
Assessing the Potential of 1,2,3-Triazole-Dihydropyrimidinone Hybrids Against Cholinesterases: In Silico, In Vitro, and In Vivo Studies

 ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
, 

Abstract
1. Introduction
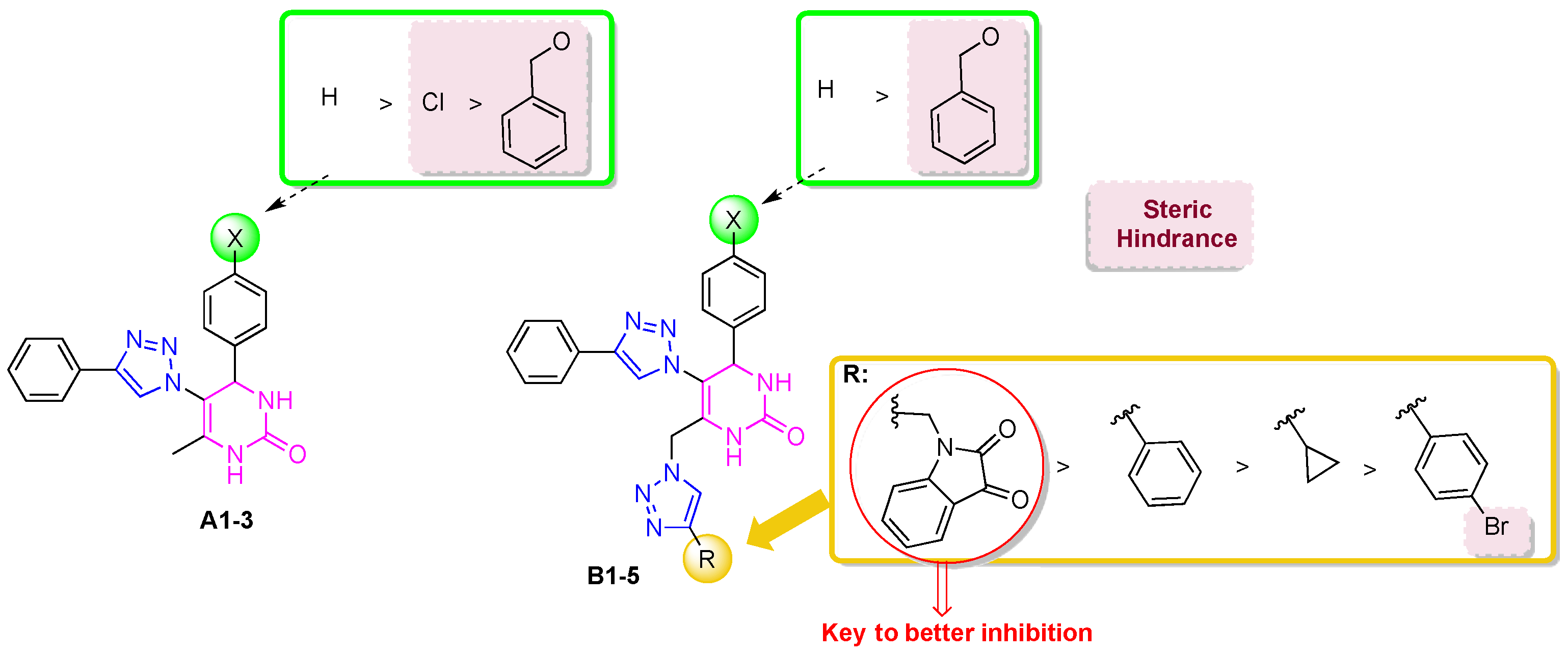
2. Results and Discussion
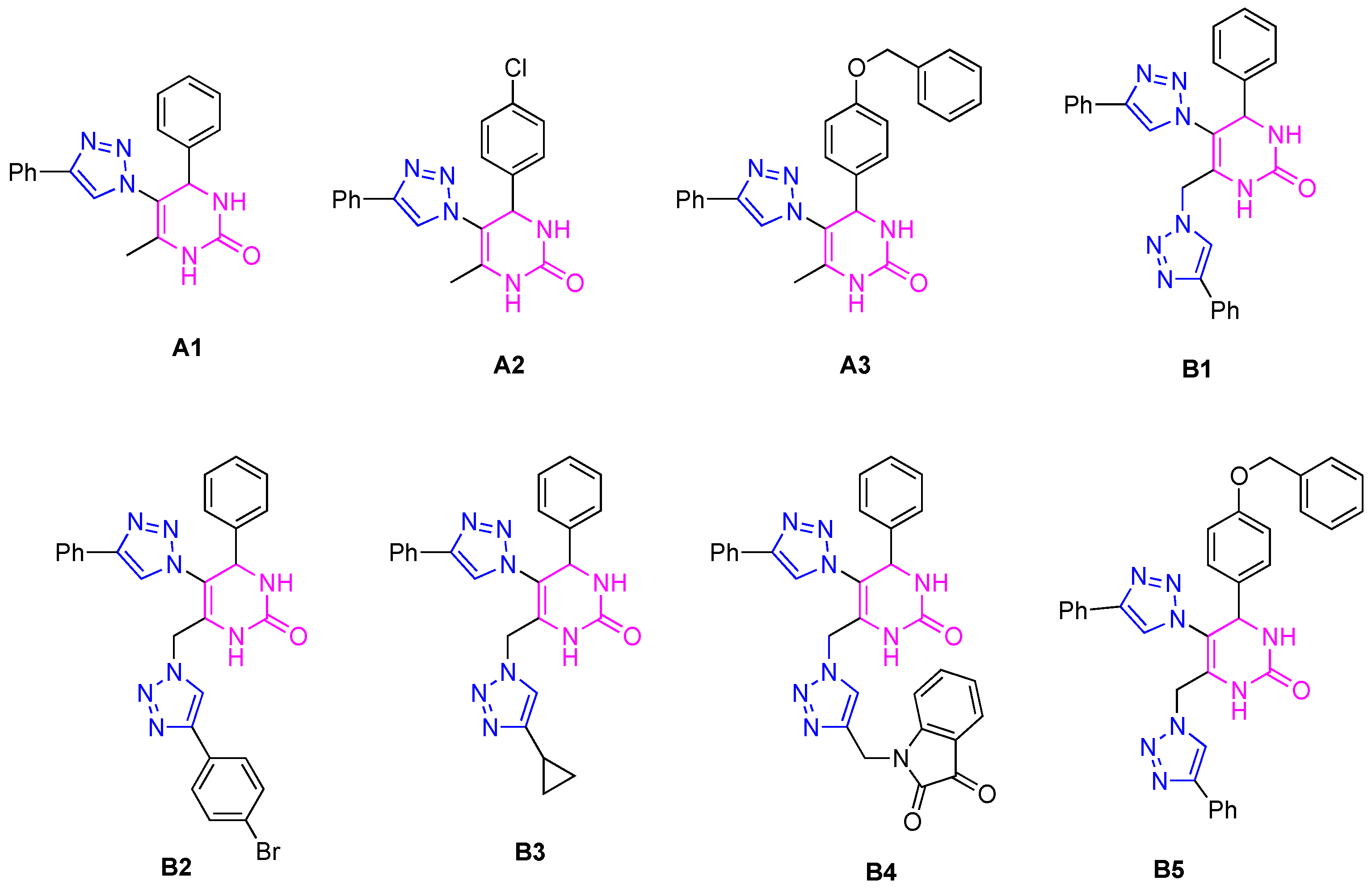
2.1. Anticholinesterase Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 ± SD (µM) a | ||
|---|---|---|
| Compound | eeAChE | eqBuChE |
| A1 | >100 | 12 ± 2 |
| A2 | >100 | 24 ± 1 |
| A3 | >100 | >100 |
| B1 | >100 | 53 ± 4 |
| B2 | >100 | >100 |
| B3 | >100 | 58 ± 3 |
| B4 | >100 | 1 ± 0.1 Mixed inhibition b Kia = 1.1 ± 0.3 µM Kib = 1.4 ± 0.2 µM |
| B5 | >100 | >100 |
| Galantamine | 2.7 ± 0.2 | 10.0 |
2.2. In Silico Studies
2.3. STD-NMR Studies
2.3.1. STD-NMR of Hybrid A1
2.3.2. STD-NMR of Hybrid B4
2.4. In Vitro Antioxidant Assays
2.5. Toxicity Assay In Vivo
2.6. SwissADME Calculations
3. Materials and Methods
3.1. General Remarks
3.2. 1,2,3-Triazole-Dihydropyrimidinone Hybrids A and B
3.3. Cholinesterase Inhibitory Assays
3.4. In Silico Studies
3.5. STD-NMR Experiments
3.6. Antioxidant Assays
3.6.1. DPPH Antioxidant Assay
3.6.2. ABTS Antioxidant Assay
3.6.3. FRAP Antioxidant Assay
3.7. Artemia Salina Lethal Toxicity Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Barbosa, D.J.; Remião, F.; Silva, R. Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochem. Pharmacol. 2023, 211, 115522. [Google Scholar] [CrossRef]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1502, 139–144. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef]
- Dorababu, A. Critical evaluation of current Alzheimer’s drug discovery (2018–2019) & futuristic Alzheimer drug model approach. Bioorg. Chem. 2019, 93, 103299. [Google Scholar] [CrossRef]
- Matos, L.H.S.; Masson, F.T.; Simeoni, L.A.; Homem-de-Mello, M. Biological activity of dihydropyrimidinone (DHPM) derivatives: A systematic review. Eur. J. Med. Chem. 2018, 143, 1779. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30. [Google Scholar] [CrossRef]
- Singh, A.; Singh, K.; Kaur, J.; Kaur, R.; Sharma, A.; Kaur, J.; Kaur, U.; Chadha, R.; Bedi, P.M.S. Pathogenesis of Alzheimer’s Disease and Diversity of 1,2,3-Triazole Scaffold in Drug Development: Design Strategies, Structural Insights, and Therapeutic Potential. ACS Chem. Neurosci. 2023, 14, 3291–3317. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3477–3744. [Google Scholar] [CrossRef]
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef]
- Xu, M.; Peng, Y.; Zhu, L.; Wang, S.; Ji, J.; Rakesh, K.P. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019, 180, 656–672. [Google Scholar] [CrossRef]
- Oh, J.M.; Kang, Y.; Hwang, J.H.; Park, J.-H.; Shin, W.-H.; Mun, S.-K.; Lee, J.U.; Yee, S.-T.; Kim, H. Synthesis of 4-substituted benzyl-2-triazole-linked-tryptamine-paeonol derivatives and evaluation of their selective inhibitions against butyrylcholinesterase and monoamine oxidase-B. Int. J. Biol. Macromol. 2022, 217, 910–921. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Edraki, N.; Sharifzadeh, M.; Khanavi, M.; Akbarzadeh, T. Novel tacrine-coumarin hybrids linked to 1,2,3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorg. Chem. 2019, 83, 303–316. [Google Scholar] [CrossRef]
- Marques, C.S.; López, Ó.; Bagetta, D.; Carreiro, E.P.; Petralla, S.; Alcaro, S.; Monti, B.; Bartolini, M.; Hoffmann, M.; Bolognesi, M.L.; et al. N-1,2,3-triazole-isatin derivatives for cholinesterase and β-amyloid aggregation inhibition: A comprehensive bioassay study. Bioorg. Chem. 2020, 98, 103753. [Google Scholar] [CrossRef]
- Carreiro, E.P.; Costa, A.R.; Antunes, C.M.; Ernesto, S.; Pinto, F.; Rodrigues, B.; Burke, A.J. Quercetin-1,2,3-Triazole Hybrids as Multifunctional Anti-Alzheimer’s Agents. Molecules 2023, 28, 7495. [Google Scholar] [CrossRef]
- Arunkhamkaew, S.; Athipornchai, A.; Apiratikul, N.; Suksamrarn, A.; Ajavakom, V. Novel racemic tetrahydrocurcuminoid dihydropyrimidinone analogues as potent acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2880–2882. [Google Scholar] [CrossRef]
- Barbosa, F.A.R.; Canto, R.F.S.; Saba, S.; Rafique, J.; Braga, A.L. Synthesis and evaluation of dihydropyrimidinone-derived selenoesters as multi-targeted directed compounds against Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 5762–5770. [Google Scholar] [CrossRef]
- Carreiro, E.P.; Sena, A.M.; Puerta, A.; Padrón, J.M.; Burke, A.J. Synthesis of Novel 1,2,3-Triazole-Dihyropyrimidinone Hybrids using Multicomponent 1,3-Dipolar Cycloaddition(Click)-Biginelli Reactions. Synlett 2020, 31, 615–621. [Google Scholar] [CrossRef]
- Laghchioua, F.E.; da Silva, C.F.M.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Mendes, R.F.; Paz, F.A.A.; Faustino, M.A.F.; Rakib, E.M.; Neves, M.G.P.M.S.; Pereira, F.; et al. Design of Promising Thiazoloindazole-Based Acetylcholinesterase Inhibitors Guided by Molecular Docking and Experimental Insights. ACS Chem. Neurosci. 2024, 15, 2853–2869. [Google Scholar] [CrossRef]
- Costa, L.D.; Silva, C.F.M.; Pinto, D.C.G.A.; Silva, A.M.S.; Pereira, F.; Faustino, M.A.F.; Tomé, A.C. Discovery of thiazolo [5,4-c]isoquinoline based compounds as acetylcholinesterase inhibitors through computational target prediction, molecular docking and bioassay. J. Mol. Struct. 2023, 1291, 136088. [Google Scholar] [CrossRef]
- Malafaia, D.; Oliveira, A.; Fernandes, P.A.; Ramos, M.J.; Albuquerque, H.M.T.; Silva, A.M.S. Chromeno [3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors. Int. J. Mol. Sci. 2021, 22, 4145. [Google Scholar] [CrossRef]
- Hameed, A.; Zehra, S.T.; Shah, S.J.A.; Khan, K.M.; Alharthy, R.D.; Furtmann, N.; Bajorath, J.; Tahir, M.N.; Iqbal, J. Syntheses, Cholinesterases Inhibition, and Molecular Docking Studies of Pyrido[2,3-b]pyrazine Derivatives. Chem. Biol. Drug Des. 2015, 86, 1115–1120. [Google Scholar] [CrossRef]
- Başaran, E.; Çakmak, R.; Şentürk, M.; Taskin-Tok, T. Biological activity and molecular docking studies of some N-phenylsulfonamides against cholinesterases and carbonic anhydrase isoenzymes. J. Mol. Recognit. 2022, 35, e2982. [Google Scholar] [CrossRef]
- Zilbeyaz, K.; Stellenboom, N.; Guney, M.; Oztekin, A.; Senturk, M. Effects of aryl methanesulfonate derivatives on acetylcholinesterase and butyrylcholinesterase. J. Biochem. Mol. Toxicol. 2018, 32, e22210. [Google Scholar] [CrossRef]
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Stephen Chan, S.; Parat, M.-O.; Brazzolotto, X.; Ross, B.P. Discovery and Structure–Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef]
- Ellman, G.; Courtney, K.; Andres, V.; Featherstone, R. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Hofmanova, T.; Marques, C.; García-Sosa, A.T.; López, Ó.; Leitzbach, L.; Carreiro, E.P.; González-Bakker, A.; Puerta, A.; Stark, H.; Padrón, J.M.; et al. N-Substituted 3-Aminooxindoles and N-Propargyl Derivatives: Potential Biological Activities against Alzheimer’s Disease. Results Chem. 2023, 6, 101032. [Google Scholar] [CrossRef]
- Asghar, A.; Yousuf, M.; Fareed, G.; Nazir, R.; Hassan, A.; Maalik, A.; Noor, T.; Iqbal, N.; Rasheed, L. Synthesis, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) activities, and molecular docking studies of a novel compound based on combination of flurbiprofen and isoniazide. RSC Adv. 2020, 10, 19346–19352. [Google Scholar] [CrossRef]
- Pourshojaei, Y.; Abiri, A.; Eskandari, K.; Haghighijoo, Z.; Najmeh Edraki, N.; Asadipour, A. Phenoxyethyl Piperidine/Morpholine Derivatives as PAS and CAS Inhibitors of Cholinesterases: Insights for Future Drug Design. Sci. Rep. 2019, 9, 19855. [Google Scholar] [CrossRef]
- Bacalhau, P.; Fernandes, L.; Martins, M.R.; Candeias, F.; Carreiro, E.P.; Lopez, O.; Caldeira, A.T.; Totobenazara, J.; Guedes, R.C.; Burke, A.J. In silico, NMR and pharmacological evaluation of an hydroxyoxindole cholinesterase inhibitor. Bioorg. Med. Chem. 2019, 27, 354–363. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ayub, A.; Wettig, S. An Overview of Nanotechnologies for Drug Delivery to the Brain. Pharmaceutics 2022, 14, 224. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Wootton-Beard, P.C.; Moran, A.; Ryan, L. Stability of the total antioxidant capacity and total polyphenol content of 23 commercially available vegetable juices before and after in vitro digestion measured by FRAP, DPPH, ABTS and Folin–Ciocalteu methods. Food Res. Int. 2011, 44, 217–224. [Google Scholar] [CrossRef]
- Rumpf, J.; Burger, R.; Schulze, M. Statistical evaluation of DPPH, ABTS, FRAP, and Folin-Ciocalteu assays to assess the antioxidant capacity of lignins. Int. J. Biol. Macromol. 2023, 233, 123470. [Google Scholar] [CrossRef]



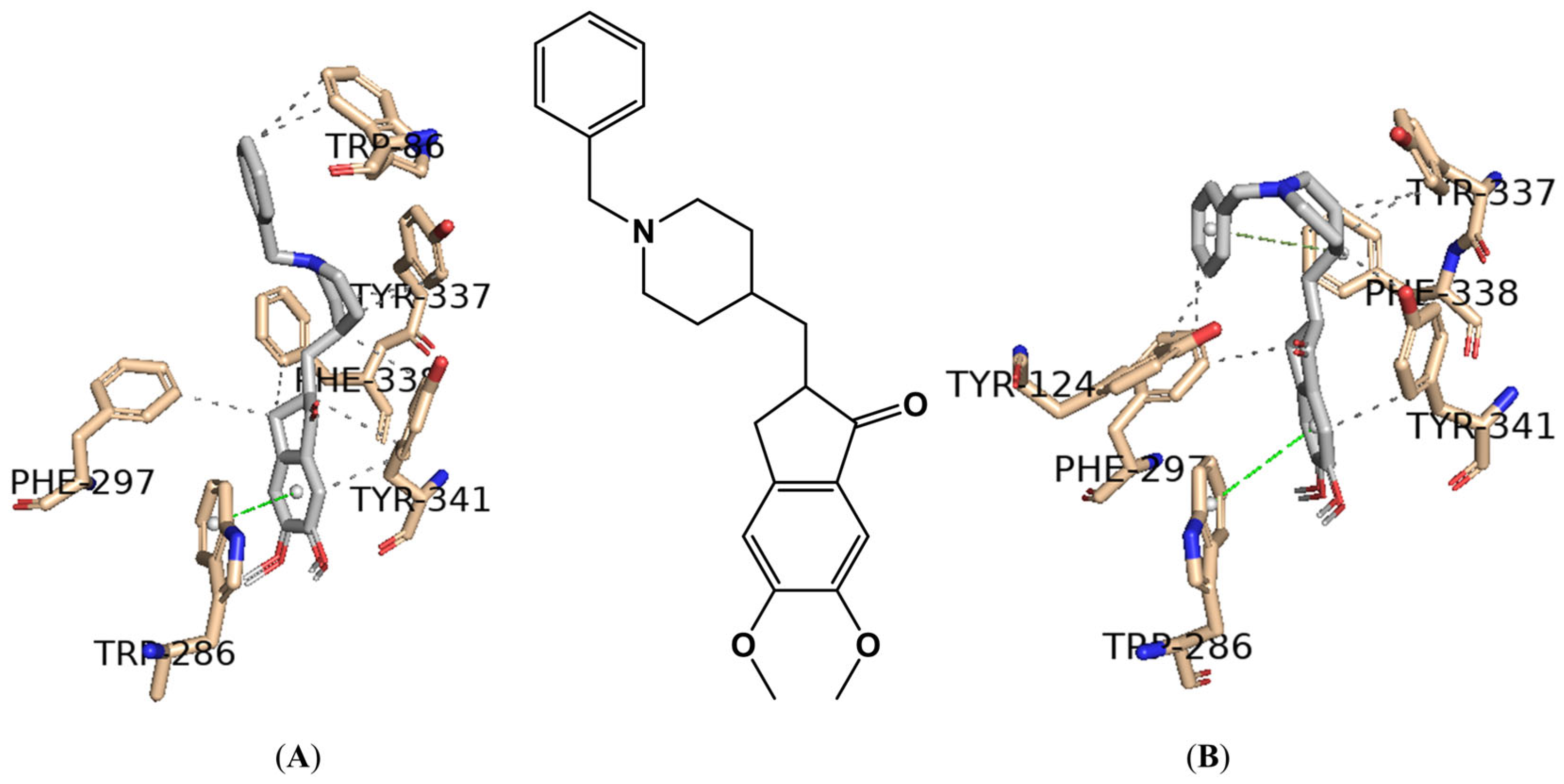
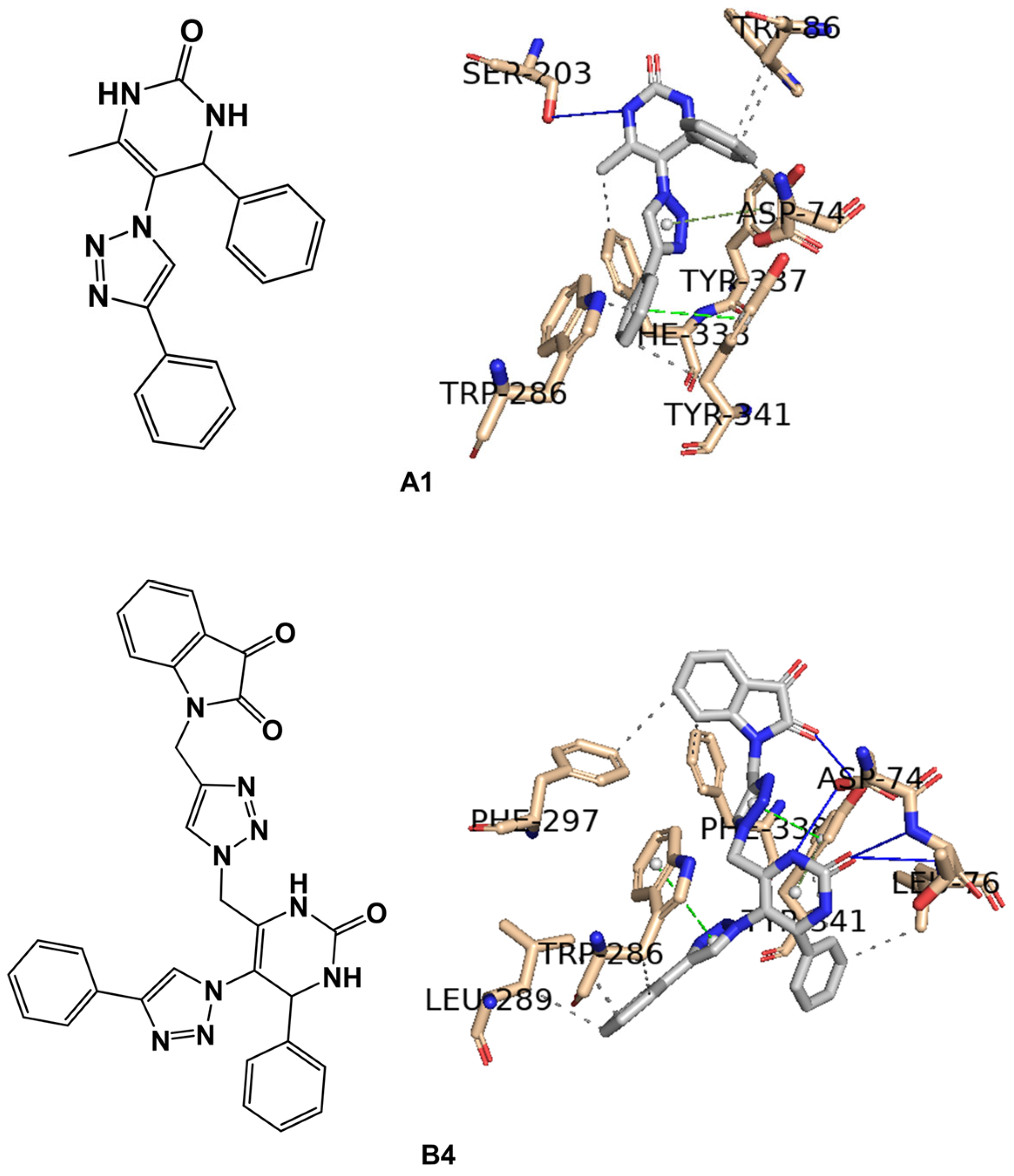
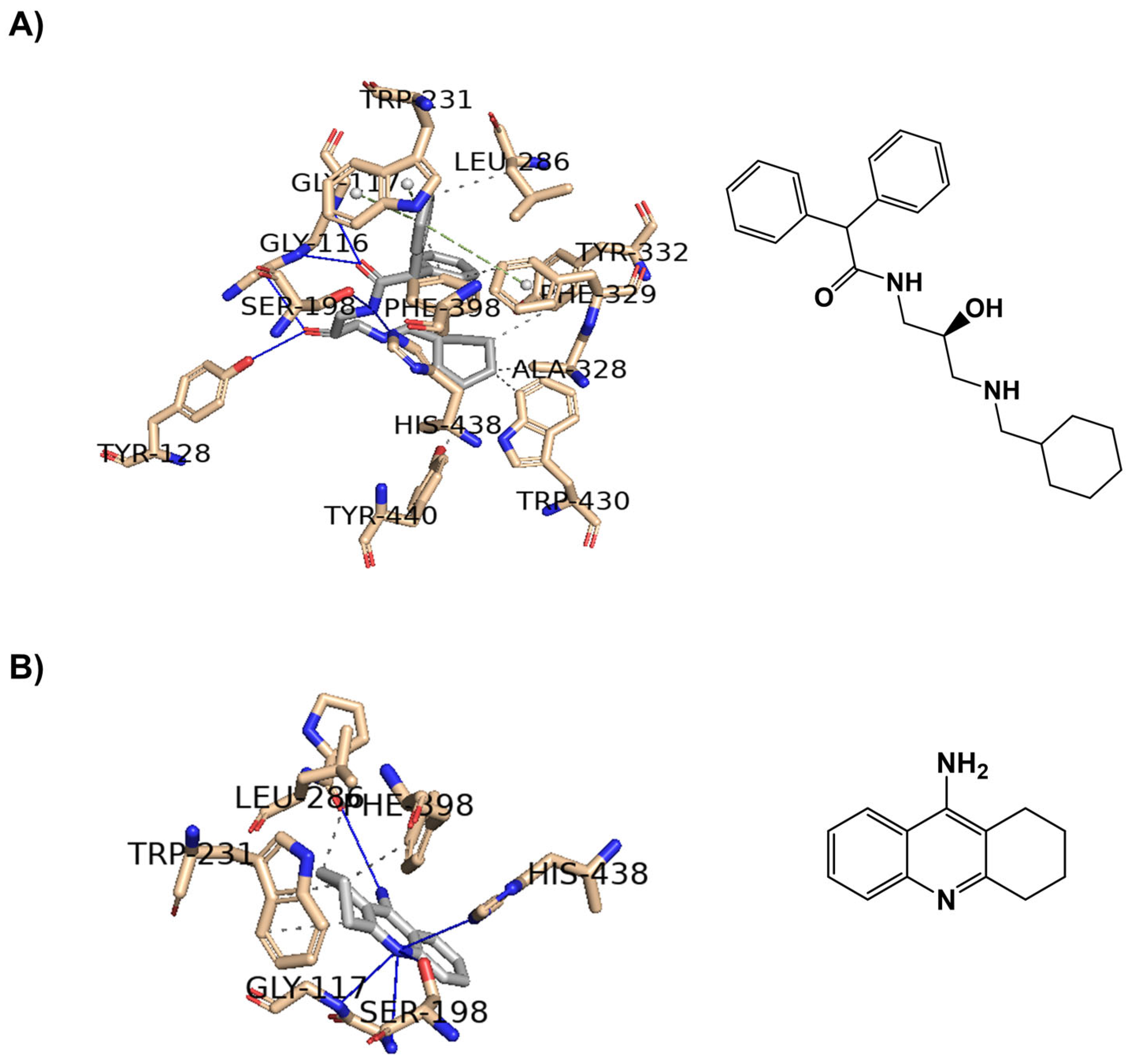
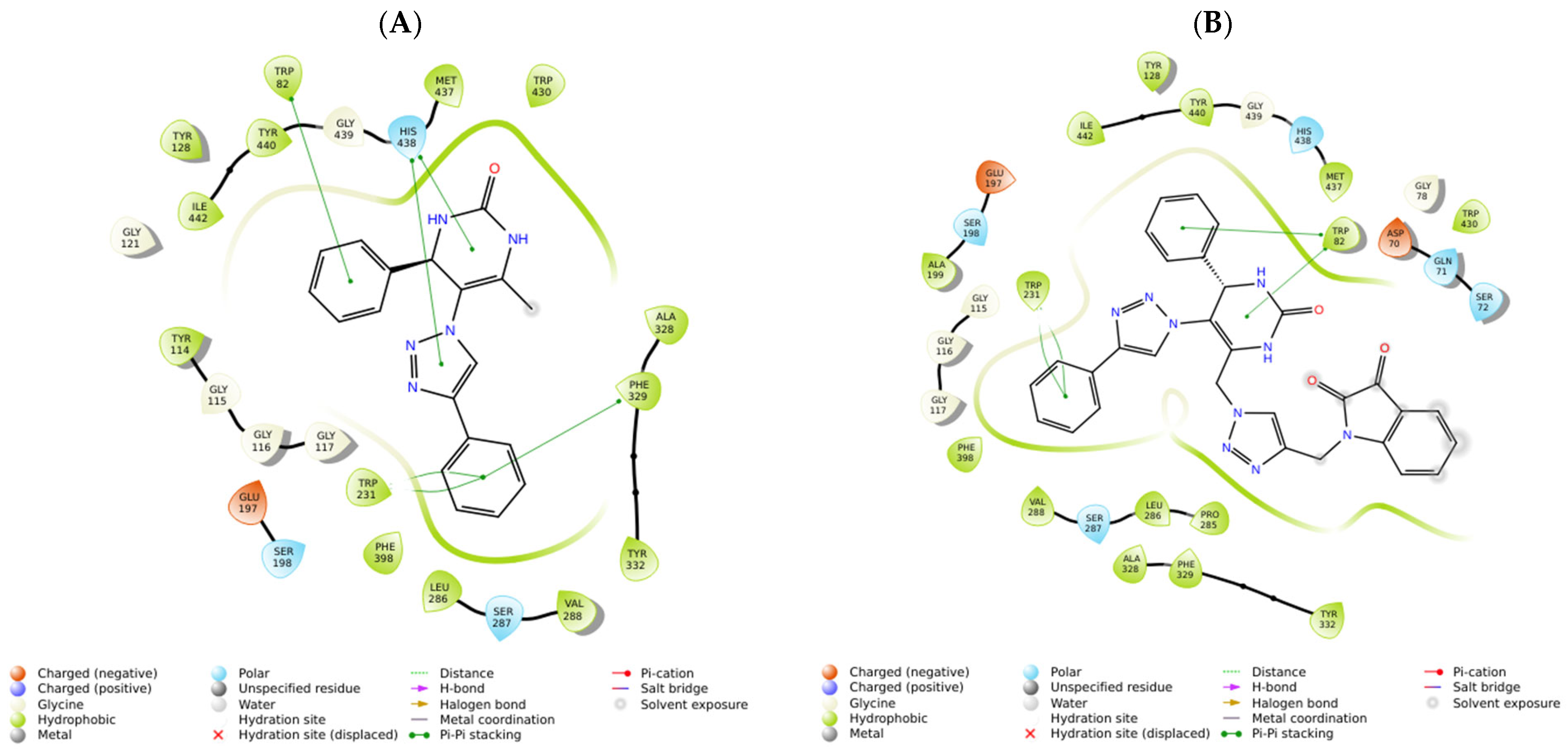
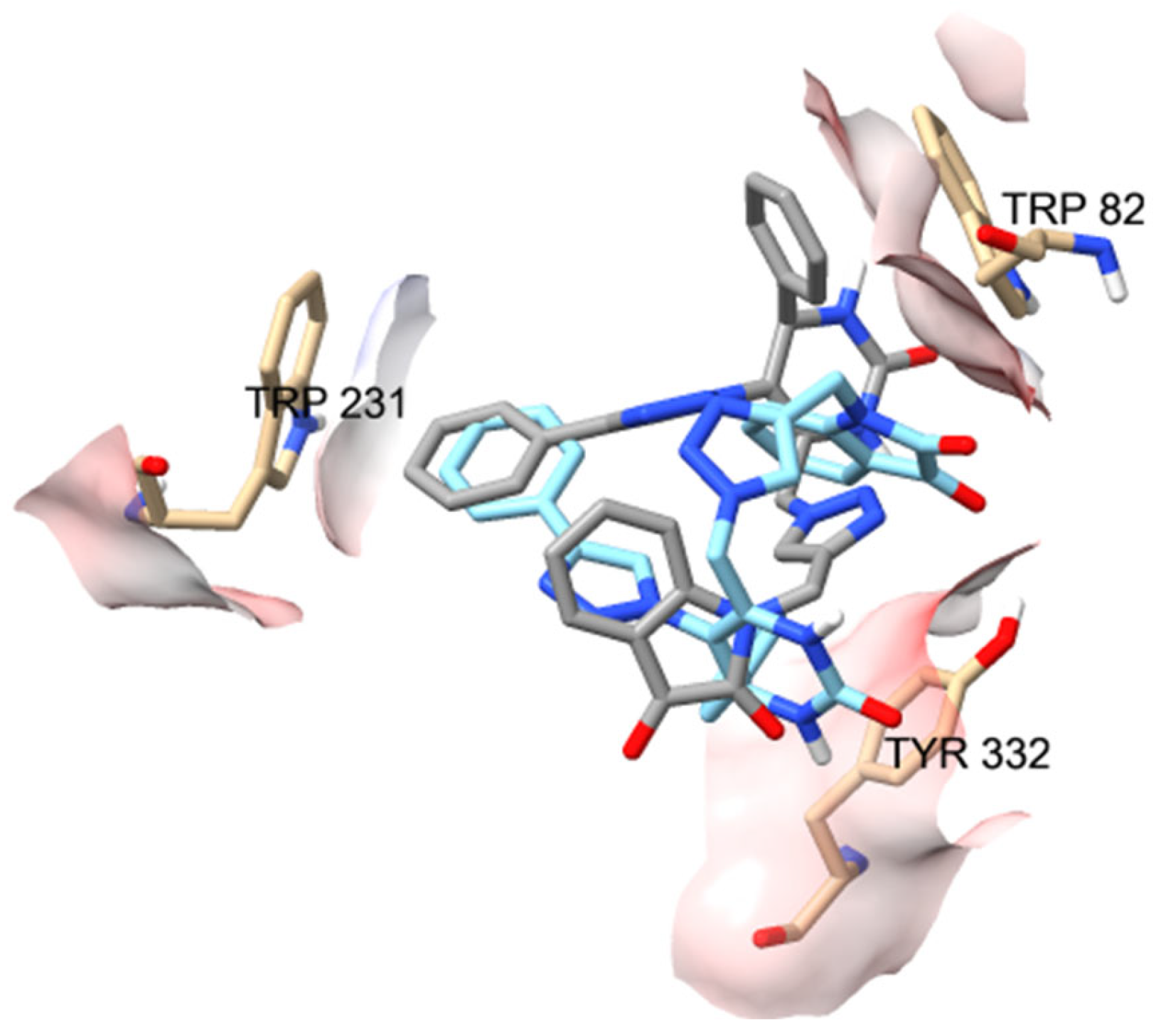



| Compounds | Positive Controls | |||||||
|---|---|---|---|---|---|---|---|---|
| A1 | B4 | Donepezil 2 | β-alanine 3 | HPD 3 | Tacrine 3 | Galantamine 2,3 | ||
| Software 1 | ChE (PDB ID) | Score (kcal/mol) 4 | ||||||
| Glide Xp | AChE (6O4W) | −3.72 | −8.99 | −15.08 | −5.29 | - | - | - |
| BuChE (4AQD) | −7.80 | −9.95 | −7.04 | −5.00 | - | - | - | |
| AutoDock Vina | AChE (6O4W) | −11.75 | −11.13 | −11.07 | - | - | - | −9.89 |
| AChE (4EY7) | −11.63 | −11.21 | −11.63 | - | - | - | −9.76 | |
| BuChE (4AQD) | −10.58 | −12.57 | - | −3.967 | - | - | −8.60 | |
| BuChE (7Q1M) | −10.62 | −13.28 | - | - | −9.81 | - | −8.72 | |
| BuChE (4BDS) | −10.96 | −12.57 | - | - | - | −8.28 | −8.98 | |
| K2Cr2O7 | A1 | A2 | A3 | B1 | B2 | B3 | B4 | B5 | |
|---|---|---|---|---|---|---|---|---|---|
| LC50 | 23.6 mg/L | - | - | - | - | >200 μM | - | - | - |
| NOEC | 10 mg/L | 200 μM | 200 μM | 200 μM | 200 μM | 1 μM | 200 μM | 200 μM | 200 μM |
| LOEC | 18 mg/L | - | - | - | - | 100 μM | - | - | - |
| Hybrid | MW 1 (g/mol) | MLOGP | LogS | HBA 2 | HBD 3 | TPSA 4 | GI | BBB | PAINS # Alerts | CYP2D6/CYP3A4 5 |
|---|---|---|---|---|---|---|---|---|---|---|
| A1 | 331.37 | 2.36 | −3.62 | 3 | 2 | 71.84 | High | Yes | 0 | No 6 |
| A2 | 365.82 | 2.86 | −4.21 | 3 | 2 | 71.84 | high | yes | 0 | No 6 |
| A3 | 437.49 | 3.13 | −5.01 | 4 | 2 | 81.07 | high | no | 0 | Yes 7 |
| B1 | 474.52 | 2.95 | −4.79 | 5 | 2 | 102.55 | high | no | 0 | No 6 |
| B2 | 553.41 | 3.51 | −5.70 | 5 | 2 | 102.55 | high | no | 0 | No 6 |
| B3 | 438.48 | 2.59 | −3.80 | 5 | 2 | 102.55 | high | no | 0 | No 6 |
| B4 | 557.56 | 1.79 | −4.43 | 7 | 2 | 139.93 | high | no | 0 | No 6 |
| B5 | 580.64 | 3.59 | −6.17 | 6 | 2 | 111.78 | high | no | 0 | No 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gastalho, C.M.; Sena, A.M.; López, Ó.; Fernández-Bolaños, J.G.; García-Sosa, A.T.; Pereira, F.; Antunes, C.M.; Costa, A.R.; Burke, A.J.; Carreiro, E.P. Assessing the Potential of 1,2,3-Triazole-Dihydropyrimidinone Hybrids Against Cholinesterases: In Silico, In Vitro, and In Vivo Studies. Int. J. Mol. Sci. 2024, 25, 11153. https://doi.org/10.3390/ijms252011153
Gastalho CM, Sena AM, López Ó, Fernández-Bolaños JG, García-Sosa AT, Pereira F, Antunes CM, Costa AR, Burke AJ, Carreiro EP. Assessing the Potential of 1,2,3-Triazole-Dihydropyrimidinone Hybrids Against Cholinesterases: In Silico, In Vitro, and In Vivo Studies. International Journal of Molecular Sciences. 2024; 25(20):11153. https://doi.org/10.3390/ijms252011153
Chicago/Turabian StyleGastalho, Carlos M., Ana M. Sena, Óscar López, José G. Fernández-Bolaños, Alfonso T. García-Sosa, Florbela Pereira, Célia M. Antunes, Ana R. Costa, Anthony J. Burke, and Elisabete P. Carreiro. 2024. "Assessing the Potential of 1,2,3-Triazole-Dihydropyrimidinone Hybrids Against Cholinesterases: In Silico, In Vitro, and In Vivo Studies" International Journal of Molecular Sciences 25, no. 20: 11153. https://doi.org/10.3390/ijms252011153
APA StyleGastalho, C. M., Sena, A. M., López, Ó., Fernández-Bolaños, J. G., García-Sosa, A. T., Pereira, F., Antunes, C. M., Costa, A. R., Burke, A. J., & Carreiro, E. P. (2024). Assessing the Potential of 1,2,3-Triazole-Dihydropyrimidinone Hybrids Against Cholinesterases: In Silico, In Vitro, and In Vivo Studies. International Journal of Molecular Sciences, 25(20), 11153. https://doi.org/10.3390/ijms252011153