
Unmasking Neuroendocrine Prostate Cancer with a Machine Learning-Driven Seven-Gene Stemness Signature That Predicts Progression

 , , , , , , , , ,
, , , , , , , , ,  and
and
Abstract
1. Introduction
2. Results
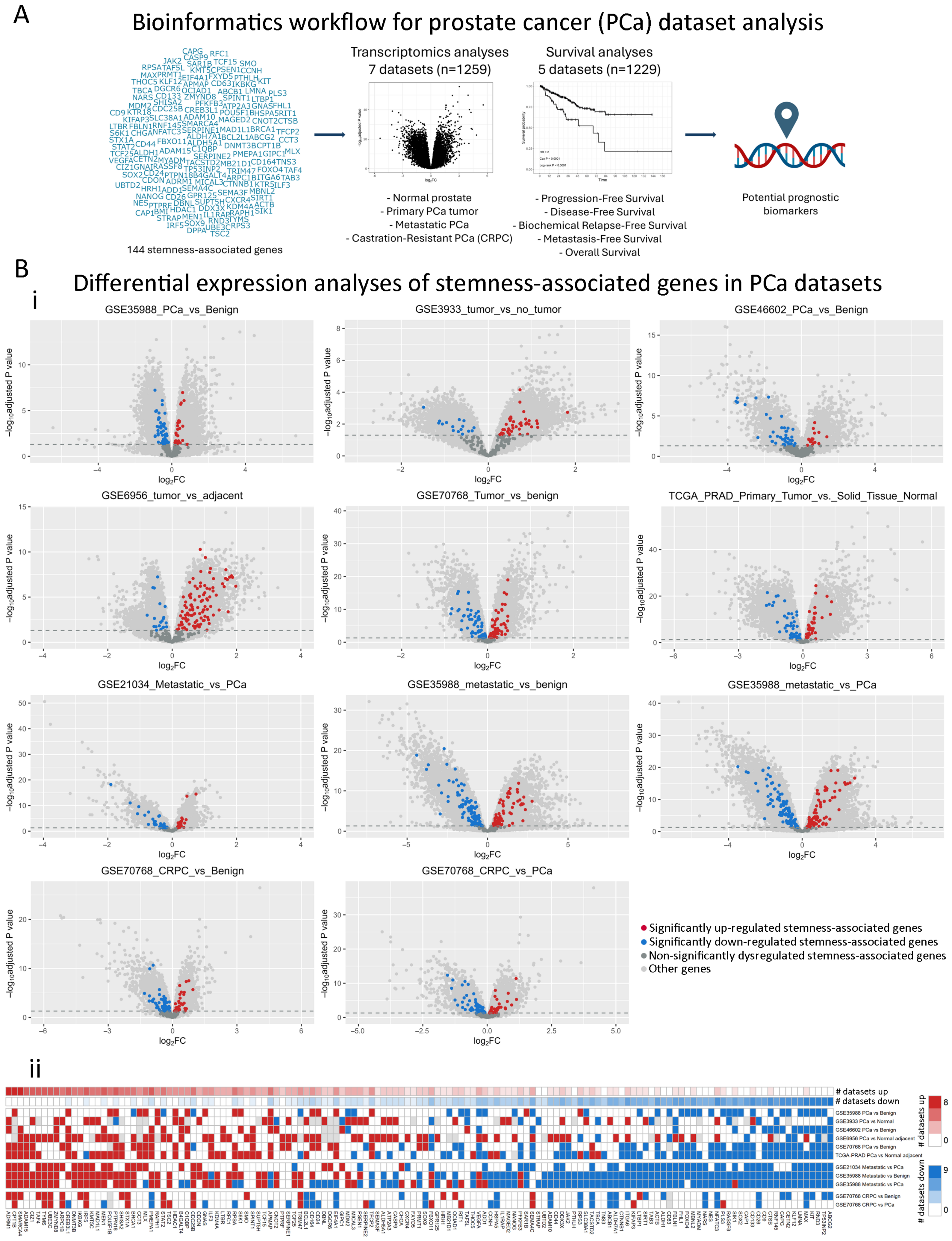
2.1. Dysregulation of Stemness-Associated Genes Across Multiple PCa Comparisons
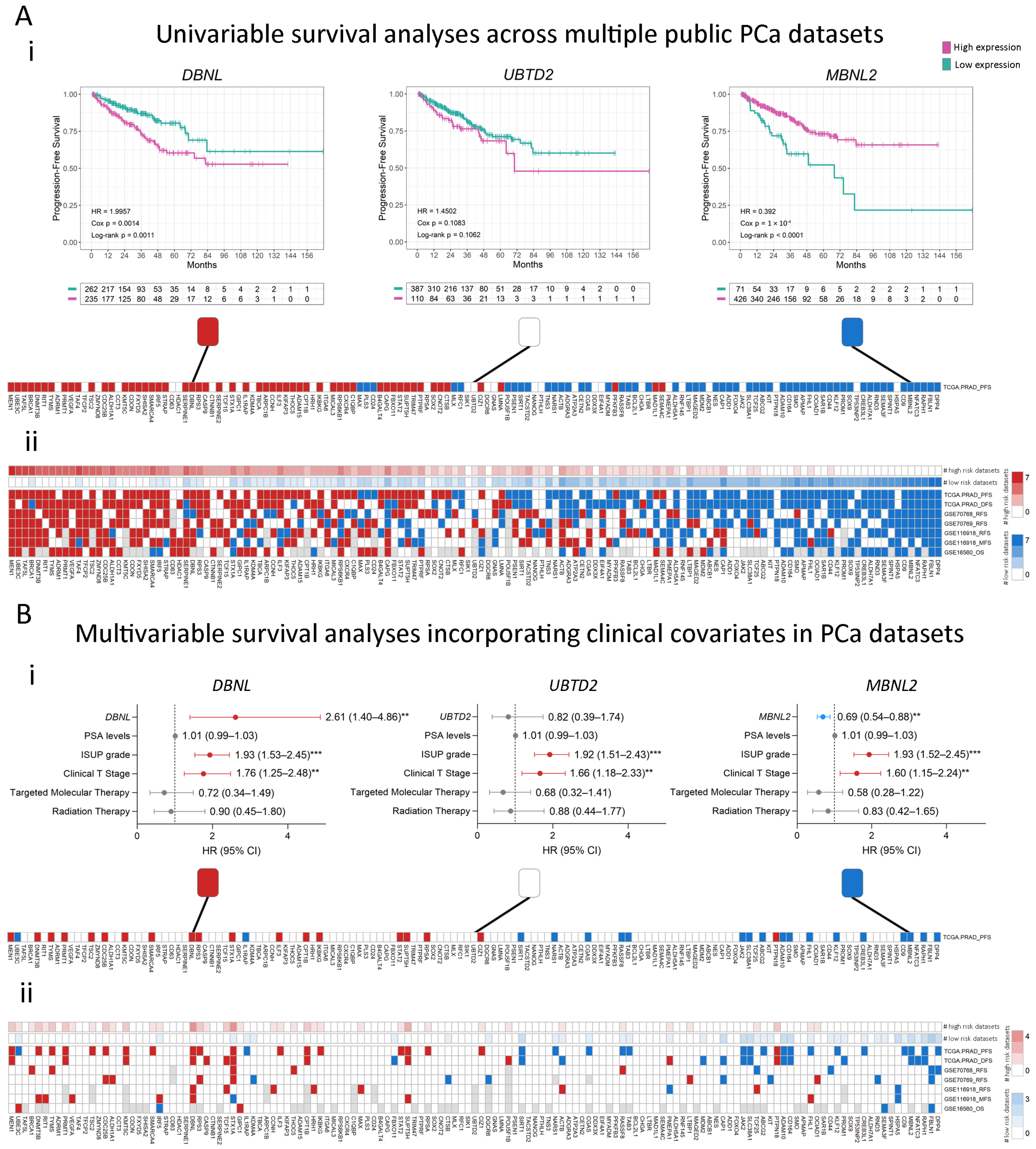
2.2. Association of Stemness Markers with PCa Patients’ Survival
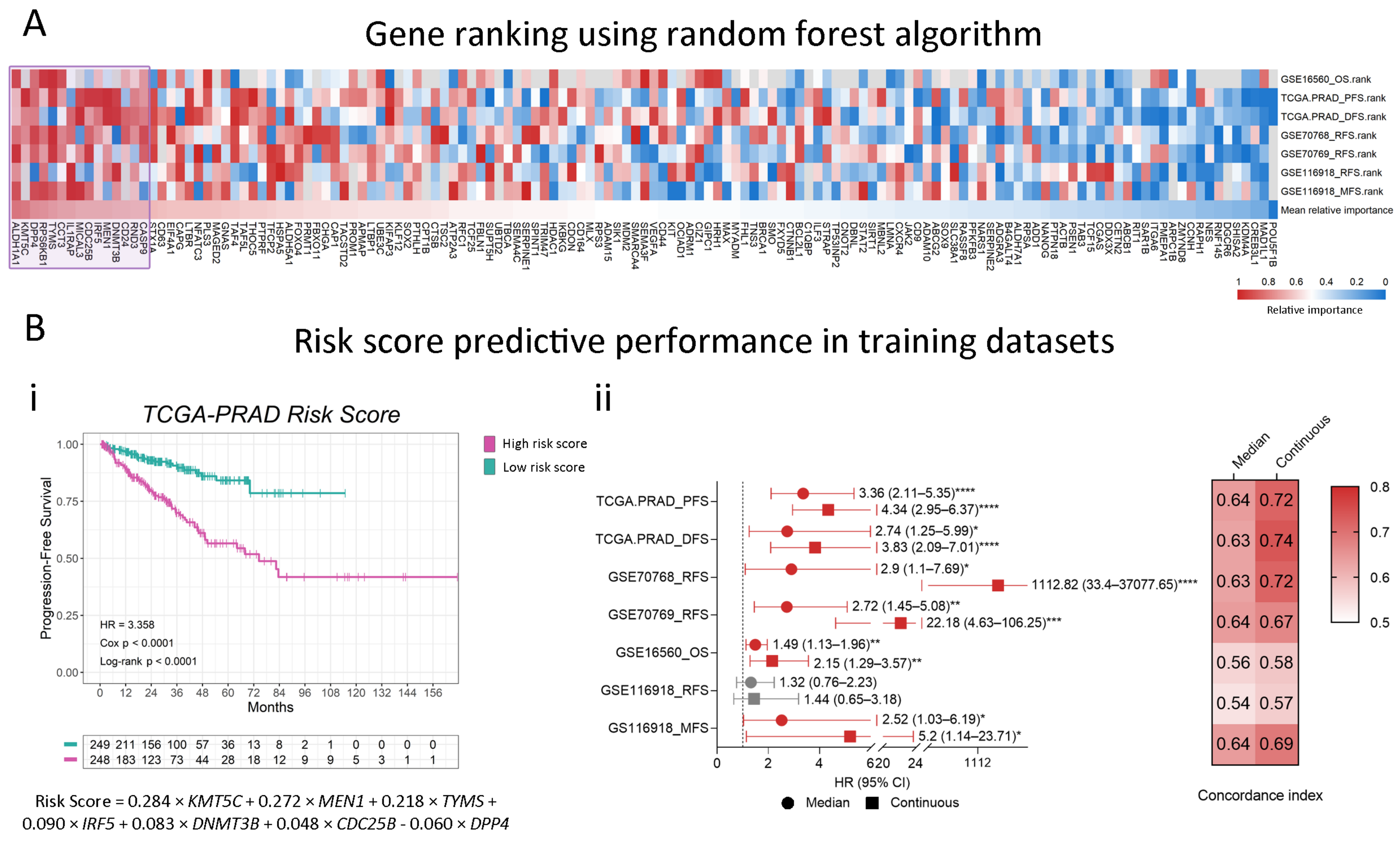
2.3. Modeling a Stemness-Associated Signature with Prognostic Value
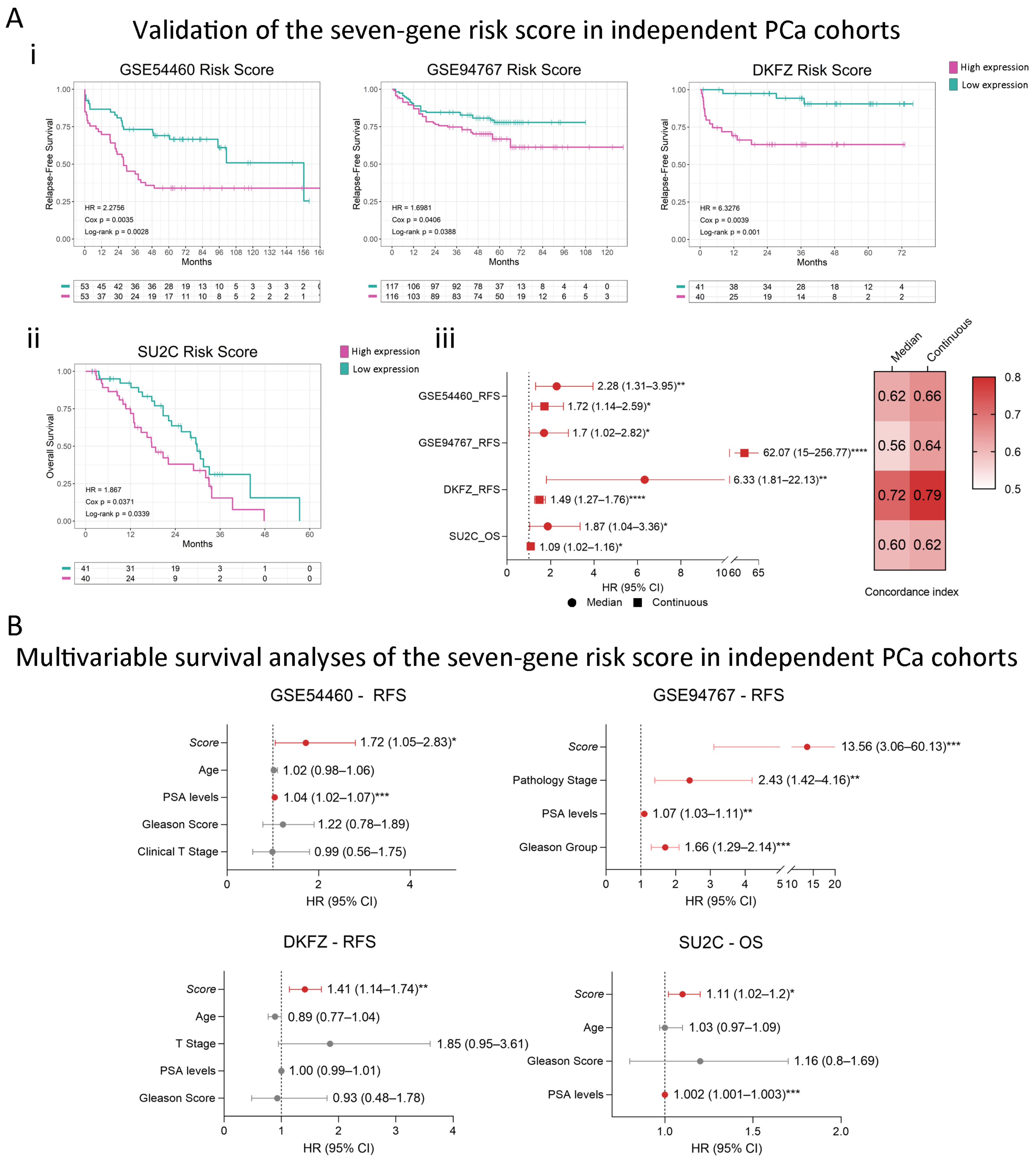
2.4. Consistent Performance Across Validation Datasets
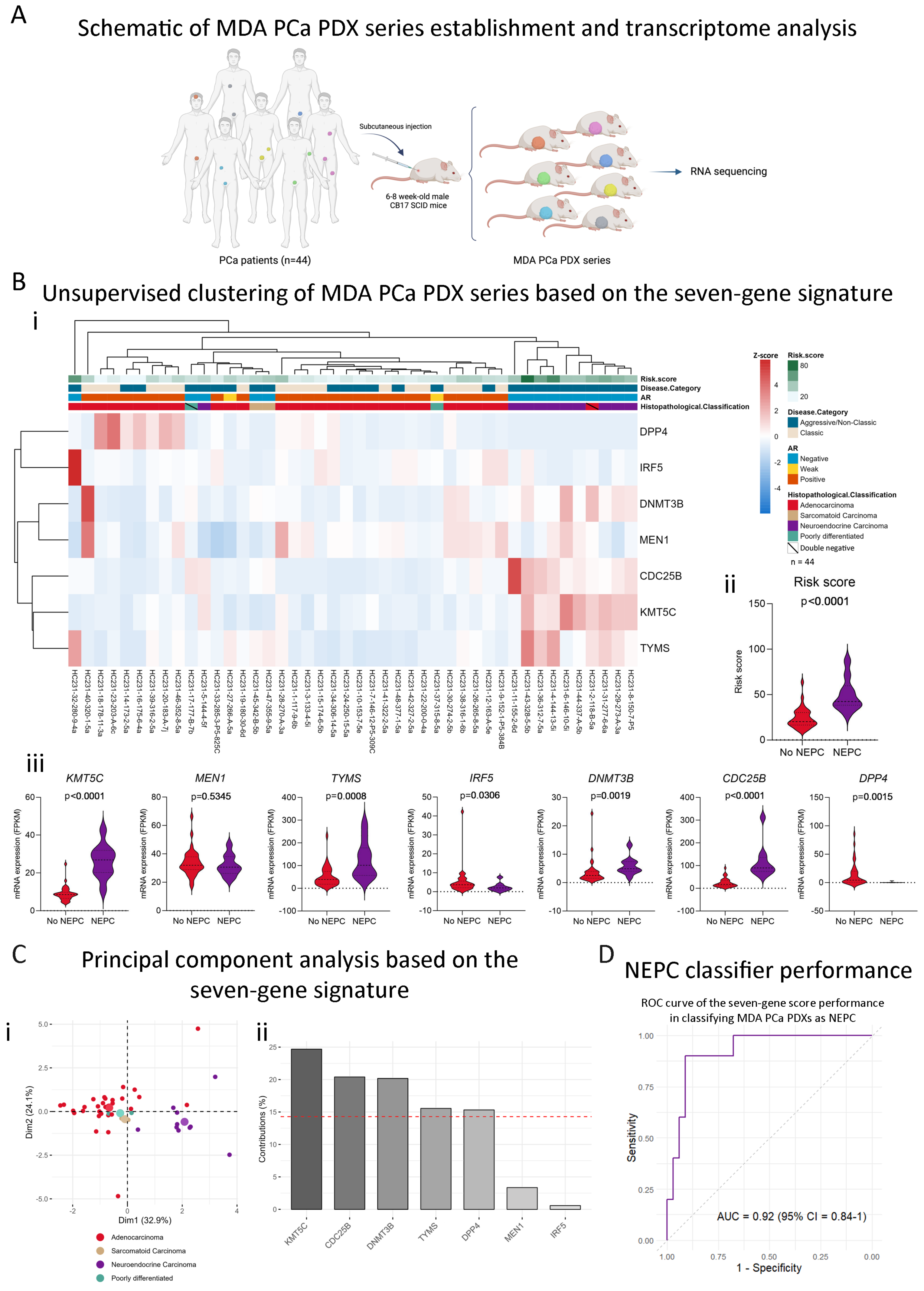
2.5. The Stemness-Associated Gene Signature Captures Neuroendocrine Disease Heterogeneity in the MDA PCa PDX Series
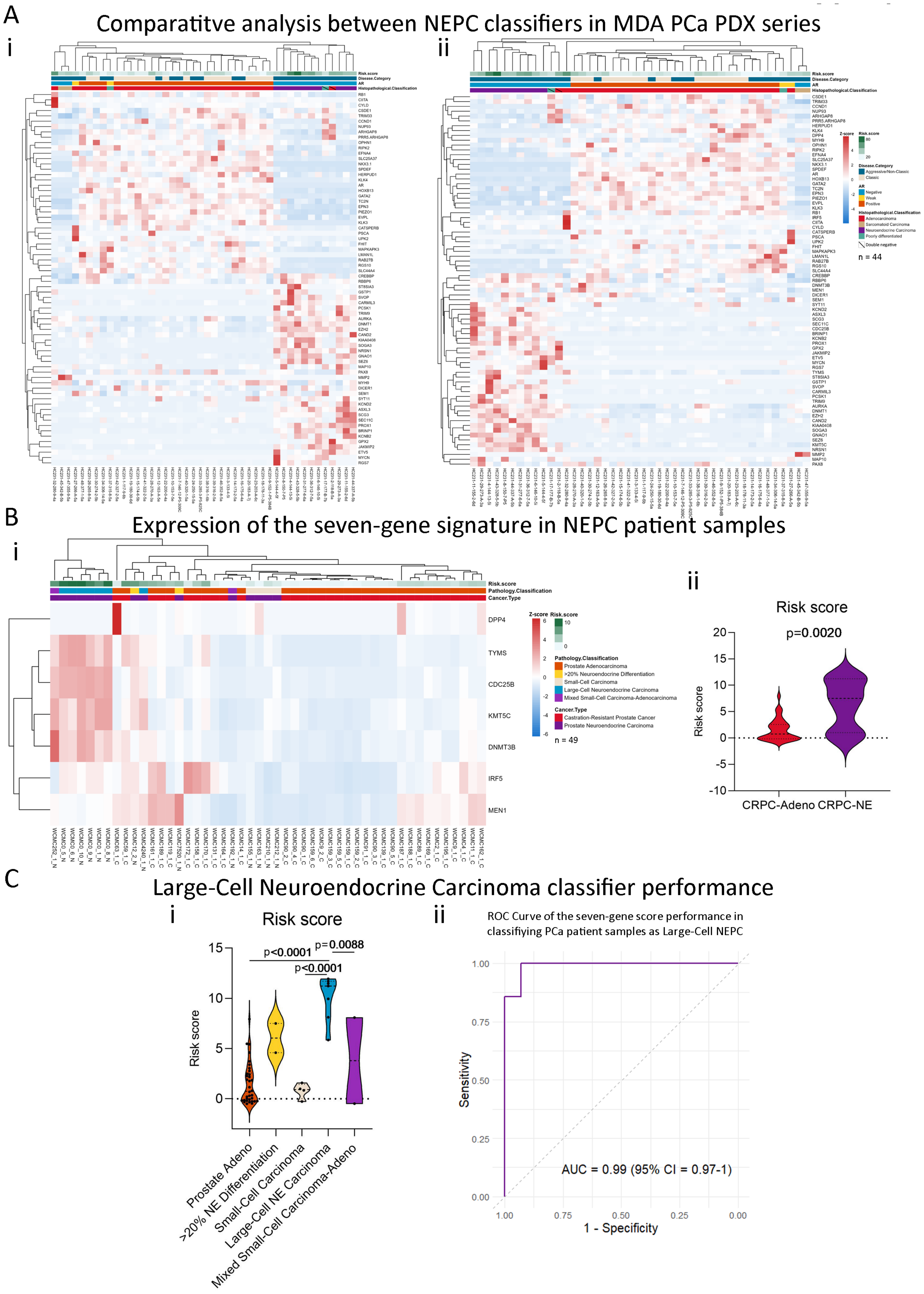
2.6. Our Stemness Score Adds Value to Pre-Existing NEPC Score
2.7. The Seven-Gene Signature Effectively Classifies Large-Cell Neuroendocrine Carcinomas
3. Discussion
4. Materials and Methods
4.1. Stemness-Associated Genes
4.2. Gene Expression Analyses in Human Patients
4.2.1. Dataset Selection Criteria
4.2.2. Differential Gene Expression Analyses
4.3. Association Between Gene Expression and Patients’ Outcomes
4.3.1. Dataset Selection Criteria
4.3.2. Survival Analyses
4.4. Selection of Candidate Genes for Modeling a Risk Score
4.5. Gene Signature and Risk Score Calculation
4.6. Transcriptome Analysis of MDA PCa PDX Series
4.7. Unsupervised Clustering and Principal Component Analysis (PCA)
4.8. Receiver Operating Characteristic (ROC) Curve for NEPC Classification
4.9. NEPC Patients’ Samples Dataset
4.10. Statistical Analyses
5. Conclusions
Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. Available online: https://acsjournals.onlinelibrary.wiley.com/doi/10.3322/caac.21834 (accessed on 18 July 2024).
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Al Salhi, Y.; Sequi, M.B.; Valenzi, F.M.; Fuschi, A.; Martoccia, A.; Suraci, P.P.; Carbone, A.; Tema, G.; Lombardo, R.; Cicione, A.; et al. Cancer Stem Cells and Prostate Cancer: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 7746. [Google Scholar] [CrossRef]
- Maitland, N.J.; Collins, A.T. Prostate Cancer Stem Cells: A New Target for Therapy. J. Clin. Oncol. 2008, 26, 2862–2870. [Google Scholar] [CrossRef]
- Banerjee, P.; Kapse, P.; Siddique, S.; Kundu, M.; Choudhari, J.; Mohanty, V.; Malhotra, D.; Gosavi, S.W.; Gacche, R.N.; Kundu, G.C. Therapeutic Implications of Cancer Stem Cells in Prostate Cancer. Cancer Biol. Med. 2023, 20, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-Institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, S.; Wang, N.; Zheng, Y.; Zhou, J.; Yang, B.; Wang, X.; Zhang, J.; Guo, L.; Wang, S.; et al. CCL5 Derived from Tumor-Associated Macrophages Promotes Prostate Cancer Stem Cells and Metastasis via Activating β-Catenin/STAT3 Signaling. Cell Death Dis. 2020, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, B.; Beresford, M.; Bowen, R.; Mitchard, J.; Chalmers, A.D. Searching for Prostate Cancer Stem Cells: Markers and Methods. Stem Cell Rev. Rep. 2013, 9, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Frame, F.M.; Polson, E.S.; Lewis, J.L.; Collins, A.T. Prostate Cancer Stem Cells: Do They Have a Basal or Luminal Phenotype? Horm. Cancer 2011, 2, 47–61. [Google Scholar] [CrossRef]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a Prostate from a Single Adult Stem Cell. Nature 2008, 456, 804–810. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a Cell of Origin for Human Prostate Cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef]
- Palanisamy, N.; Yang, J.; Shepherd, P.D.A.; Li-Ning-Tapia, E.M.; Labanca, E.; Manyam, G.C.; Ravoori, M.K.; Kundra, V.; Araujo, J.C.; Efstathiou, E.; et al. The MD Anderson Prostate Cancer Patient-Derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-Driven Therapy Development. Clin. Cancer Res. 2020, 26, 4933–4946. [Google Scholar] [CrossRef]
- Anselmino, N.; Labanca, E.; Shepherd, P.D.A.; Dong, J.; Yang, J.; Song, X.; Nandakumar, S.; Kundra, R.; Lee, C.; Schultz, N.; et al. Integrative Molecular Analyses of the MD Anderson Prostate Cancer Patient-Derived Xenograft (MDA PCa PDX) Series. Clin. Cancer Res. 2024, 30, 2272–2285. [Google Scholar] [CrossRef]
- Serritella, A.V.; Beltran, H.; Lotan, T.L.; VanderWeele, D.J.; Karzai, F.; Madan, R.A.; Hussain, M. Large Cell Neuroendocrine Prostate Cancer: Large Is Not Small. Oncologist 2024, 29, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.-M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed Morphologic Classification of Prostate Cancer With Neuroendocrine Differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Moch, H.; Cubilla, A.L.; Ulbright, T.M.; Reuter, V.E. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part B: Prostate and Bladder Tumours. Eur. Urol. 2016, 70, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.J.; Humphrey, P.A.; Belani, J.; van der Kwast, T.H.; Srigley, J.R. Large Cell Neuroendocrine Carcinoma of Prostate: A Clinicopathologic Summary of 7 Cases of a Rare Manifestation of Advanced Prostate Cancer. Am. J. Surg. Pathol. 2006, 30, 684–693. [Google Scholar] [CrossRef]
- Nguyen, N.; Ronald Dean Franz, I.I.; Mohammed, O.; Huynh, R.; Son, C.K.; Khan, R.N.; Ahmed, B. A Systematic Review of Primary Large Cell Neuroendocrine Carcinoma of the Prostate. Front. Oncol. 2024, 14, 1341794. [Google Scholar] [CrossRef]
- Aggarwal, R.; Zhang, T.; Small, E.J.; Armstrong, A.J. Neuroendocrine Prostate Cancer: Subtypes, Biology, and Clinical Outcomes. J. Natl. Compr. Cancer Netw. 2014, 12, 719–726. [Google Scholar] [CrossRef]
- Bhagirath, D.; Liston, M.; Akoto, T.; Lui, B.; Bensing, B.A.; Sharma, A.; Saini, S. Novel, Non-Invasive Markers for Detecting Therapy Induced Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer Patients. Sci. Rep. 2021, 11, 8279. [Google Scholar] [CrossRef]
- Dang, Q.; Li, L.; Xie, H.; He, D.; Chen, J.; Song, W.; Chang, L.S.; Chang, H.-C.; Yeh, S.; Chang, C. Anti-Androgen Enzalutamide Enhances Prostate Cancer Neuroendocrine (NE) Differentiation via Altering the Infiltrated Mast Cells → Androgen Receptor (AR) → miRNA32 Signals. Mol. Oncol. 2015, 9, 1241–1251. [Google Scholar] [CrossRef]
- Ou, Y.-H.; Jiang, Y.-D.; Li, Q.; Zhuang, Y.-J.; Dang, Q.; Tan, W.-L. Infiltrating mast cells promote neuroendocrine differentiation and increase docetaxel resistance of prostate cancer cells by up-regulating p21. Nan Fang Yi Ke Da Xue Xue Bao 2018, 38, 723–730. [Google Scholar] [CrossRef]
- Maimaitiyiming, A.; An, H.; Xing, C.; Li, X.; Li, Z.; Bai, J.; Luo, C.; Zhuo, T.; Huang, X.; Maimaiti, A.; et al. Machine Learning-Driven Mast Cell Gene Signatures for Prognostic and Therapeutic Prediction in Prostate Cancer. Heliyon 2024, 10, e35157. [Google Scholar] [CrossRef]
- Aller, M.-A.; Arias, A.; Arias, J.-I.; Arias, J. Carcinogenesis: The Cancer Cell–Mast Cell Connection. Inflamm. Res. 2019, 68, 103–116. [Google Scholar] [CrossRef]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical Features of Neuroendocrine Prostate Cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef]
- Cherif, C.; Nguyen, D.T.; Paris, C.; Le, T.K.; Sefiane, T.; Carbuccia, N.; Finetti, P.; Chaffanet, M.; Kaoutari, A.E.; Vernerey, J.; et al. Menin Inhibition Suppresses Castration-Resistant Prostate Cancer and Enhances Chemosensitivity. Oncogene 2022, 41, 125–137. [Google Scholar] [CrossRef]
- Quan, Y.; Zhang, X.; Wang, M.; Ping, H. Histone Lysine Methylation Patterns in Prostate Cancer Microenvironment Infiltration: Integrated Bioinformatic Analysis and Histological Validation. Front. Oncol. 2022, 12, 981226. [Google Scholar] [CrossRef] [PubMed]
- Tzelepi, V.; Logotheti, S.; Efstathiou, E.; Troncoso, P.; Aparicio, A.; Sakellakis, M.; Hoang, A.; Perimenis, P.; Melachrinou, M.; Logothetis, C.; et al. Epigenetics and Prostate Cancer: Defining the Timing of DNA Methyltransferase Deregulation during Prostate Cancer Progression. Pathology 2020, 52, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4—A Potential Biomarker and Target for Cancer Therapy. Pharmacol. Ther. 2019, 198, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Burdelski, C.; Strauss, C.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Melling, N.; Lebok, P.; Minner, S.; Koop, C.; Graefen, M.; et al. Overexpression of Thymidylate Synthase (TYMS) Is Associated with Aggressive Tumor Features and Early PSA Recurrence in Prostate Cancer. Oncotarget 2015, 6, 8377–8387. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.W.; Hashimoto, Y.; Ma, Z.-Q.; Tsai, M.-J.; Tsai, S.Y. Overexpression of Cdc25B, an Androgen Receptor Coactivator, in Prostate Cancer. Oncogene 2003, 22, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.K.; Collado, G.; Barnes, B.J. Role of Interferon Regulatory Factor 5 (IRF5) in Tumor Progression: Prognostic and Therapeutic Potential. Biochim. Biophys. Acta BBA—Rev. Cancer 2024, 1879, 189061. [Google Scholar] [CrossRef]
- Chen, H.; Fang, S.; Zhu, X.; Liu, H. Cancer-Associated Fibroblasts and Prostate Cancer Stem Cells: Crosstalk Mechanisms and Implications for Disease Progression. Front. Cell Dev. Biol. 2024, 12, 1412337. [Google Scholar] [CrossRef]
- Ellis, L.; Loda, M. Advanced Neuroendocrine Prostate Tumors Regress to Stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 14406–14407. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Gupta, K.; Kyprianou, N. Epigenetic Mechanisms Underlying Subtype Heterogeneity and Tumor Recurrence in Prostate Cancer. Nat. Commun. 2023, 14, 567. [Google Scholar] [CrossRef]
- Guijarro, M.V.; Nawab, A.; Dib, P.; Burkett, S.; Luo, X.; Feely, M.; Nasri, E.; Seifert, R.P.; Kaye, F.J.; Zajac-Kaye, M. TYMS Promotes Genomic Instability and Tumor Progression in Ink4a/Arf Null Background. Oncogene 2023, 42, 1926–1939. [Google Scholar] [CrossRef]
- Ibe, T.; Shimizu, K.; Nakano, T.; Kakegawa, S.; Kamiyoshihara, M.; Nakajima, T.; Kaira, K.; Takeyoshi, I. High-Grade Neuroendocrine Carcinoma of the Lung Shows Increased Thymidylate Synthase Expression Compared to Other Histotypes. J. Surg. Oncol. 2010, 102, 11–17. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-Throughput Screening Using Patient-Derived Tumor Xenografts to Predict Clinical Trial Drug Response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Li, C.; Higgins, J.P.; Van De Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene Expression Profiling Identifies Clinically Relevant Subtypes of Prostate Cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef]
- Malhotra, S.; Lapointe, J.; Salari, K.; Higgins, J.P.; Ferrari, M.; Montgomery, K.; van de Rijn, M.; Brooks, J.D.; Pollack, J.R. A Tri-Marker Proliferation Index Predicts Biochemical Recurrence after Surgery for Prostate Cancer. PLoS ONE 2011, 6, e20293. [Google Scholar] [CrossRef]
- Mortensen, M.M.; Høyer, S.; Lynnerup, A.S.; Ørntoft, T.F.; Sørensen, K.D.; Borre, M.; Dyrskjøt, L. Expression Profiling of Prostate Cancer Tissue Delineates Genes Associated with Recurrence after Prostatectomy. Sci. Rep. 2015, 5, 16018. [Google Scholar] [CrossRef]
- Wallace, T.A.; Prueitt, R.L.; Yi, M.; Howe, T.M.; Gillespie, J.W.; Yfantis, H.G.; Stephens, R.M.; Caporaso, N.E.; Loffredo, C.A.; Ambs, S. Tumor Immunobiological Differences in Prostate Cancer between African-American and European-American Men. Cancer Res. 2008, 68, 927–936. [Google Scholar] [CrossRef]
- Ross-Adams, H.; Lamb, A.; Dunning, M.; Halim, S.; Lindberg, J.; Massie, C.; Egevad, L.; Russell, R.; Ramos-Montoya, A.; Vowler, S.; et al. Integration of Copy Number and Transcriptomics Provides Risk Stratification in Prostate Cancer: A Discovery and Validation Cohort Study. eBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- TCGA-PRAD. Available online: https://portal.gdc.cancer.gov/projects/TCGA-PRAD (accessed on 4 August 2021).
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Oshlack, A.; Smyth, G.K. Optimizing the Noise versus Bias Trade-off for Illumina Whole Genome Expression BeadChips. Nucleic Acids Res. 2010, 38, e204. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Lyons, C.A.; Walker, S.M.; McQuaid, S.; Hynes, S.O.; Mitchell, D.M.; Pang, B.; Logan, G.E.; McCavigan, A.M.; O’Rourke, D.; et al. Validation of a Metastatic Assay Using Biopsies to Improve Risk Stratification in Patients with Prostate Cancer Treated with Radical Radiation Therapy. Ann. Oncol. 2018, 29, 215–222. [Google Scholar] [CrossRef]
- Sboner, A.; Demichelis, F.; Calza, S.; Pawitan, Y.; Setlur, S.R.; Hoshida, Y.; Perner, S.; Adami, H.O.; Fall, K.; Mucci, L.A.; et al. Molecular Sampling of Prostate Cancer: A Dilemma for Predicting Disease Progression. BMC Med. Genom. 2010, 3, 8. [Google Scholar] [CrossRef]
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global Transcriptome Analysis of Formalin-Fixed Prostate Cancer Specimens Identifies Biomarkers of Disease Recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef]
- Luca, B.-A.; Brewer, D.S.; Edwards, D.R.; Edwards, S.; Whitaker, H.C.; Merson, S.; Dennis, N.; Cooper, R.A.; Hazell, S.; Warren, A.Y.; et al. DESNT: A Poor Prognosis Category of Human Prostate Cancer. Eur. Urol. Focus 2018, 4, 842–850. [Google Scholar] [CrossRef]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. The Logrank Test. BMJ 2004, 328, 1073. [Google Scholar] [CrossRef]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Gyorffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A Comprehensive and Straightforward Web Application Enabling Rapid Biomarker Cutoff Optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Breslow, N.E. Analysis of Survival Data under the Proportional Hazards Model. Int. Stat. Rev. Rev. Int. Stat. 1975, 43, 45–57. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in S. R Package Version. 1999. Available online: https://www.mayo.edu/research/documents/tr53pdf/doc-10027379 (accessed on 22 September 2024).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S.; Package ‘Survminer’. Drawing Survival Curves Using ‘ggplot2’. R Package Version 0.3.1. 2014. Available online: https://cran.r-project.org/web/packages/survminer/index.html (accessed on 22 September 2024).
- Ishwaran, H.; Kogalur, U.B. randomForestSRC: Fast Unified Random Forests for Survival, Regression, and Classification (RF-SRC). 2024. Available online: https://cran.r-project.org/web/packages/randomForestSRC/index.html (accessed on 22 September 2024).
- Ishwaran, H.; Lu, M. Standard Errors and Confidence Intervals for Variable Importance in Random Forest Regression, Classification, and Survival. Stat. Med. 2019, 38, 558–582. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 22 September 2024).
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R Package. 2020. Available online: https://cran.r-project.org/web/packages/factoextra/index.html (accessed on 22 September 2024).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: Display and Analyze ROC Curves; 2010; 1.18.5. Available online: https://cran.r-project.org/web/packages/pROC/pROC.pdf (accessed on 22 September 2024).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Dexter, T.A. R: A Language and Environment for Statistical Computing. Quat. Res. 2014, 81, 114–124. [Google Scholar] [CrossRef]
- RStudio. RStudio|Open Source & Professional Software for Data Science Teams—RStudio. Available online: https://www.rstudio.com/ (accessed on 22 September 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Hadley, W. Ggplot2. Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots. R Package Version 0.4.0.999. 2020. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 22 September 2024).
- Neuwirth, E.; Maindonald, J. Package “RColorBrewer”. 2015. Available online: http://cran.nexr.com/web/packages/RColorBrewer/RColorBrewer.pdf (accessed on 22 September 2024).
- Davis, S.; Meltzer, P.S. GEOquery: A Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Samples |
|---|---|
| GSE35988 [46] | Localized PCa (n = 59), matched benign prostate tissues (n = 28), and metastatic CRPC (n = 35). |
| GSE3933 [47,48] | Localized PCa (n = 62) and normal prostate (n = 41). |
| GSE46602 [49] | PCa (n = 36) and benign tissue (n = 14). |
| GSE6956 [50] | Primary PCa (n = 69) and normal adjacent prostate (n = 18). |
| GSE70768 [51] | Primary PCa (n = 112), benign tissue (n = 74) and CRPC (n = 13). |
| TCGA-PRAD [52] | Primary PCa (n = 497) and normal adjacent tissue samples (n = 51). |
| GSE21034 [53] | Primary PCa (n = 131) and metastatic tissue samples (n = 19). |
| Dataset | Samples | Survival Endpoint | Covariates | Cohort |
|---|---|---|---|---|
| TCGA-PRAD [52] | 497 PCa (RNAseq) | Disease Progression Disease-Free Time (n = 337) | Gleason Group, PSA levels, Clinical T Stage, Targeted Molecular/Radiation Therapy | Training |
| GSE70768 [51] | 111 PCa (Microarray) | Biochemical Relapse | Age, Gleason Group, PSA levels, T Stage | Training |
| GSE70769 [51] | 92 PCa (Microarray) | Biochemical Relapse | Gleason Group, PSA levels, T Stage | Training |
| GSE116918 [56] | 248 PCa (Microarray) | Metastasis Development Relapse | Age, Gleason Score, PSA levels, T Stage | Training |
| GSE16560 [57] | 281 PCa (Microarray) | Death | Age, Gleason Group | Training |
| GSE54460 [58] | 106 PCa (RNA-seq) | Biochemical Relapse | Age, Gleason Score, PSA levels, T Stage | Validation |
| GSE94767 [59] | 233 PCa (Microarray) | Biochemical Relapse | Gleason Group, PSA levels, T Stage | Validation |
| DKFZ [60] | 81 PCa (RNA-seq) | Biochemical Relapse | Age, Gleason Score, PSA levels, T Stage | Validation |
| SU2C-PCF [61] | 81 metastatic CRPC (RNA-seq) | Death | Age, Gleason Score, PSA levels | Validation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabater, A.; Sanchis, P.; Seniuk, R.; Pascual, G.; Anselmino, N.; Alonso, D.F.; Cayol, F.; Vazquez, E.; Marti, M.; Cotignola, J.; et al. Unmasking Neuroendocrine Prostate Cancer with a Machine Learning-Driven Seven-Gene Stemness Signature That Predicts Progression. Int. J. Mol. Sci. 2024, 25, 11356. https://doi.org/10.3390/ijms252111356
Sabater A, Sanchis P, Seniuk R, Pascual G, Anselmino N, Alonso DF, Cayol F, Vazquez E, Marti M, Cotignola J, et al. Unmasking Neuroendocrine Prostate Cancer with a Machine Learning-Driven Seven-Gene Stemness Signature That Predicts Progression. International Journal of Molecular Sciences. 2024; 25(21):11356. https://doi.org/10.3390/ijms252111356
Chicago/Turabian StyleSabater, Agustina, Pablo Sanchis, Rocio Seniuk, Gaston Pascual, Nicolas Anselmino, Daniel F. Alonso, Federico Cayol, Elba Vazquez, Marcelo Marti, Javier Cotignola, and et al. 2024. "Unmasking Neuroendocrine Prostate Cancer with a Machine Learning-Driven Seven-Gene Stemness Signature That Predicts Progression" International Journal of Molecular Sciences 25, no. 21: 11356. https://doi.org/10.3390/ijms252111356
APA StyleSabater, A., Sanchis, P., Seniuk, R., Pascual, G., Anselmino, N., Alonso, D. F., Cayol, F., Vazquez, E., Marti, M., Cotignola, J., Toro, A., Labanca, E., Bizzotto, J., & Gueron, G. (2024). Unmasking Neuroendocrine Prostate Cancer with a Machine Learning-Driven Seven-Gene Stemness Signature That Predicts Progression. International Journal of Molecular Sciences, 25(21), 11356. https://doi.org/10.3390/ijms252111356