (Nicotiana tabacum L. cv. TN90). Plants 2021, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Ma, J.; Wang, S.; Tang, M.; Zhang, B.; Ma, Y.; Li, L.; Sun, G.; Dong, S.; Liu, Y.; et al. Liverwort bHLH transcription factors and the origin of stomata in plants. Curr. Biol. 2023, 333, 2806–2813. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, J.; Liu, D.; Wei, Y.L.; Wang, Y.; Li, X.B. Characterization of bHLH/HLH genes that are involved in brassinosteroid (BR) signaling in fiber development of cotton (Gossypium hirsutum). BMC Plant Biol. 2018, 18, 304. [Google Scholar] [CrossRef]
- Groszmann, M.; Paicu, T.; Smyth, D.R. Functional domains of SPATULA, a bHLH transcription factor involved in carpel and fruit development in Arabidopsis. Plant J. 2008, 55, 40–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, J.; Zhang, L.; Jin, H.; Fang, S. Genome-wide identification of bHLH transcription factors and their response to salt stress in Cyclocarya paliurus. Front. Plant Sci. 2023, 14, 1117246. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, Y.R.; Wang, Q.J.; Yao, Y.X.; You, C.X.; Hao, Y.J. Overexpression of MdbHLH104 gene enhances the tolerance to iron deficiency in apple. Plant Biotechnol. J. 2016, 14, 1633–1645. [Google Scholar] [CrossRef]
- Qiu, J.R.; Huang, Z.; Xiang, X.Y.; Xu, W.X.; Wang, J.T.; Chen, J.; Song, L.; Xiao, Y.; Li, X.; Ma, J.; et al. MfbHLH38, a Myrothamnus flabellifolia bHLH transcription factor, confers tolerance to drought and salinity stresses in Arabidopsis. BMC Plant Biol. 2020, 20, 542. [Google Scholar] [CrossRef]
- Liao, W.; Cai, J.; Xu, H.; Wang, Y.; Cao, Y.; Ruan, M.; Chen, S.; Peng, M. The transcription factor MebHLH18 in cassava functions in decreasing low temperature-induced leaf abscission to promote low-temperature tolerance. Front. Plant Sci. 2023, 13, 1101821. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Song, Z.; Zhang, Y.; Li, Z.; Wang, Y.; Liu, X.; Ma, J.; Quan, J.; Wu, X.; Liu, M.; et al. The bHLH transcription factor PPLS1 regulates the color of pulvinus and leaf sheath in foxtail millet (Setaria italica). Theor. Appl. Genet. 2020, 133, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yue, M.; Liu, Y.; Zhang, N.; Lin, Y.; Zhang, Y.; Wang, Y.; Li, M.; Luo, Y.; Zhang, Y.; et al. A novel R2R3-MYB transcription factor FaMYB5 positively regulates anthocyanin and proanthocyanidin biosynthesis in cultivated strawberries (Fragaria × ananassa). Plant Biotechnol. J. 2023, 21, 1140–1158. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Wang, Y.; Chen, M.; Shi, X.; Zhou, X.; Zhang, Z. Phytochemistry and antioxidant activities of the rhizome and radix of Millettia speciosa based on UHPLC-Q-Exactive Orbitrap-MS. Molecules 2022, 27, 7398. [Google Scholar] [CrossRef]
- Lam, V.Q.; Anh, H.; Quan, N.V.; Xuan, T.D.; Hanamura, I.; Uchino, K.; Karnan, S.; Takami, A. Cytotoxicity of Callerya speciosa fractions against myeloma and lymphoma cell lines. Molecules 2022, 27, 2322. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, M.; Yang, Q.; Wang, Q.; Ma, B.; Li, Z.; Cheng, W.; Tang, H.; Feng, S.; Wang, Z. Metabolomic profiling of M. speciosa champ at different growth stages. Food Chem. 2021, 376, 131941. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, C.; Lin, Y.; Cai, J. Ameliorating effect on glycolipid metabolism and chemical profile of Millettia speciosa champ. extract. J. Ethnopharmacol. 2021, 279, 114360. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Yu, L.; Ming, R.; Tan, X.; Li, L.; Huang, R.; Tan, Y.; Yao, S. A chromosome-level genome assembly of Callerya speciosa sheds new light on the biosynthesis of root-specific isoflavonoids. Ind. Crops Prod. 2023, 200, 116877. [Google Scholar] [CrossRef]
- Doerge, R.W. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 2002, 3, 43–52. [Google Scholar] [CrossRef]
- Zernova, O.V.; Lygin, A.V.; Widholm, J.M.; Lozovaya, V.V. Modification of isoflavones in soybean seeds via expression of multiple phenolic biosynthetic genes. Plant Physiol. Biochem. 2009, 47, 769–777. [Google Scholar] [CrossRef]
- Huang, W.; Lv, H.; Wang, Y. Functional characterization of a novel R2R3-MYB transcription factor modulating the flavonoid biosynthetic pathway from Epimedium sagittatum. Front. Plant Sci. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Wu, R.; Xia, Y.; Pang, Y. Identification of Transcription Factor Genes and Functional Characterization of PlMYB1 From Pueraria lobata. Front. Plant Sci. 2021, 12, 743518. [Google Scholar] [CrossRef]
- Yu, L.; Chen, L.; Qin, C.; Ming, R.; Huang, D.; Huang, R.; Yao, S. Effects of methyl jasmonateon the accumulation of active components and related gene expressions in Callerya speciosa. Chin. Med. Mat. 2023, 46, 2929–2937. [Google Scholar]
- Sohn, S.I.; Pandian, S.; Oh, Y.J.; Kang, H.J.; Cho, W.S.; Cho, Y.S. Metabolic engineering of isoflavones: An updated overview. Front. Plant Sci. 2021, 12, 670103. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Qi, T.; Fan, M.; Zhang, X.; Gao, H.; Huang, H.; Wu, D.; Guo, H.; Xie, D. The bHLH subgroup IIId factors negatively regulate jasmonate-mediated plant defense and development. PLoS Genet. 2013, 9, e1003653. [Google Scholar] [CrossRef]
- Hao, Y.; Zong, X.; Ren, P.; Qian, Y.; Fu, A. Basic helix-loop-helix (bHLH) transcription factors regulate a wide range of functions in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 7152. [Google Scholar] [CrossRef]
- Xu, P.; Wu, L.; Cao, M.; Ma, C.; Xiao, K.; Li, Y.; Lian, H. Identification of MBW complex components implicated in the biosynthesis of flavonoids in woodland strawberry. Front. Plant Sci. 2021, 12, 774943. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, L.; Zhang, N.; Zhang, L.; Liu, Y.; Lin, Y.; Zhang, Y.; Luo, Y.; Zhang, Y.; Wang, Y.; et al. Regulation of flavonoids in strawberry fruits by FaMYB5/FaMYB10 dominated MYB-bHLH-WD40 ternary complexes. Front. Plant Sci. 2023, 14, 1145670. [Google Scholar] [CrossRef]
- Hu, X.; Liang, Z.; Sun, T.; Huang, L.; Wang, Y.; Chan, Z.; Xiang, L. The R2R3-MYB transcriptional repressor TgMYB4 negatively regulates anthocyanin biosynthesis in tulips (Tulipa gesneriana L.). Int. J. Mol. Sci. 2024, 25, 563. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, Y.; Li, L.; Meng, H.; Yang, Y.; Dong, Z.; Wang, L.; Wu, G. Genome-wide identification and characterization of bHLH transcription factors related to anthocyanin biosynthesis in red walnut (Juglans regia L.). Front. Genet. 2021, 12, 632509. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, C.L.; Cui, M.; Zhou, W.J.; Wu, H.L.; Ling, H.Q. Four IVa bHLH transcription factors are novel interactors of FIT and mediate JA inhibition of iron uptake in Arabidopsis. Mol. Plant 2018, 11, 1166–1183. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; An, X.H.; Liu, X.; Hu, D.G.; Wang, X.F.; You, C.X.; Hao, Y.J. MdSnRK1.1 interacts with MdJAZ18 to regulate sucrose-induced anthocyanin and proanthocyanidin accumulation in apple. J. Exp. Bot. 2017, 68, 2977–2990. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Yingqi, H.; Ahmad, N.; Yuanyuan, T.; Jianyu, L.; Liyan, W.; Gang, W.; Xiuming, L.; Yuanyuan, D.; Fawei, W.; Weican, L.; et al. Genome-wide identification, expression analysis, and subcellular localization of Carthamus tinctorius bHLH transcription factors. Int. J. Mol. Sci. 2019, 20, 3044. [Google Scholar] [CrossRef] [PubMed]
- Vision, T.J.; Brown, D.G.; Tanksley, S.D. The origins of genomic duplications in Arabidopsis. Science 2000, 290, 2114–2117. [Google Scholar] [CrossRef]
- Fan, Y.; Yang, H.; Lai, D.; He, A.; Xue, G.; Feng, L.; Chen, L.; Cheng, X.B.; Ruan, J.; Yan, J.; et al. Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in sorghum [Sorghum bicolor (L.) Moench]. BMC Genom. 2021, 22, 415. [Google Scholar] [CrossRef]
- Wu, Z.; Zeng, W.; Li, C.; Wang, J.; Shang, X.; Xiao, L.; Cao, S.; Zhang, Y.; Xu, S.; Yan, H. Genome-wide identification and expression pattern analysis of R2R3-MYB transcription factor gene family involved in puerarin biosynthesis and response to hormone in Pueraria lobata var. thomsonii. BMC Plant Biol. 2023, 23, 107. [Google Scholar] [CrossRef]
- Yao, S.; Lan, Z.; Huang, R.; Tan, Y.; Huang, D.; Gu, J.; Pan, C. Hormonal and transcriptional analyses provides new insights into the molecular mechanisms underlying root thickening and isoflavonoid biosynthesis in Callerya speciosa (Champ. ex Benth.) Schot. Sci. Rep. 2021, 11, 9. [Google Scholar] [CrossRef]
- Yu, L.; Huang, D.; Gu, J.; Pan, D.; Tan, Y.; Huang, R.; Yao, S. Identification of isoflavonoid biosynthesis-related R2R3-MYB transcription factors in Callerya speciosa (Champ. ex Benth.) Schot using transcriptome-based gene coexpression analysis. Int. J. Genom. 2021, 2021, 9939403. [Google Scholar] [CrossRef]
- Zhou, W.; Shi, M.; Deng, C.; Lu, S.; Huang, F.; Wang, Y.; Kai, G. The methyl jasmonate-responsive transcription factor SmMYB1 promotes phenolic acid biosynthesis in Salvia miltiorrhiza. Hortic. Res. 2021, 8, 10. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Chao, J.; Li, Z.; Sun, Y.; Aluko, O.O.; Wu, X.; Wang, Q.; Liu, G. MG2C: A user-friendly online tool for drawing genetic maps. Mol. Hortic. 2021, 1, 16. [Google Scholar] [CrossRef]
- Yu, L.; Ming, R.; Huang, D.; Qin, C.; Li, L.; Tan, Y.; Huang, R.; Yao, S. Selection and validation of suitable reference genes for gene expression studies in Callerya speciosa (Champ. ex Benth.) Schot under different experimental conditions. ACS Agric. Sci. Technol. 2022, 2, 1276–1284. [Google Scholar] [CrossRef]
- He, J.; Xu, Y.; Huang, D.; Fu, J.; Liu, Z.; Wang, L.; Zhang, Y.; Xu, R.; Li, L.; Deng, X.; et al. TRIPTYCHON-LIKE regulates aspects of both fruit flavor and color in citrus. J. Exp. Bot. 2022, 73, 3610–3624. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Tan, X.; Ming, R.; Huang, D.; Tan, Y.; Li, L.; Huang, R.; Yao, S. Genome-Wide Identification of the bHLH Gene Family in Callerya speciosa Reveals Its Potential Role in the Regulation of Isoflavonoid Biosynthesis. Int. J. Mol. Sci. 2024, 25, 11900. https://doi.org/10.3390/ijms252211900
Chen L, Tan X, Ming R, Huang D, Tan Y, Li L, Huang R, Yao S. Genome-Wide Identification of the bHLH Gene Family in Callerya speciosa Reveals Its Potential Role in the Regulation of Isoflavonoid Biosynthesis. International Journal of Molecular Sciences. 2024; 25(22):11900. https://doi.org/10.3390/ijms252211900
Chicago/Turabian StyleChen, Liuping, Xiaoming Tan, Ruhong Ming, Ding Huang, Yong Tan, Liangbo Li, Rongshao Huang, and Shaochang Yao. 2024. "Genome-Wide Identification of the bHLH Gene Family in Callerya speciosa Reveals Its Potential Role in the Regulation of Isoflavonoid Biosynthesis" International Journal of Molecular Sciences 25, no. 22: 11900. https://doi.org/10.3390/ijms252211900
APA StyleChen, L., Tan, X., Ming, R., Huang, D., Tan, Y., Li, L., Huang, R., & Yao, S. (2024). Genome-Wide Identification of the bHLH Gene Family in Callerya speciosa Reveals Its Potential Role in the Regulation of Isoflavonoid Biosynthesis. International Journal of Molecular Sciences, 25(22), 11900. https://doi.org/10.3390/ijms252211900