
The Interaction Mechanism Between C14-Polyacetylene Compounds and the Rat TRPA1 Receptor: An In Silico Study

Abstract
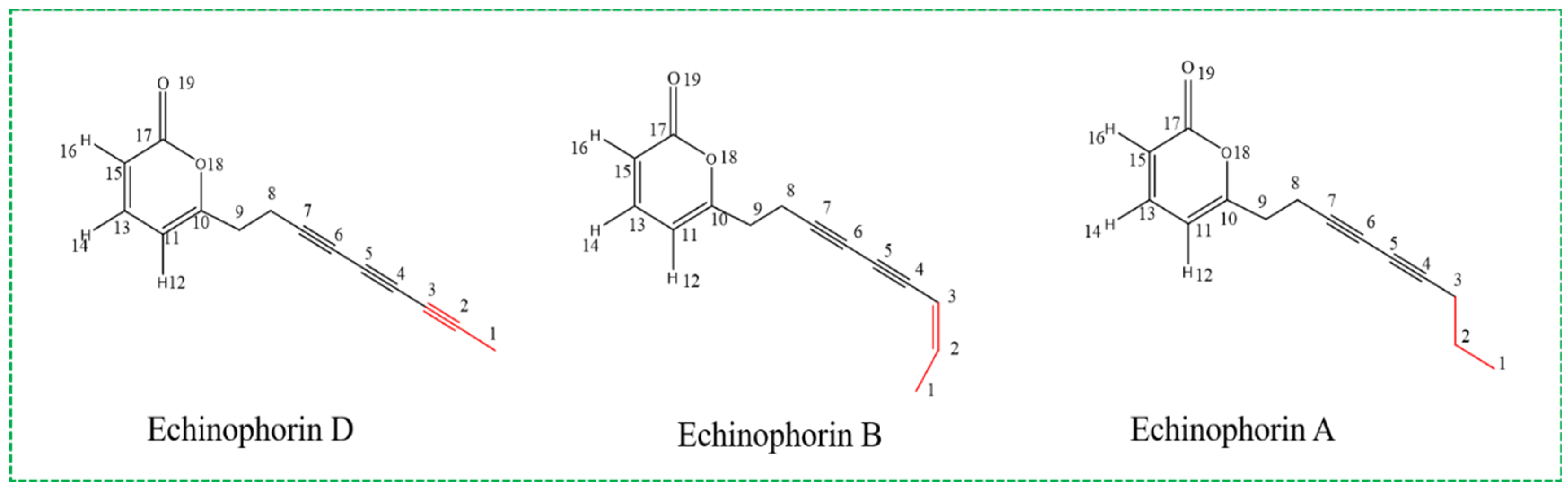
1. Introduction
2. Results
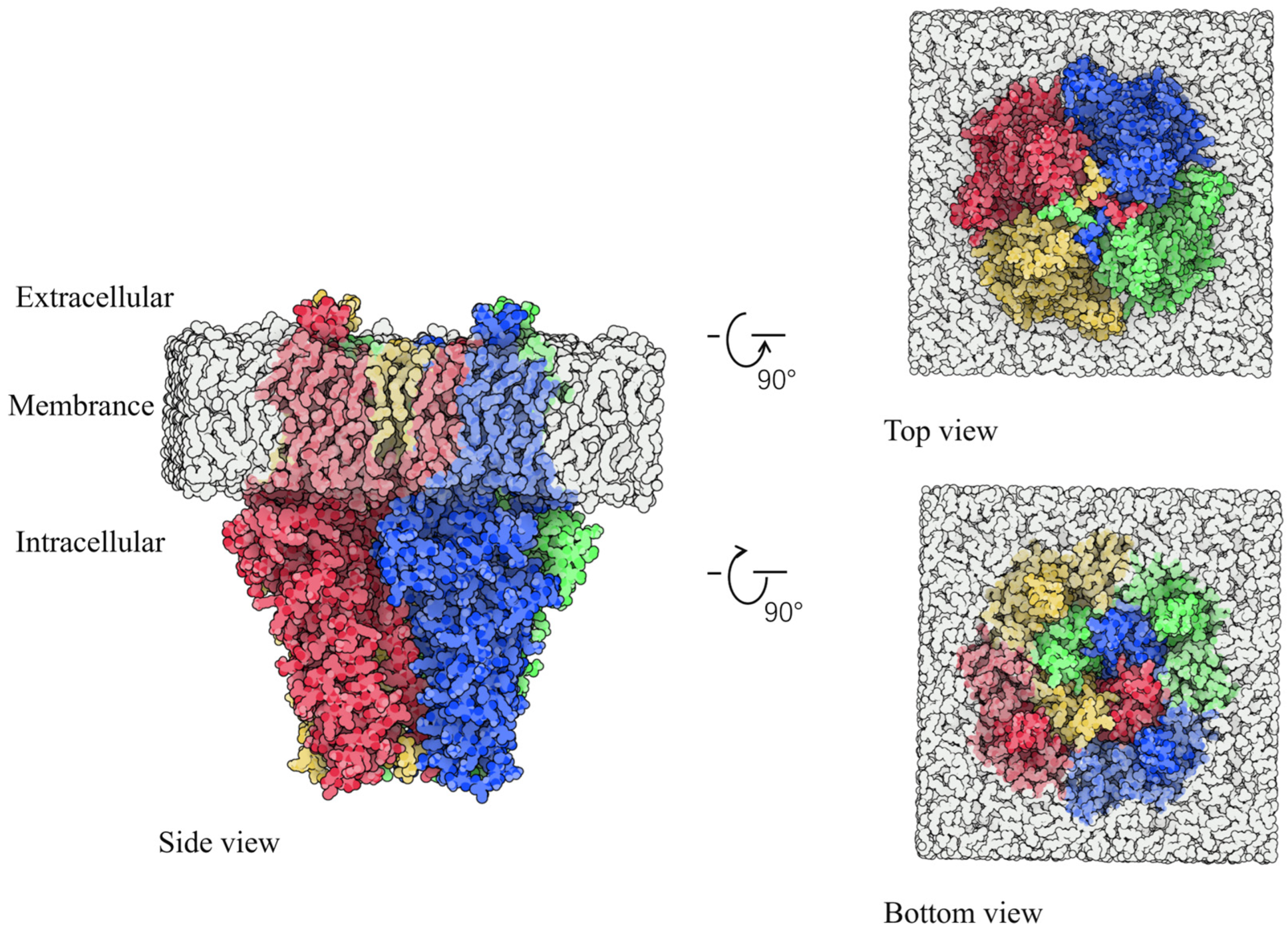
2.1. Identification of Binding Sites
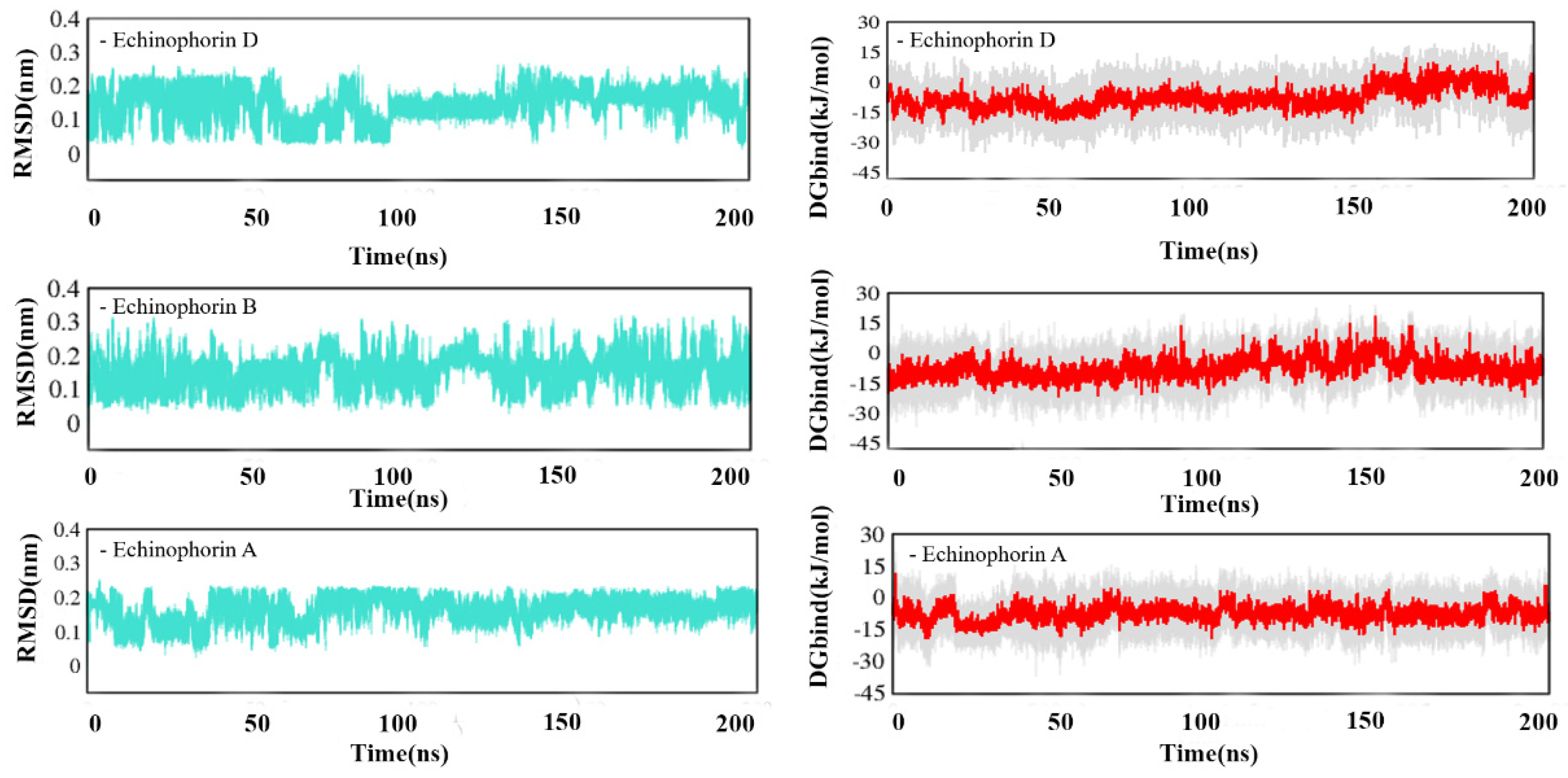
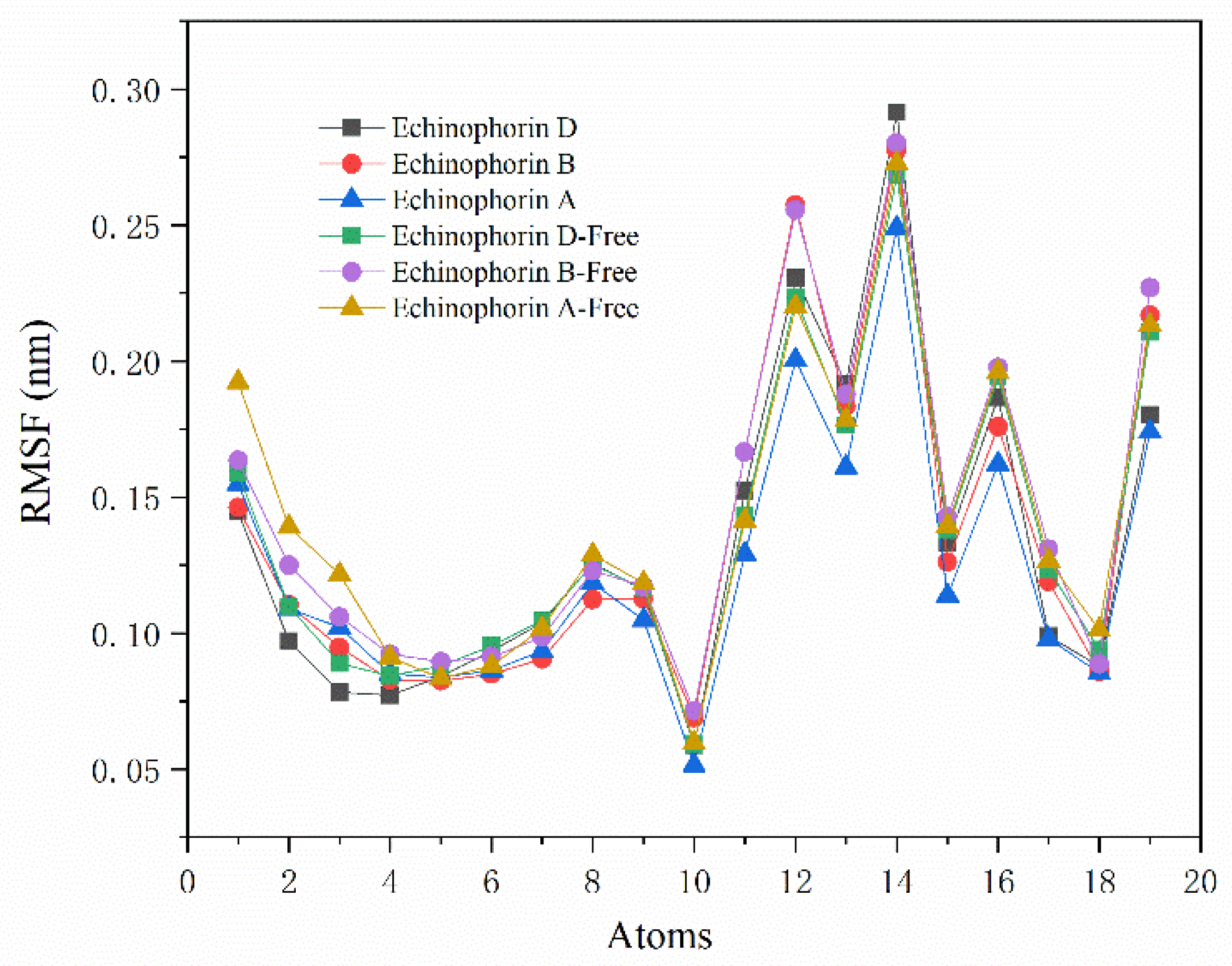
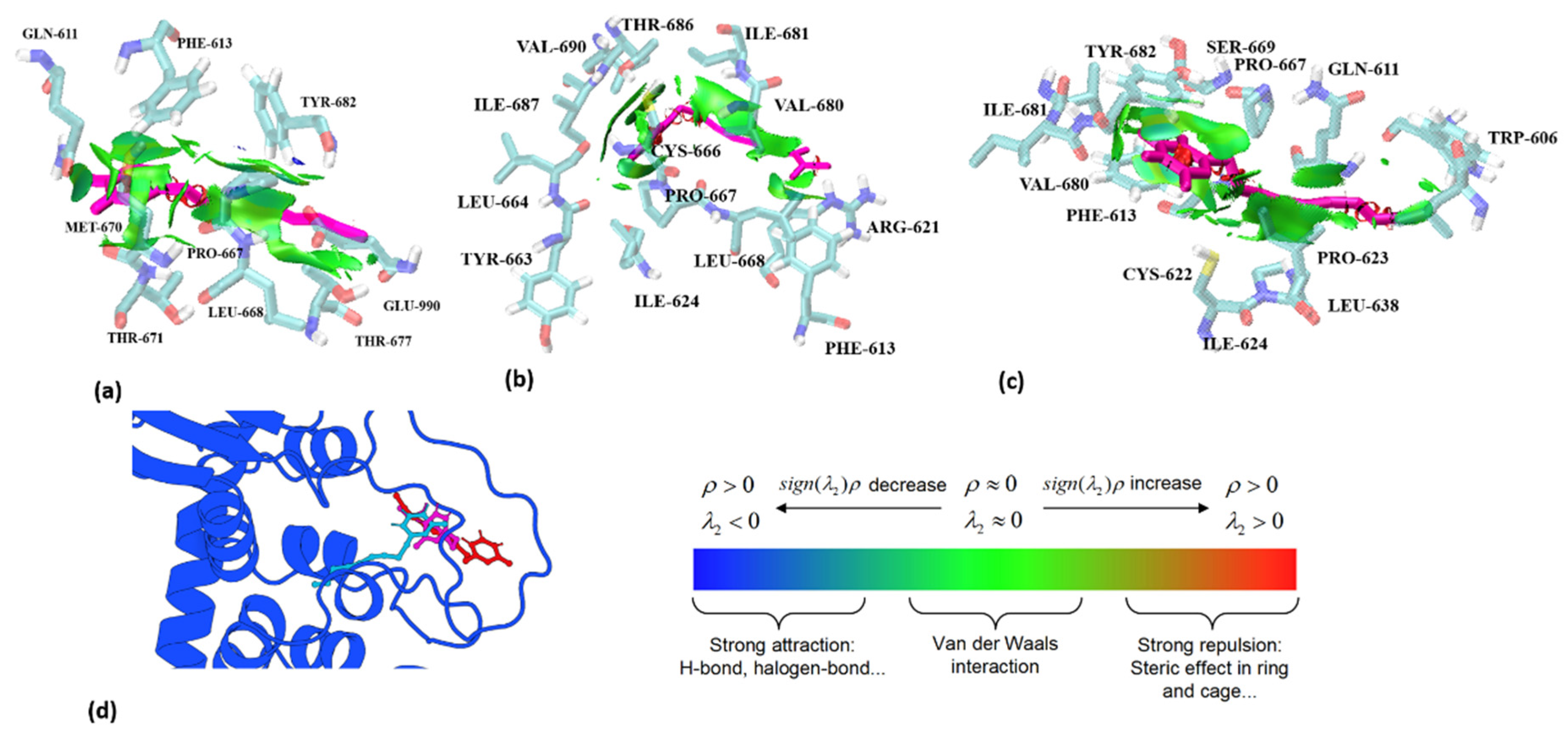
2.2. Interaction Analysis
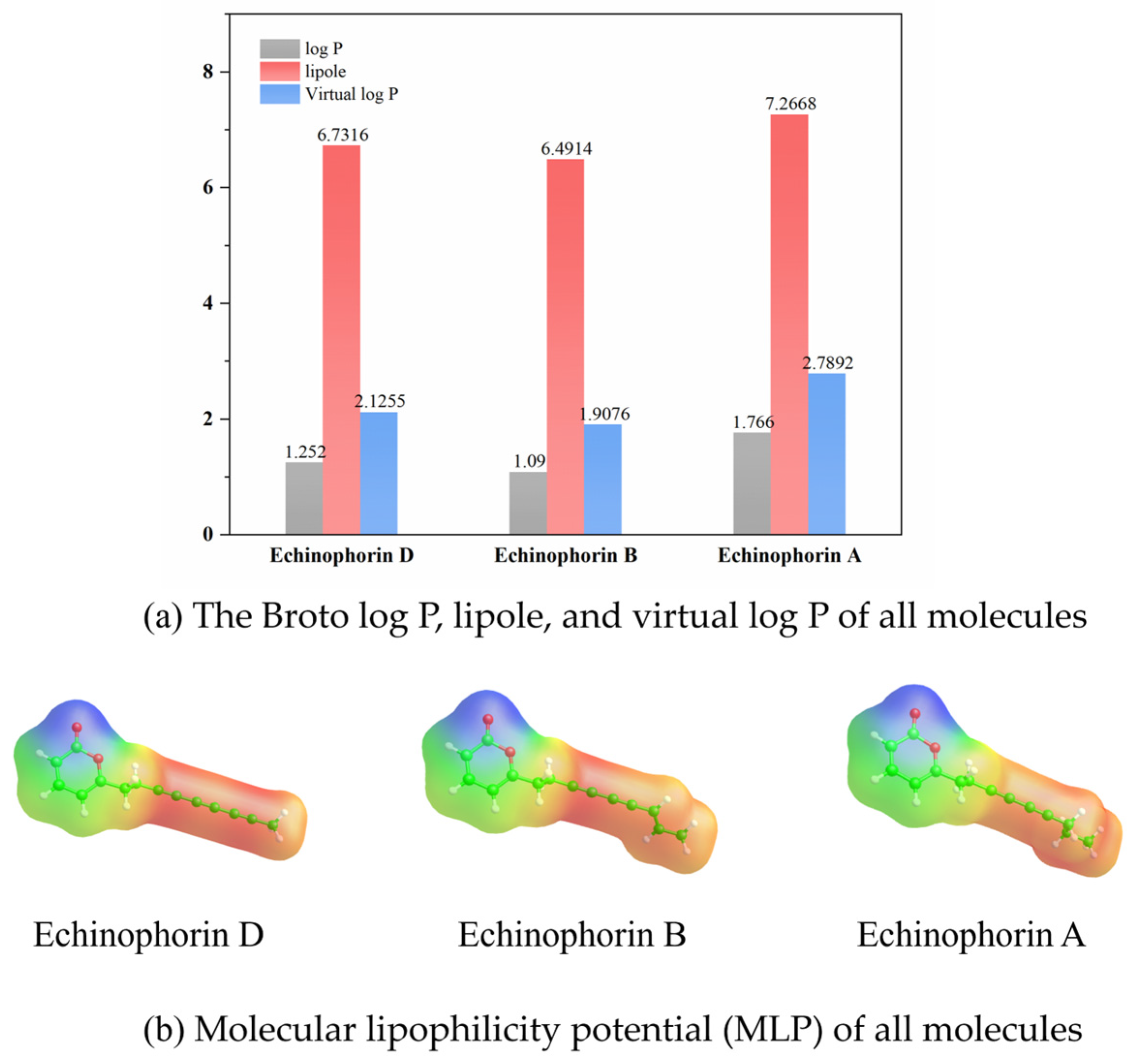
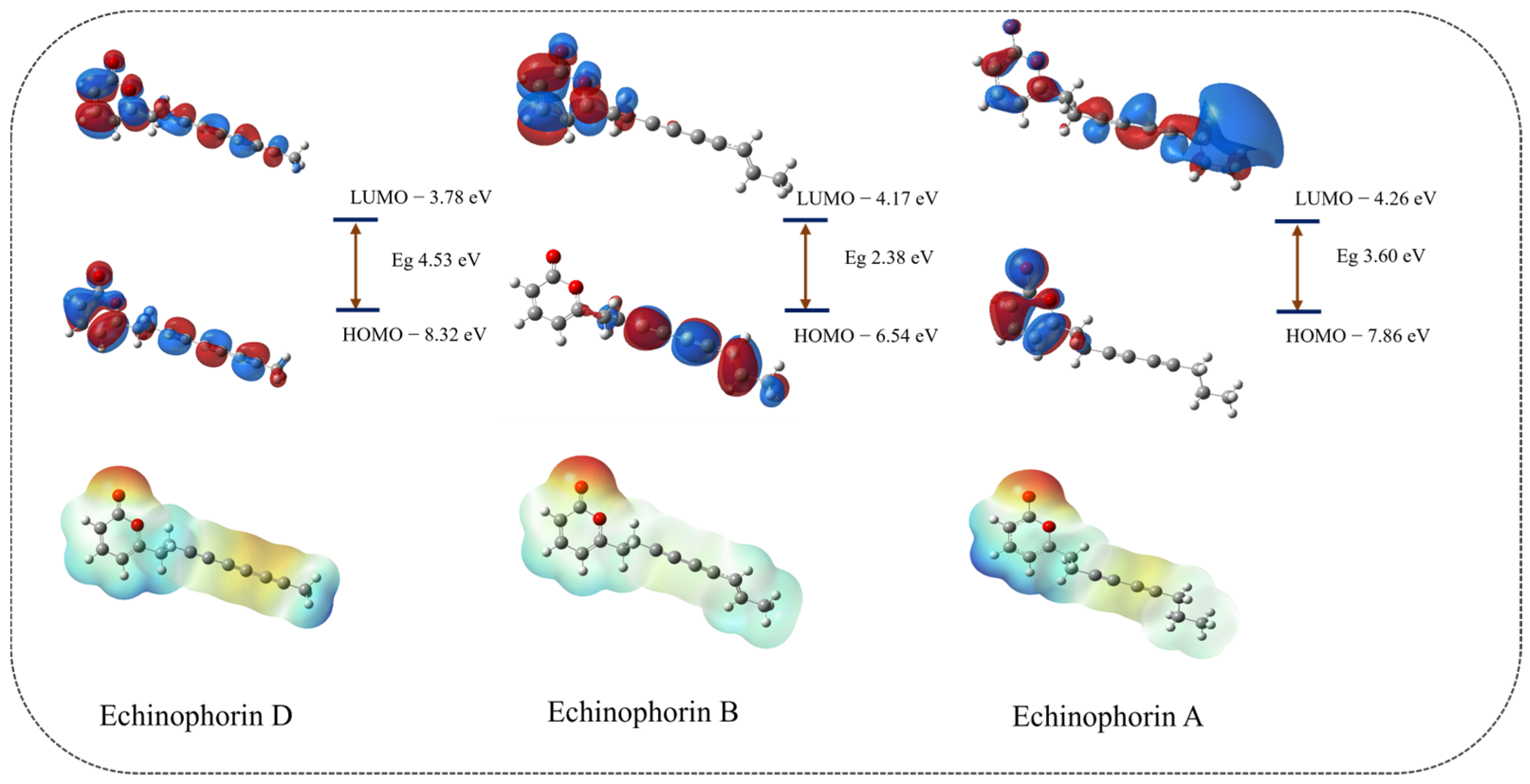
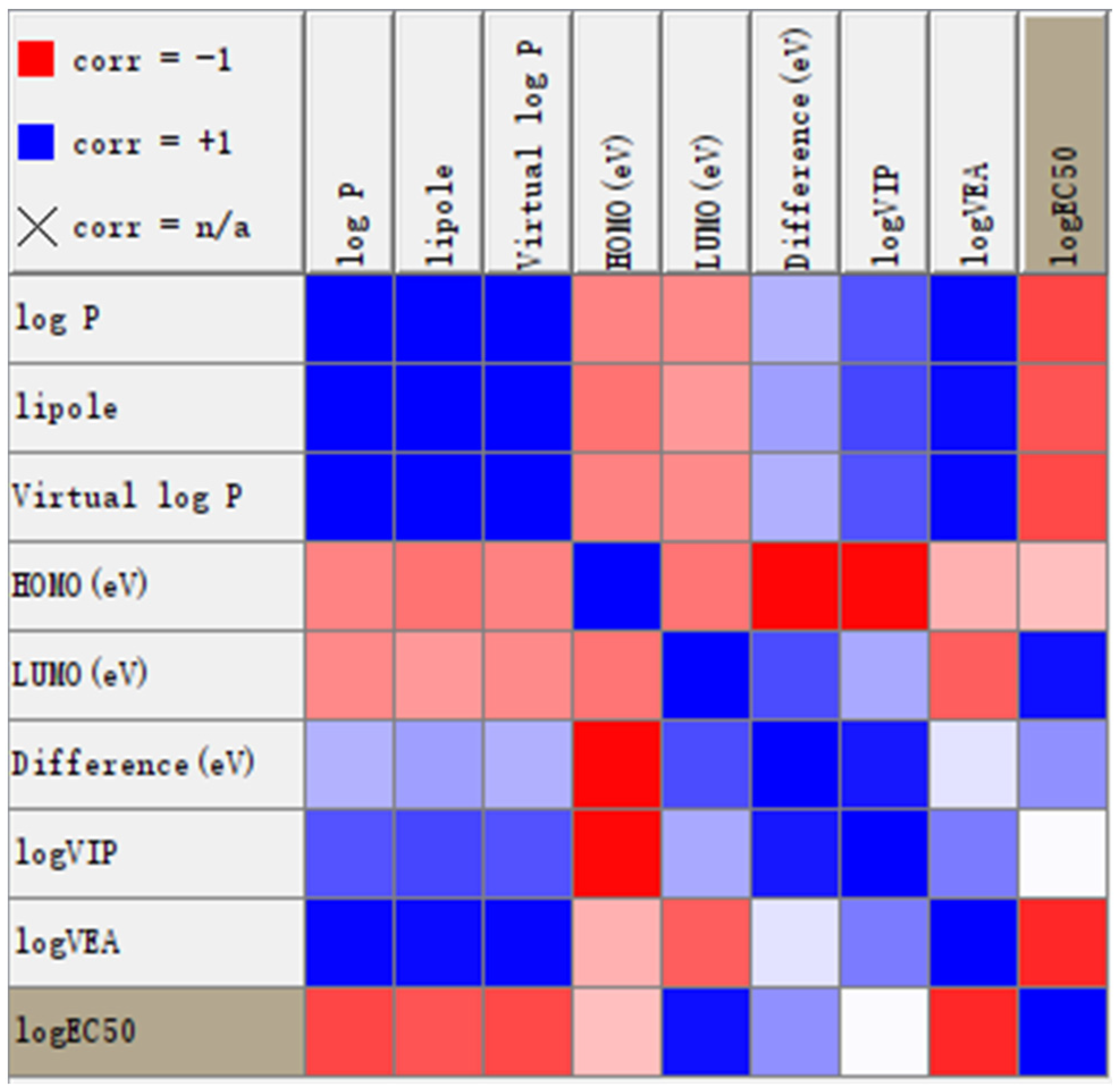
2.3. Molecular Properties
3. Discussion
4. Materials and Methods
4.1. Structure Preparation and Binding Site Prediction
4.2. Molecular Dynamics Simulation and Binding Energy Calculation
4.3. Interaction Analysis
4.4. Molecular Property Calculation and Correlation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Negri, R. Polyacetylenes from terrestrial plants and fungi: Recent phytochemical and biological advances. Fitoterapia 2015, 106, 92–109. [Google Scholar] [CrossRef] [PubMed]
- Uwai, K.; Ohashi, K.; Takaya, Y.; Ohta, T.; Tadano, T.; Kisara, K.; Shibusawa, K.; Sakakibara, R.; Oshima, Y. Exploring the Structural Basis of Neurotoxicity in C17-Polyacetylenes Isolated from Water Hemlock. J. Med. Chem. 2000, 43, 4508–4515. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Pollastro, F.; Verotta, L.; Ballero, M.; Romano, A.; Wyrembek, P.; Szczuraszek, K.; Mozrzymas, J.W.; Taglialatela-Scafati, O. Polyacetylenes from Sardinian Oenanthe fistulosa: A Molecular Clue to risus sardonicus. J. Nat. Prod. 2009, 72, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Dawid, C.; Dunemann, F.; Schwab, W.; Nothnagel, T.; Hofmann, T. Bioactive C17-Polyacetylenes in Carrots (Daucus carota L.): Current Knowledge and Future Perspectives. J. Agric. Food Chem. 2015, 63, 9211–9222. [Google Scholar] [CrossRef]
- Chianese, G.; Sirignano, C.; Shokoohinia, Y.; Mohammadi, Z.; Bazvandi, L.; Jafari, F.; Jalilian, F.; Moriello, A.S.; De Petrocellis, L.; Taglialatela-Scafati, O.; et al. TRPA1 Modulating C14 Polyacetylenes from the Iranian Endemic Plant Echinophora platyloba. Molecules 2018, 23, 1750. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Preti, D.; Materazzi, S.; Geppetti, P. Transient receptor potential ankyrin 1 (TRPA1) channel as emerging target for novel analgesics and anti-inflammatory agents. J. Med. Chem. 2010, 53, 5085–5107. [Google Scholar] [CrossRef]
- Hinman, A.; Chuang, H.-H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568. [Google Scholar] [CrossRef]
- Li, H. TRP Channel Classification. Adv. Exp. Med. Biol. 2017, 976, 1–8. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Yuan, P. Structural biology of thermoTRPV channels. Cell Calcium 2019, 84, 102106. [Google Scholar] [CrossRef]
- Khairatkar-Joshi, N.; Shah, D.M.; Mukhopadhyay, I.; Lingam, V.P.; Thomas, A.; A Horne, G.; Kinstrie, R.; Copland, M.; Bartolomé-Nebreda, J.-M.; Conde-Ceide, S.; et al. TRPC channel modulators and their potential therapeutic applications. Pharm. Pat. Anal. 2015, 4, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Peier, A.M.; Reeve, A.J.; Eid, S.R.; Mosbacher, J.; Hricik, T.R.; Earley, T.J.; Hergarden, A.C.; Andersson, D.A.; Hwang, S.W.; et al. ANKTM1, a TRP-like Channel Expressed in Nociceptive Neurons, Is Activated by Cold Temperatures. Cell 2003, 112, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, M.; Siemens, J.; Materazzi, S.; Bautista, D.M.; Nassini, R.; Campi, B.; Imamachi, N.; Andrè, E.; Patacchini, R.; Cottrell, G.S.; et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. USA 2007, 104, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Benemei, S.; Fusi, C.; Trevisan, G.; Geppetti, P. The TRPA1 channel in migraine mechanism and treatment. Br. J. Pharmacol. 2014, 171, 2552–2567. [Google Scholar] [CrossRef]
- Ryckmans, T.; Aubdool, A.A.; Bodkin, J.V.; Cox, P.; Brain, S.D.; Dupont, T.; Fairman, E.; Hashizume, Y.; Ishii, N.; Kato, T.; et al. Design and pharmacological evaluation of PF-4840154, a non-electrophilic reference agonist of the TrpA1 channel. Bioorg. Med. Chem. Lett. 2011, 21, 4857–4859. [Google Scholar] [CrossRef]
- Suo, Y.; Wang, Z.; Zubcevic, L.; Hsu, A.L.; He, Q.; Borgnia, M.J.; Ji, R.-R.; Lee, S.-Y. Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 2020, 105, 882–894.e5. [Google Scholar] [CrossRef]
- Zhao, J.; King, J.V.L.; Paulsen, C.E.; Cheng, Y.; Julius, D. Irritant-evoked activation and calcium modulation of the TRPA1 receptor. Nature 2020, 585, 141–145. [Google Scholar] [CrossRef]
- Fischer, M.J.; Leffler, A.; Niedermirtl, F.; Kistner, K.; Eberhardt, M.; Reeh, P.W.; Nau, C. The general anesthetic propofol excites nociceptors by activating TRPV1 and TRPA1 rather than GABAA receptors. J. Biol. Chem. 2010, 285, 34781–34792. [Google Scholar] [CrossRef]
- Liu, C.; Reese, R.; Vu, S.; Rougé, L.; Shields, S.D.; Kakiuchi-Kiyota, S.; Chen, H.; Johnson, K.; Shi, Y.P.; Chernov-Rogan, T.; et al. A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2020, 109, 273–284.e4. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Antonczak, S. Electronic description of four flavonoids revisited by DFT method. J. Mol. Struct. 2008, 856, 38–45. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef]
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Almlöf, M.; Carlsson, J.; Åqvist, J. Improving the Accuracy of the Linear Interaction Energy Method for Solvation Free Energies. J. Chem. Theory Comput. 2007, 3, 2162–2175. [Google Scholar] [CrossRef]
- Boukharta, L.; Keränen, H.; Stary-Weinzinger, A.; Wallin, G.; de Groot, B.L.; Åqvist, J. Computer simulations of structure-activity relationships for HERG channel blockers. Biochemistry 2011, 50, 6146–6156. [Google Scholar] [CrossRef]
- Rifai, E.A.; van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1–Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef]
- Capoferri, L.; Verkade-Vreeker, M.C.A.; Buitenhuis, D.; Commandeur, J.N.M.; Pastor, M.; Vermeulen, N.P.E.; Geerke, D.P. Linear Interaction Energy Based Prediction of Cytochrome P450 1A2 Binding Affinities with Reliability Estimation. PLoS ONE 2015, 10, e0142232. [Google Scholar] [CrossRef]
- van Dijk, M.; ter Laak, A.M.; Wichard, J.D.; Capoferri, L.; Vermeulen, N.P.E.; Geerke, D.P. Comprehensive and Automated Linear Interaction Energy Based Binding-Affinity Prediction for Multifarious Cytochrome P450 Aromatase Inhibitors. J. Chem. Inf. Model. 2017, 57, 2294–2308. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M.A. Medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent Interaction Analysis in Fluctuating Environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Götze, J.P.; Pi, Y.; Petry, S.; Langkabel, F.; Witte, J.F.; Lemke, O. A user-friendly, Python-based quantum mechanics/Gromacs interface: gmx2qmmm. Int. J. Quantum Chem. 2021, 121, e26486. [Google Scholar] [CrossRef]
- Naik, A.; Samant, L. Correlation Review of Classification Algorithm Using Data Mining Tool: WEKA, Rapidminer, Tanagra, Orange and Knime. Procedia Comput. Sci. 2016, 85, 662–668. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | [Uel]P-L | [Uvdw]P-L | [Uel]L-Rest | [Uvdw]L-Rest | [Uel]L-water | [Uvdw]L-water | ΔGbind | ΔGEXP | γ/ΔGbind |
|---|---|---|---|---|---|---|---|---|---|
| Ⅱ-D | −54.2478 ± 2.3 | −103.834 ± 2.5 | −16.0441 ± 2.8 | −19.4748 ± 1.3 | −73.246 ± 0.051 | −74.9464 ± 0.095 | −24.506 | −23.320 | 69.465% |
| Ⅱ-B | −37.1776 ± 2.9 | −82.3549 ± 5.8 | −36.6222 ± 1.9 | −32.7639 ± 3.7 | −72.6516 ± 0.052 | −75.9172 ± 0.095 | −24.612 | −25.439 | 69.166% |
| Ⅱ-A | −29.4323 ± 3.0 | −133.438 ± 2.2 | −33.4841 ± 3.2 | −5.97493 ± 1.3 | −71.5446 ± 0.045 | −77.2402 ± 0.180 | −24.566 | −24.926 | 69.295% |
| Residue ID | 606 | 611 | 613 | 621 | 622 | 623 | 624 | 638 | 663 | 664 | 666 | 667 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (a) | GLN | PHE | PRO | |||||||||
| (b) | PHE | ARG | CYS | PRO | ILE | LEU | TYR | LEU | CYS | PRO | ||
| (c) | TRP | GLN | PHE | ILE | PRO | |||||||
| Residue ID | 668 | 669 | 670 | 671 | 677 | 680 | 681 | 682 | 686 | 687 | 690 | 990 |
| (a) | LEU | MET | THR | THR | TYR | GLU | ||||||
| (b) | LEU | VAL | ILE | THR | ILE | VAL | ||||||
| (c) | SER | VAL | ILE | TYR |
| Molecule | Echinophorin D | Echinophorin B | Echinophorin A |
|---|---|---|---|
| HOMO (eV) | −8.315 | −6.543 | −7.861 |
| LUMO (eV) | −3.784 | −4.166 | −4.264 |
| Difference (eV) | 4.531 | 2.377 | 3.597 |
| VIP (kcal mol−1) | 223.44 | 190.688 | 222.778 |
| VEA (kcal mol−1) | 55.672 | 54.917 | 77.785 |
| Potency EC50 μM | 30.9 ± 2.8 | 25.0 ± 3.0 | 20.3 ± 3.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Gao, D.; Yang, Y.; Liu, L.; Zhao, X.; Na, R. The Interaction Mechanism Between C14-Polyacetylene Compounds and the Rat TRPA1 Receptor: An In Silico Study. Int. J. Mol. Sci. 2024, 25, 11290. https://doi.org/10.3390/ijms252011290
Yu H, Gao D, Yang Y, Liu L, Zhao X, Na R. The Interaction Mechanism Between C14-Polyacetylene Compounds and the Rat TRPA1 Receptor: An In Silico Study. International Journal of Molecular Sciences. 2024; 25(20):11290. https://doi.org/10.3390/ijms252011290
Chicago/Turabian StyleYu, Hui, Denghui Gao, Ying Yang, Lu Liu, Xi Zhao, and Risong Na. 2024. "The Interaction Mechanism Between C14-Polyacetylene Compounds and the Rat TRPA1 Receptor: An In Silico Study" International Journal of Molecular Sciences 25, no. 20: 11290. https://doi.org/10.3390/ijms252011290
APA StyleYu, H., Gao, D., Yang, Y., Liu, L., Zhao, X., & Na, R. (2024). The Interaction Mechanism Between C14-Polyacetylene Compounds and the Rat TRPA1 Receptor: An In Silico Study. International Journal of Molecular Sciences, 25(20), 11290. https://doi.org/10.3390/ijms252011290