
Genome-Wide Association Study of Sweet Potato Storage Root Traits Using GWASpoly, a Gene Dosage-Sensitive Model
 , and
, and 
Abstract
1. Introduction
2. Results
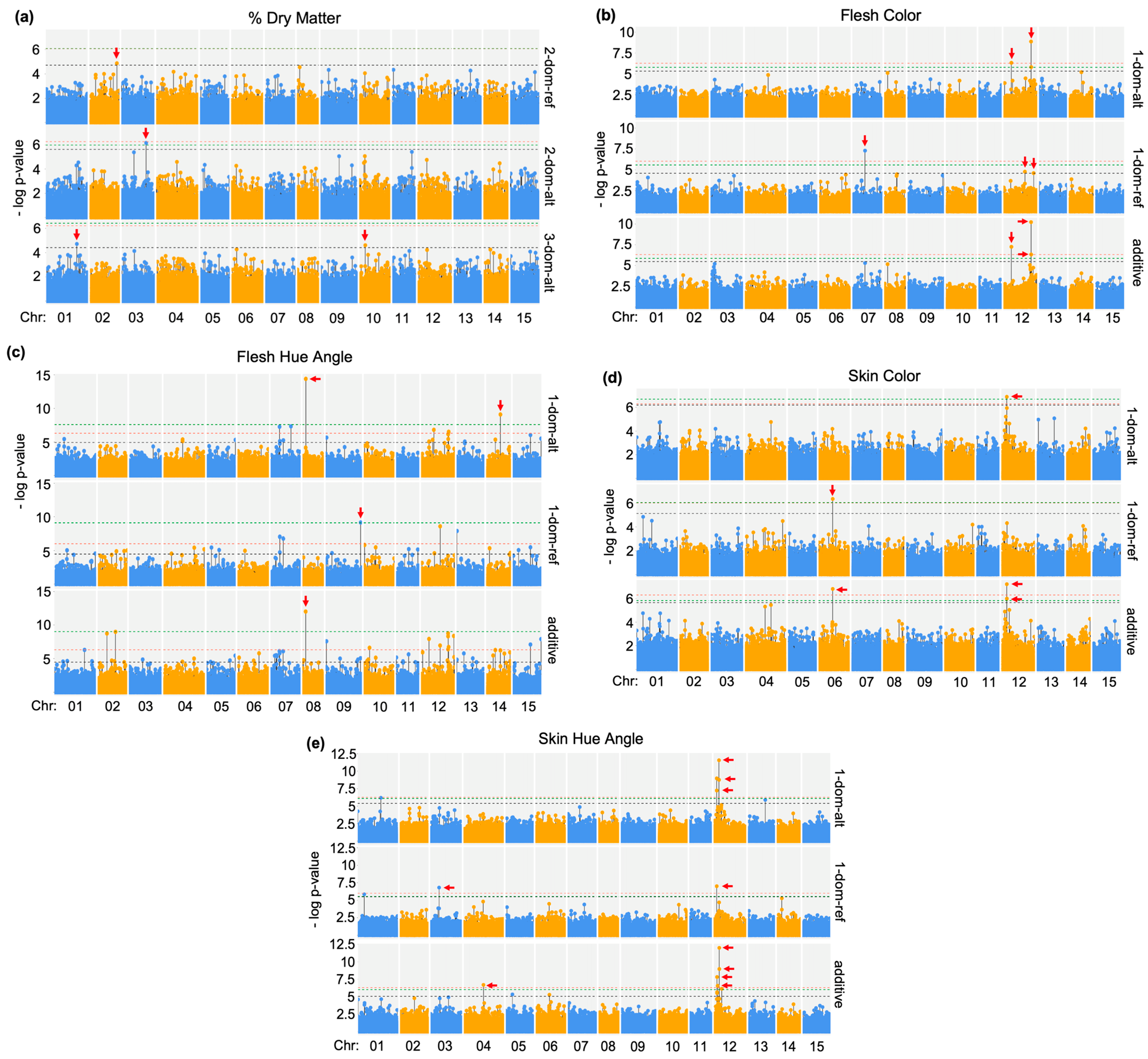
2.1. Significant SNPs Associated with Storage Root Traits
2.2. I. trifida and I. batatas Genes Near Root Trait SNPs
2.3. Candidate Genes Associated with Dry Matter SNPs
2.4. Candidate Genes from Sweet Potato Flesh Color-Associated SNPs
2.5. Candidate Genes from Sweet Potato Skin Color-Associated SNPs
3. Discussion
3.1. Genetic Regulation of Sweet Potato Storage Root Traits
3.2. Metabolism of Sweet Potato Storage Root Starch, Carotenoids, and Anthocyanins
3.3. Transmembrane Transporters Affect Sweet Potato Metabolism
3.4. Transcription Factors Implicated in Expression of Sweet Potato Storage Root Traits
3.5. Sweet Potato Genomic Resources
4. Materials and Methods
4.1. Plant Material
4.2. Storage Root Phenotypic Data
4.3. DNA Isolation and Genotyping
4.4. Genome-Wide Association Study Analyses
4.5. Identification of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motsa, N.M.; Modi, A.T.; Mabhaudhi, T. Sweet potato (Ipomoea batatas L.) as a drought tolerant and food security crop. S. Afr. J. Sci. 2015, 111, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sapakhova, Z.; Raissova, N.; Daurov, D.; Zhapar, K.; Daurova, A.; Zhigailov, A.; Zhambakin, K.; Shamekova, M. Sweet Potato as a Key Crop for Food Security under the Conditions of Global Climate Change: A Review. Plants 2023, 12, 2516. [Google Scholar] [CrossRef] [PubMed]
- Laveriano-Santos, E.P.; López-Yerena, A.; Jaime-Rodríguez, C.; González-Coria, J.; Lamuela-Raventós, R.M.; Vallverdú-Queralt, A.; Romanyà, J.; Pérez, M. Sweet Potato Is Not Simply an Abundant Food Crop: A Comprehensive Review of Its Phytochemical Constituents, Biological Activities, and the Effects of Processing. Antioxidants 2022, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Ziska, L.H.; Runion, G.B.; Tomecek, M.; Prior, S.A.; Torbet, H.A.; Sicher, R. An evaluation of cassava, sweet potato and field corn as potential carbohydrate sources for bioethanol production in Alabama and Maryland. Biomass Bioenergy 2009, 33, 1503–1508. [Google Scholar] [CrossRef]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Report. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Yan, M.; Nie, H.; Wang, Y.; Wang, X.; Jarret, R.; Zhao, J.; Wang, H.; Yang, J. Exploring and exploiting genetics and genomics for sweetpotato improvement: Status and perspectives. Plant Commun. 2022, 3, 100332. [Google Scholar] [CrossRef]
- Wu, S.; Lau, K.H.; Cao, Q.; Hamilton, J.P.; Sun, H.; Zhou, C.; Eserman, L.; Gemenet, D.C.; Olukolu, B.A.; Wang, H.; et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 2018, 9, 4580. [Google Scholar] [CrossRef]
- Wadl, P.A.; Olukolu, B.A.; Branham, S.E.; Jarret, R.L.; Yencho, G.C.; Jackson, D.M. Genetic Diversity and Population Structure of the USDA Sweetpotato (Ipomoea batatas) Germplasm Collections Using GBSpoly. Front. Plant Sci. 2018, 9, 1166. [Google Scholar] [CrossRef]
- Slonecki, T.J.; Rutter, W.B.; Olukolu, B.A.; Yencho, G.C.; Jackson, D.M.; Wadl, P.A. Genetic diversity, population structure, and selection of breeder germplasm subsets from the USDA sweetpotato (Ipomoea batatas) collection. Front. Plant Sci. 2023, 13, 1022555. [Google Scholar] [CrossRef]
- Mollinari, M.; Olukolu, B.A.; Pereira, G.d.S.; Khan, A.; Gemenet, D.; Yencho, G.C.; Zeng, Z.-B. Unraveling the Hexaploid Sweetpotato Inheritance Using Ultra-Dense Multilocus Mapping. G3 Genes Genomes Genet. 2020, 10, 281–292. [Google Scholar] [CrossRef]
- Cervantes-Flores, J.C.; Sosinski, B.; Pecota, K.V.; Mwanga, R.O.M.; Catignani, G.L.; Truong, V.D.; Watkins, R.H.; Ulmer, M.R.; Yencho, G.C. Identification of quantitative trait loci for dry-matter, starch, and β-carotene content in sweetpotato. Mol. Breed. 2010, 28, 201–216. [Google Scholar] [CrossRef]
- Haque, E.; Tabuchi, H.; Monden, Y.; Suematsu, K.; Shirasawa, K.; Isobe, S.; Tanaka, M. QTL analysis and GWAS of agronomic traits in sweetpotato (Ipomoea batatas L.) using genome wide SNPs. Breed. Sci. 2020, 70, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Gemenet, D.C.; da Silva Pereira, G.; De Boeck, B.; Wood, J.C.; Mollinari, M.; Olukolu, B.A.; Diaz, F.; Mosquera, V.; Ssali, R.T.; David, M.; et al. Quantitative trait loci and differential gene expression analyses reveal the genetic basis for negatively associated β-carotene and starch content in hexaploid sweetpotato [Ipomoea batatas (L.) Lam.]. Theor. Appl. Genet. 2019, 133, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, J.; Dong, X.; Han, Y.; Ma, Q.; Ding, Y.; Zhao, F.; Zhang, J.; Chen, H.; Xu, Q.; et al. Carotenoid accumulation affects redox status, starch metabolism, and flavonoid/anthocyanin accumulation in citrus. BMC Plant Biol. 2015, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Allen, L.H.; Bhutta, Z.A.; Caulfield, L.E.; de Onis, M.; Ezzati, M.; Mathers, C.; Rivera, J. Maternal and child undernutrition: Global and regional exposures and health consequences. Lancet 2008, 371, 243–260. [Google Scholar] [CrossRef]
- Low, J.W.; Mwanga, R.O.M.; Andrade, M.; Carey, E.; Ball, A.-M. Tackling vitamin A deficiency with biofortified sweetpotato in sub-Saharan Africa. Glob. Food Secur. 2017, 14, 23–30. [Google Scholar] [CrossRef]
- Torres, A.; Aguilar-Osorio, G.; Camacho, M.; Basurto, F.; Navarro-Ocana, A. Characterization of polyphenol oxidase from purple sweet potato (Ipomoea batatas L. Lam) and its affinity towards acylated anthocyanins and caffeoylquinic acid derivatives. Food Chem. 2021, 356, 129709. [Google Scholar] [CrossRef]
- Yang, J.; Moeinzadeh, M.H.; Kuhl, H.; Helmuth, J.; Xiao, P.; Haas, S.; Liu, G.; Zheng, J.; Sun, Z.; Fan, W.; et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants 2017, 3, 696–703. [Google Scholar] [CrossRef]
- Molitor, C.; Mauracher, S.G.; Rompel, A. Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases. Proc. Natl. Acad. Sci. USA 2016, 113, E1806–E1815. [Google Scholar] [CrossRef]
- Araji, S.; Grammer, T.A.; Gertzen, R.; Anderson, S.D.; Mikulic-Petkovsek, M.; Veberic, R.; Phu, M.L.; Solar, A.; Leslie, C.A.; Dandekar, A.M.; et al. Novel Roles for the Polyphenol Oxidase Enzyme in Secondary Metabolism and the Regulation of Cell Death in Walnut. Plant Physiol. 2014, 164, 1191–1203. [Google Scholar] [CrossRef]
- Borghi, L.; Kang, J.; de Brito Francisco, R. Filling the Gap: Functional Clustering of ABC Proteins for the Investigation of Hormonal Transport in planta. Front. Plant Sci. 2019, 10, 422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lai, Z.; Shi, J.; Xiao, Y.; Chen, Z.; Xu, X. Roles of arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Hadley, B.; Litfin, T.; Day, C.J.; Haselhorst, T.; Zhou, Y.; Tiralongo, J. Nucleotide Sugar Transporter SLC35 Family Structure and Function. Comput. Struct. Biotechnol. J. 2019, 17, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zhang, J.; Wang, X.; Han, X.; Wei, B.; Wang, J.; Li, B.; Yu, H.; Huang, Q.; Gu, H.; et al. The WRKY Transcription Factor WRKY71/EXB1 Controls Shoot Branching by Transcriptionally Regulating RAX Genes in Arabidopsis. Plant Cell 2015, 27, 3112–3127. [Google Scholar] [CrossRef]
- Tan, C.; Yang, J.; Xue, X.; Wei, J.; Li, H.; Li, Z.; Duan, Y. MsMYB62-like as a negative regulator of anthocyanin biosynthesis in Malus spectabilis. Plant Signal. Behav. 2024, 19, 2318509. [Google Scholar] [CrossRef]
- Oshima, Y.; Mitsuda, N. The MIXTA-like transcription factor MYB16 is a major regulator of cuticle formation in vegetative organs. Plant Signal. Behav. 2013, 8, e26826. [Google Scholar] [CrossRef]
- Zhao, S.; Zhao, X.; Xu, X.; Han, Z.; Qiu, C. Transcription Factor IAA27 Positively Regulates P Uptake through Promoted Adventitious Root Development in Apple Plants. Int. J. Mol. Sci. 2022, 23, 14029. [Google Scholar] [CrossRef]
- Kodama, M.; Brinch-Pedersen, H.; Sharma, S.; Holme, I.B.; Joernsgaard, B.; Dzhanfezova, T.; Amby, D.B.; Vieira, F.G.; Liu, S.; Gilbert, M.T.P. Identification of transcription factor genes involved in anthocyanin biosynthesis in carrot (Daucus carota L.) using RNA-Seq. BMC Genom. 2018, 19, 811. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B.; Yan, D.; Dong, W.; Yang, W.; Li, Q.; Zeng, L.; Wang, J.; Wang, L.; Hicks, L.M.; et al. Two Arabidopsis cytochrome P450 monooxygenases, CYP714A1 and CYP714A2, function redundantly in plant development through gibberellin deactivation. Plant J. 2011, 67, 342–353. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Palva, E.T. The WRKY70 transcription factor: A node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell 2004, 16, 319–331. [Google Scholar] [CrossRef]
- Wang, X.C.; Wu, J.; Guan, M.L.; Zhao, C.H.; Geng, P.; Zhao, Q. Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 2019, 101, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Van Eck, J.; Zhou, X.; Lopez, A.B.; O’Halloran, D.M.; Cosman, K.M.; Conlin, B.J.; Paolillo, D.J.; Garvin, D.F.; Vrebalov, J.; et al. The CauliflowerOrGene Encodes a DnaJ Cysteine-Rich Domain-Containing Protein That Mediates High Levels of β-Carotene Accumulation. Plant Cell 2006, 18, 3594–3605. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Yuan, H.; Chen, C.; Kadirjan-Kalbach, D.K.; Mazourek, M.; Osteryoung, K.W.; Li, L. ORHis, a Natural Variant of OR, Specifically Interacts with Plastid Division Factor ARC3 to Regulate Chromoplast Number and Carotenoid Accumulation. Mol. Plant 2020, 13, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, D.X. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl. Acad. Sci. USA 2008, 105, 13679–13684. [Google Scholar] [CrossRef] [PubMed]
- Soderman, E.; Mattsson, J.; Engstrom, P. The Arabidopsis homeobox gene ATHB-7 is induced by water deficit and by abscisic acid. Plant J. 1996, 10, 375–381. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.L.; Zhou, Y.K.; Guo, D.; Zhu, J.H.; Peng, S.Q. Transcriptomes analysis reveals novel insight into the molecular mechanisms of somatic embryogenesis in Hevea brasiliensis. BMC Genom. 2021, 22, 183. [Google Scholar] [CrossRef]
- Ballicora, M.A.; Iglesias, A.A.; Preiss, J. ADP-Glucose Pyrophosphorylase: A Regulatory Enzyme for Plant Starch Synthesis. Photosynth. Res. 2004, 79, 1–24. [Google Scholar] [CrossRef]
- Routaboul, J.M.; Dubos, C.; Beck, G.; Marquis, C.; Bidzinski, P.; Loudet, O.; Lepiniec, L. Metabolite profiling and quantitative genetics of natural variation for flavonoids in Arabidopsis. J. Exp. Bot. 2012, 63, 3749–3764. [Google Scholar] [CrossRef]
- Rehman, H.M.; Nawaz, M.A.; Shah, Z.H.; Ludwig-Müller, J.; Chung, G.; Ahmad, M.Q.; Yang, S.H.; Lee, S.I. Comparative genomic and transcriptomic analyses of Family-1 UDP glycosyltransferase in three Brassica species and Arabidopsis indicates stress-responsive regulation. Sci. Rep. 2018, 8, 1875. [Google Scholar] [CrossRef]
- Makkena, S.; Lamb, R.S. The bHLH transcription factor SPATULA regulates root growth by controlling the size of the root meristem. BMC Plant Biol. 2013, 13, 1. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Li, J.; Yang, Z.; Huang, H.; Xu, L. Imitation Switch chromatin remodeling factors and their interacting RINGLET proteins act together in controlling the plant vegetative phase in Arabidopsis. Plant J. 2012, 72, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.-T.; Xu, P.; Zhao, P.-X.; Liu, R.; Yu, L.-H.; Xiang, C.-B. Arabidopsis ERF109 mediates cross-talk between jasmonic acid and auxin biosynthesis during lateral root formation. Nat. Commun. 2014, 5, 5833. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, S.; Liu, D.; Patil, G.B.; Zhai, H.; Wang, F.; Stephenson, T.J.; Wang, Y.; Wang, B.; Valliyodan, B.; et al. A Sweetpotato Geranylgeranyl Pyrophosphate Synthase Gene, IbGGPS, Increases Carotenoid Content and Enhances Osmotic Stress Tolerance in Arabidopsis thaliana. PLoS ONE 2015, 10, e0137623. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ahn, Y.O.; Ahn, M.J.; Lee, H.S.; Kwak, S.S. Down-regulation of beta-carotene hydroxylase increases beta-carotene and total carotenoids enhancing salt stress tolerance in transgenic cultured cells of sweetpotato. Phytochemistry 2012, 74, 69–78. [Google Scholar] [CrossRef]
- Gu, Y.; Han, S.; Chen, L.; Mu, J.; Duan, L.; Li, Y.; Yan, Y.; Li, X. Expression and regulation of genes involved in the reserve starch biosynthesis pathway in hexaploid wheat (Triticum aestivum L.). Crop J. 2021, 9, 440–455. [Google Scholar] [CrossRef]
- Isaioglou, I.; Podia, V.; Velentzas, A.D.; Kapolas, G.; Beris, D.; Karampelias, M.; Plitsi, P.K.; Chatzopoulos, D.; Samakovli, D.; Roussis, A.; et al. APRF1 Interactome Reveals HSP90 as a New Player in the Complex That Epigenetically Regulates Flowering Time in Arabidopsis thaliana. Int. J. Mol. Sci. 2024, 25, 1313. [Google Scholar] [CrossRef]
- Cervantes-Flores, J.C.; Yencho, G.C.; Kriegner, A.; Pecota, K.V.; Faulk, M.A.; Mwanga, R.O.M.; Sosinski, B.R. Development of a genetic linkage map and identification of homologous linkage groups in sweetpotato using multiple-dose AFLP markers. Mol. Breed. 2007, 21, 511–532. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Y.; Shi, T.; Kou, M.; Sun, J.; Xu, T.; Li, Q.; Wu, S.; Cao, Q.; Hou, W.; et al. Genome-wide analysis of expression quantitative trait loci (eQTLs) reveals the regulatory architecture of gene expression variation in the storage roots of sweet potato. Hortic. Res. 2020, 7, 90. [Google Scholar] [CrossRef]
- Ding, N.; Wang, A.; Zhang, X.; Wu, Y.; Wang, R.; Cui, H.; Huang, R.; Luo, Y. Identification and analysis of glutathione S-transferase gene family in sweet potato reveal divergent GST-mediated networks in aboveground and underground tissues in response to abiotic stresses. BMC Plant Biol. 2017, 17, 225. [Google Scholar] [CrossRef]
- Tanaka, M.; Takahata, Y.; Kurata, R.; Nakayama, H.; Yoshinaga, M. Structural and functional characterization of IbMYB1 genes in recent Japanese purple-fleshed sweetpotato cultivars. Mol. Breed. 2011, 29, 565–574. [Google Scholar] [CrossRef]
- Lloyd, J.R.; Kossmann, J. Starch Trek: The Search for Yield. Front. Plant Sci. 2019, 9, 1930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Zhang, H.; Zhai, H.; Liu, Q.; He, S. A soluble starch synthase I gene, IbSSI, alters the content, composition, granule size and structure of starch in transgenic sweet potato. Sci. Rep. 2017, 7, 2315. [Google Scholar] [CrossRef]
- Wang, H.; Wu, Y.; Zhang, Y.; Yang, J.; Fan, W.; Zhang, H.; Zhao, S.; Yuan, L.; Zhang, P. CRISPR/Cas9-Based Mutagenesis of Starch Biosynthetic Genes in Sweet Potato (Ipomoea batatas) for the Improvement of Starch Quality. Int. J. Mol. Sci. 2019, 20, 4702. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Cross, P.J.; Dobson, R.C.J.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A Three-Ring Circus: Metabolism of the Three Proteogenic Aromatic Amino Acids and Their Role in the Health of Plants and Animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef]
- Baxter, I.R.; Young, J.C.; Armstrong, G.; Foster, N.; Bogenschutz, N.; Cordova, T.; Peer, W.A.; Hazen, S.P.; Murphy, A.S.; Harper, J.F. A plasma membrane H+-ATPase is required for the formation of proanthocyanidins in the seed coat endothelium of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2005, 102, 2649–2654. [Google Scholar] [CrossRef]
- Michalak, A.; Wdowikowska, A.; Janicka, M. Plant Plasma Membrane Proton Pump: One Protein with Multiple Functions. Cells 2022, 11, 4052. [Google Scholar] [CrossRef]
- Knappe, S.; Löttgert, T.; Schneider, A.; Voll, L.; Flügge, U.I.; Fischer, K. Characterization of two functional phosphoenolpyruvate/phosphate translocator (PPT) genes in Arabidopsis–AtPPT1 may be involved in the provision of signals for correct mesophyll development. Plant J. 2003, 36, 411–420. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Wang, L.; Tang, W.; Hu, Y.; Zhang, Y.; Sun, J.; Guo, X.; Lu, H.; Yang, Y.; Fang, C.; Niu, X.; et al. A MYB/bHLH complex regulates tissue-specific anthocyanin biosynthesis in the inner pericarp of red-centered kiwifruit Actinidia chinensis cv. Hongyang. Plant J. 2019, 99, 359–378. [Google Scholar] [CrossRef]
- Bello, B.K.; Hou, Y.; Zhao, J.; Jiao, G.; Wu, Y.; Li, Z.; Wang, Y.; Tong, X.; Wang, W.; Yuan, W.; et al. NF-YB1-YC12-bHLH144 complex directly activates Wx to regulate grain quality in rice (Oryza sativa L.). Plant Biotechnol. J. 2019, 17, 1222–1235. [Google Scholar] [CrossRef]
- Lau, O.S.; Davies, K.A.; Chang, J.; Adrian, J.; Rowe, M.H.; Ballenger, C.E.; Bergmann, D.C. Direct roles of SPEECHLESS in the specification of stomatal self-renewing cells. Science 2014, 345, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Makkena, S.; Lamb, R.S. The bHLH transcription factor SPATULA is a key regulator of organ size in Arabidopsis thaliana. Plant Signal. Behav. 2013, 8, e24140. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, C.L.; Cui, M.; Zhou, W.J.; Wu, H.L.; Ling, H.Q. Four IVa bHLH Transcription Factors Are Novel Interactors of FIT and Mediate JA Inhibition of Iron Uptake in Arabidopsis. Mol. Plant 2018, 11, 1166–1183. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Choi, Y.D. Direct regulation of WRKY70 by AtMYB44 in plant defense responses. Plant Signal. Behav. 2013, 8, e20783. [Google Scholar]
- Muller, D.; Schmitz, G.; Theres, K. Blind homologous R2R3 Myb genes control the pattern of lateral meristem initiation in Arabidopsis. Plant Cell 2006, 18, 586–597. [Google Scholar] [CrossRef]
- Hirai, H.; Sherr, C.J. Interaction of D-type cyclins with a novel myb-like transcription factor, DMP1. Mol. Cell. Biol. 1996, 16, 6457–6467. [Google Scholar] [CrossRef]
- Liu, Y.; Mattila, J.; Ventela, S.; Yadav, L.; Zhang, W.; Lamichane, N.; Sundstrom, J.; Kauko, O.; Grenman, R.; Varjosalo, M.; et al. PWP1 Mediates Nutrient-Dependent Growth Control through Nucleolar Regulation of Ribosomal Gene Expression. Dev. Cell 2017, 43, 240–252.e5. [Google Scholar] [CrossRef]
- Ahn, C.S.; Cho, H.K.; Lee, D.H.; Sim, H.J.; Kim, S.G.; Pai, H.S. Functional characterization of the ribosome biogenesis factors PES, BOP1, and WDR12 (PeBoW), and mechanisms of defective cell growth and proliferation caused by PeBoW deficiency in Arabidopsis. J. Exp. Bot. 2016, 67, 5217–5232. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Yang, B.; Xu, S.; Wei, X.; Zhao, P.; Niu, F.; Sun, M.; Wang, C.; Cheng, H.; et al. WRKY55 transcription factor positively regulates leaf senescence and the defense response by modulating the transcription of genes implicated in the biosynthesis of reactive oxygen species and salicylic acid in Arabidopsis. Development 2020, 147, dev189647. [Google Scholar] [CrossRef]
- Li, R.; Liu, H.; Liu, Y.; Guo, J.; Chen, Y.; Lan, X.; Lu, C. Insights into the mechanism underlying UV-B induced flavonoid metabolism in callus of a Tibetan medicinal plant Mirabilis himalaica. J. Plant Physiol. 2023, 288, 154074. [Google Scholar] [CrossRef]
- Bahieldin, A.; Atef, A.; Edris, S.; Gadalla, N.O.; Ramadan, A.M.; Hassan, S.M.; Al Attas, S.G.; Al-Kordy, M.A.; Al-Hajar, A.S.M.; Sabir, J.S.M.; et al. Multifunctional activities of ERF109 as affected by salt stress in Arabidopsis. Sci. Rep. 2018, 8, 6403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, T.; Li, L.; Zhang, X.; Yang, T.; Liu, C.; Chu, J.; Zheng, B. The chromatin remodeling complex imitation of switch controls stamen filament elongation by promoting jasmonic acid biosynthesis in Arabidopsis. J. Genet. Genom. 2021, 48, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Tzuri, G.; Zhou, X.; Chayut, N.; Yuan, H.; Portnoy, V.; Meir, A.; Sa’ar, U.; Baumkoler, F.; Mazourek, M.; Lewinsohn, E.; et al. A ‘golden’ SNP in CmOr governs the fruit flesh color of melon (Cucumis melo). Plant J. 2015, 82, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Owsiany, K.; Sheeja, T.E.; Zhou, X.; Rodriguez, C.; Li, Y.; Welsch, R.; Chayut, N.; Yang, Y.; Thannhauser, T.W.; et al. A Single Amino Acid Substitution in an ORANGE Protein Promotes Carotenoid Overaccumulation in Arabidopsis. Plant Physiol. 2015, 169, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E.; Kim, H.S.; Wang, Z.; Ke, Q.; Lee, C.-J.; Park, S.-U.; Lim, Y.-H.; Park, W.S.; Ahn, M.-J.; Kwak, S.-S. A single amino acid change at position 96 (Arg to His) of the sweetpotato Orange protein leads to carotenoid overaccumulation. Plant Cell Rep. 2019, 38, 1393–1402. [Google Scholar] [CrossRef]
- Slater, A.T.; Cogan, N.O.I.; Forster, J.W.; Hayes, B.J.; Daetwyler, H.D. Improving Genetic Gain with Genomic Selection in Autotetraploid Potato. Plant Genome 2016, 9, plantgenome2016.02.0021. [Google Scholar] [CrossRef]
- Michael Jackson, D.; Harrison, H.F.; Jarret, R.L.; Wadl, P.A. Color analysis of storage roots from the USDA, ARS sweetpotato (Ipomoea batatas) germplasm collection. Genet. Resour. Crop Evol. 2018, 65, 1217–1236. [Google Scholar] [CrossRef]
- Huamán, Z. (Ed.) Descriptors for Sweet Potato; International Board for Plant Genetic Resources: Rome, Italy, 1991. [Google Scholar]
- Kuster, R.D.; Yencho, G.C.; Olukolu, B.A. ngsComposer: An automated pipeline for empirically based NGS data quality filtering. Brief. Bioinform. 2021, 22, bbab092. [Google Scholar] [CrossRef]
- Gerard, D. Pairwise linkage disequilibrium estimation for polyploids. Mol. Ecol. Resour. 2021, 21, 1230–1242. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for Genome-Wide Association Studies in Autopolyploids and Its Application to Potato. Plant Genome 2016, 9, plantgenome2015.08.0073. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef] [PubMed]
- Neff, M.M.; Neff, J.D.; Chory, J.; Pepper, A.E. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: Experimental applications in Arabidopsis thaliana genetics. Plant J. 1998, 14, 387–392. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Trait | SNP Name | Genomic Position |
|---|---|---|
| Dry Matter | DM1 | Chr01: 27,238,168 |
| DM2 | Chr02: 23,017,613 | |
| DM3* | Chr03: 18,888,440 * | |
| DM4 | Chr10: 3,229,129 | |
| Flesh Color | FC1 | Chr07: 5,556,076 |
| FC2 | Chr12: 3,933,566 | |
| FC3 | Chr12: 18,956,680 | |
| FC4 | Chr12: 22,042,439 | |
| FC5 | Chr12: 22,064,285 | |
| FC6 | Chr12: 23,146,784 | |
| Flesh Hue Angle | FHA1 | Chr08: 1,457,982 |
| FHA2 | Chr09: 23,213,578 | |
| FHA3 | Chr14: 11,541,393 | |
| Skin Color and Hue Angle | SC1 | Chr06: 18,709,220 |
| SC2, SHA1 | Chr12: 2,823,583 | |
| SC3, SHA2 | Chr12: 2,841,327 | |
| SHA3 | Chr01: 3,487,779 | |
| SHA4 | Chr01: 23,116,643 | |
| SHA5 | Chr03: 5,991,712 | |
| SHA6 | Chr04: 12,982,060 | |
| SHA7* | Chr12: 1,568,751 * | |
| SHA8* | Chr12: 1,854,779 * | |
| SHA9 | Chr12: 4,042,610 |
| SNP | Gene ID | Annotation | Homologs | References into Function |
|---|---|---|---|---|
| DM3* | 03E_G012960 | unknown function DUF914 | SLC35 | solute carrier family 35 nucleotide sugar transporters, drug metabolite transporter family [23] |
| DM3* | 03D_G014090 | H+—ATPase | ATPase 4 | primary active transport; overexpression increases rice yield [56] |
| FC5 | 12C_G029170 | nucleotide-diphospho-sugar transferase family | unknown | predicted, putative ADP-glucose transporter |
| SHA9 | 12C_G008210 | phosphoenolpyruvate (PEP)/phosphate translocator | PPT2 | PEP/Pi Translocator, PEP transport for shikimate pathway [57] |
| SNP | Gene ID | Annotation | Homologs | References into Function |
|---|---|---|---|---|
| DM4 | 10A_G004370 | bHLH | bHLH 144 | starch synthesis in rice [60] |
| FC3 | 12D_G020790 | bHLH | bHLH 6 | anthocyanin biosynthesis in carrot [28] |
| FC6 | 12E_G025410 | bHLH | SPEECHLESS | stomatal lineage development, stemness [61] |
| FHA1 | 08F_G003550 | bHLH | bHLH 94 | development/embryogenesis [36] |
| SC2, SHA1 | 12D_G004200 | bHLH | SPATULA | root meristem development [62] |
| SHA5 | 03F_G010380 | bHLH | bHLH 18 | jasmonic acid regulated Fe2+ transport [63] |
| FC1 | 07A_G006800 | Myb | Myb 62 | negative regulator of anthocyanin biosynthesis [25] |
| FC2 | 12C_G007940 | Myb | Myb 16 | positive regulator cutin and wax biosynthetic genes [26] |
| SHA9 | 12B_G007380 | Myb | Myb 16 | |
| FC4, FC5 | 12C_G029230 | Myb | Myb 4 | negative regulator of anthocyanin biosynthesis [31] |
| SHA5 | 03B_G012820 | Myb | Myb 44 | salicylic and jasmonic acid signaling, WRKY70 regulation [64] |
| SHA8* | 12E_G003940 | Myb | RAX3 | meristem development [65] |
| SHA9 | 12C_G008030 | Myb-family | DMTF1 | cyclin D binding Myb Transcription Factor 1;, aka DMP1 [66] |
| DM1 | 01F_G025330 | WD40 repeat | PTP1 | periodic tryptophan protein 1, chromatin-associated, ribosome biogenesis (growth) [67] |
| SHA7 | 12C_G004320 | WD40 repeat | APRF1 | anthesis promoting factor 1, epigenetic regulation of flowering time, HSP90 interaction [46] |
| SHA9 | 12C_G008110 | WD40 repeat | WDR12 | ribosome biogenesis (growth), high expression in roots [68] |
| DM2 | 02B_G028270 | WRKY | WRKY 40 | transcriptional repressor of abscisic acid signaling [22] |
| DM5 | 10C_G004950 | WRKY | WRKY 71 | meristem development [24] |
| FC4, FC5 | 12F_G025420 | WRKY | WRKY 70 | integration of salicylic and jasmonic acid signaling, Myb-44 regulates [30] |
| FC4, FC5 | 12E_G024250 | WRKY | WRKY 55 | salicylic acid and ROS signaling [69] |
| SHA5 | 03B_G012780 | WRKY | WRKY 7 | salicylic acid responsive, regulation of flavonoid metabolism [70] |
| SHA4 | 01A_G021900 | Redox responsive | ERF109 | jasmonic acid, auxin signaling [71] |
| FHA1 | 08A_G002660 | jumonji | JMJ706 | chromatin remodeling H3K9 demethylase, rice flower development [34] |
| SHA3 | 01A_G008090 | homeobox-1 | RLT1 | chromatin remodeling, jasmonic acid biosynthesis [72] |
| FC3 | 12B_G022290 | Phytochrome-associated | IAA27-like | auxin signaling, root development [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowers, R.R.; Slonecki, T.J.; Olukolu, B.A.; Yencho, G.C.; Wadl, P.A. Genome-Wide Association Study of Sweet Potato Storage Root Traits Using GWASpoly, a Gene Dosage-Sensitive Model. Int. J. Mol. Sci. 2024, 25, 11727. https://doi.org/10.3390/ijms252111727
Bowers RR, Slonecki TJ, Olukolu BA, Yencho GC, Wadl PA. Genome-Wide Association Study of Sweet Potato Storage Root Traits Using GWASpoly, a Gene Dosage-Sensitive Model. International Journal of Molecular Sciences. 2024; 25(21):11727. https://doi.org/10.3390/ijms252111727
Chicago/Turabian StyleBowers, Robert R., Tyler J. Slonecki, Bode A. Olukolu, G. Craig Yencho, and Phillip A. Wadl. 2024. "Genome-Wide Association Study of Sweet Potato Storage Root Traits Using GWASpoly, a Gene Dosage-Sensitive Model" International Journal of Molecular Sciences 25, no. 21: 11727. https://doi.org/10.3390/ijms252111727
APA StyleBowers, R. R., Slonecki, T. J., Olukolu, B. A., Yencho, G. C., & Wadl, P. A. (2024). Genome-Wide Association Study of Sweet Potato Storage Root Traits Using GWASpoly, a Gene Dosage-Sensitive Model. International Journal of Molecular Sciences, 25(21), 11727. https://doi.org/10.3390/ijms252111727