
Assessing Genetic Diversity in Endangered Plant Orchidantha chinensis: Chloroplast Genome Assembly and Simple Sequence Repeat Marker-Based Evaluation


Abstract
1. Introduction
2. Results
2.1. Assembly, Annotation, and Phylogenetic Analysis of Chloroplast Genome of O. chinensis
2.2. SSR Identification of O. chinensis and Polymorphism Detection
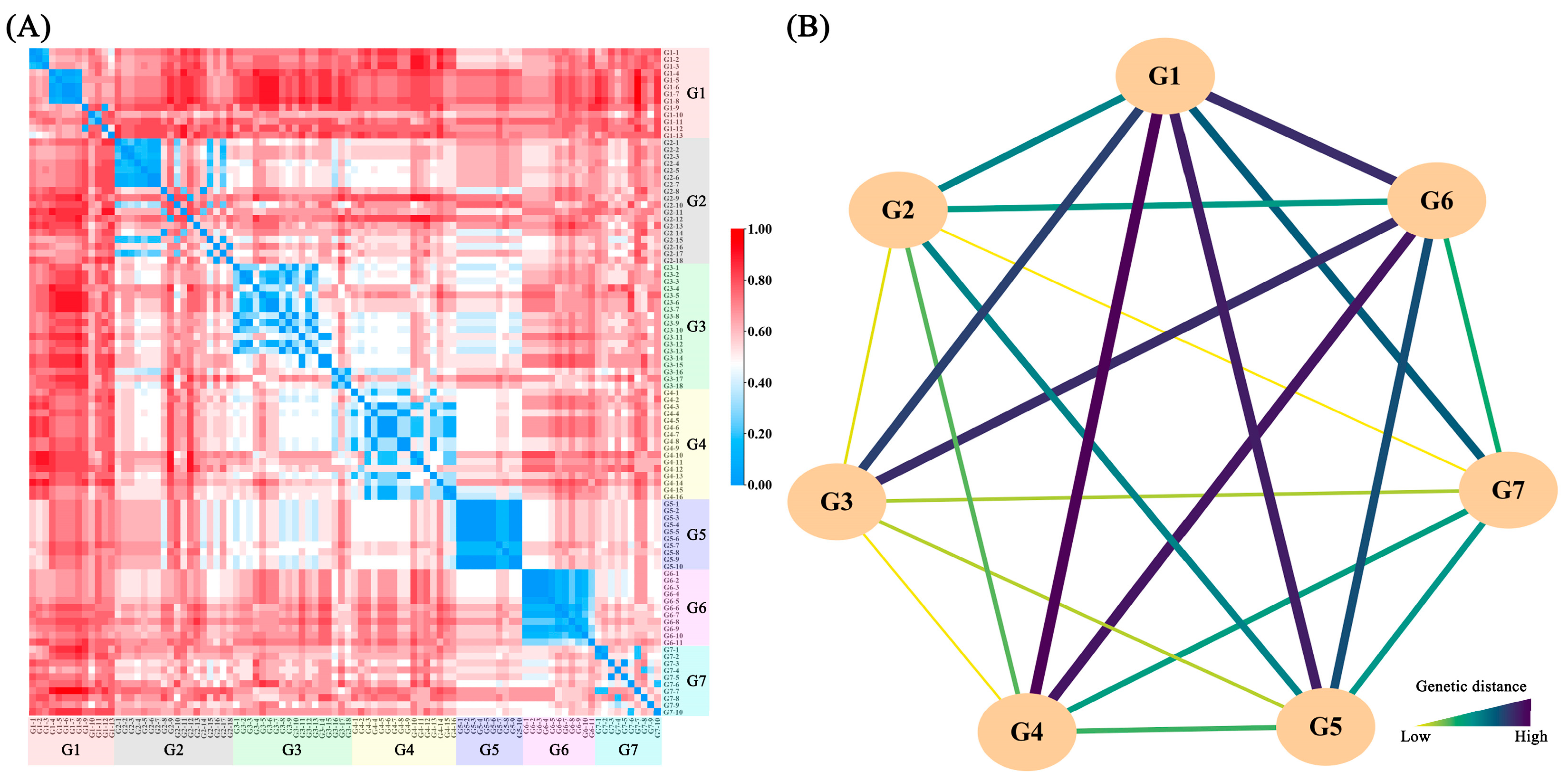
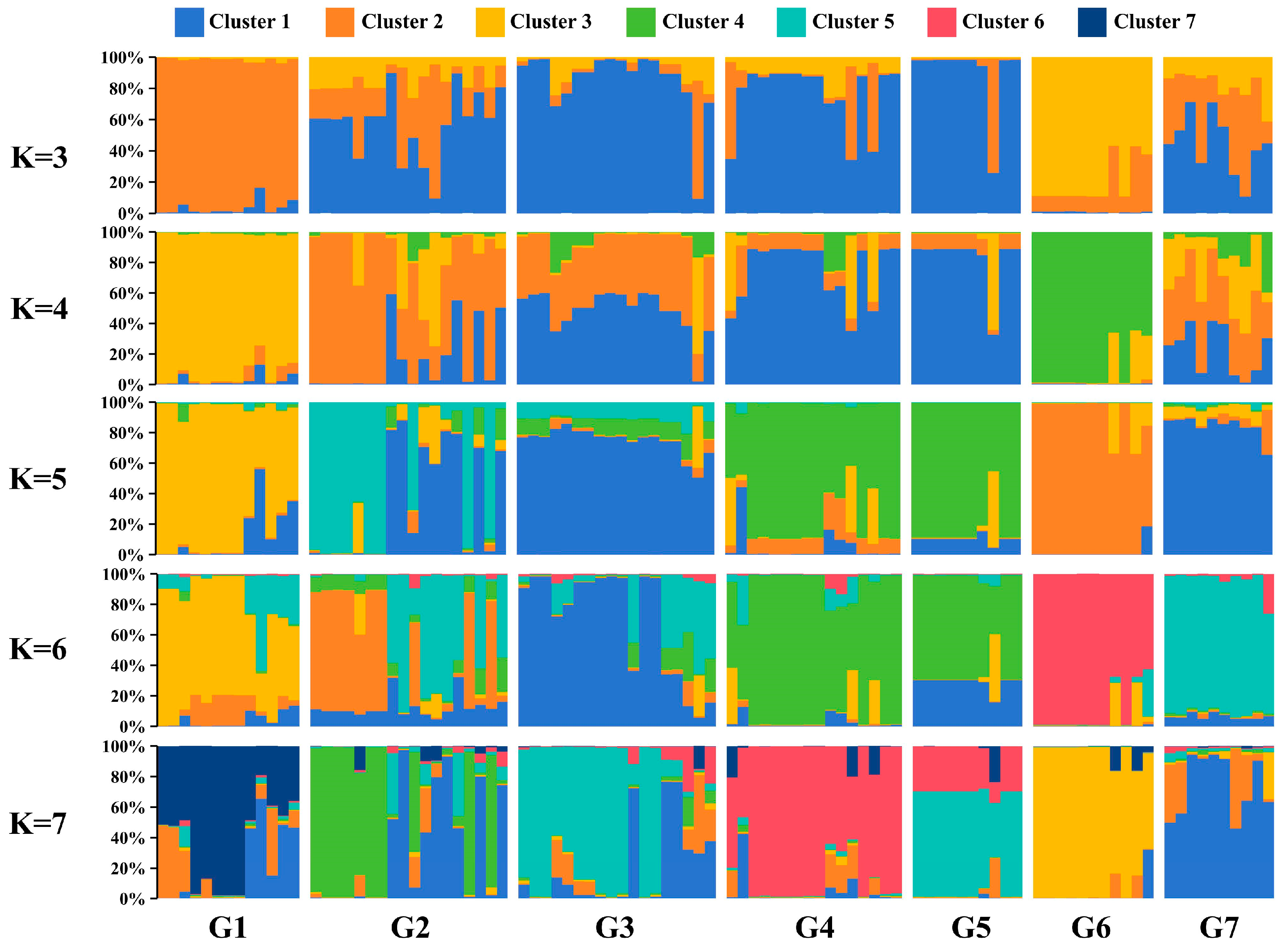
2.3. Genetic Diversity and Population Structure Analysis
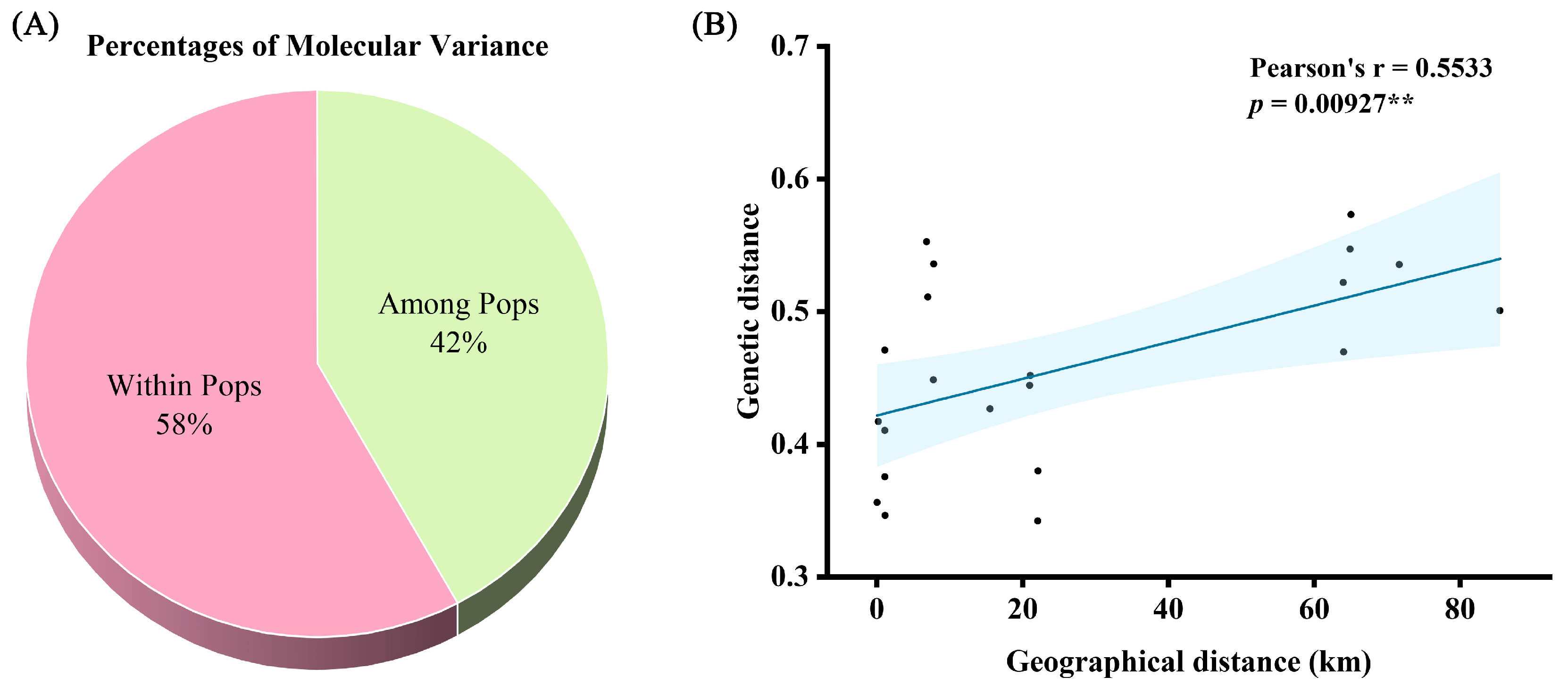
2.4. AMOVA and Correlation Analysis of Genetic and Geographic Distances
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Assembly of the O. chinensis Chloroplast Genome
4.3. RNA Sequencing and Assembly of O. chinensis Leaves
4.4. Identification of SSR Loci and Primer Design
4.5. Screening of Polymorphic SSR Primers
4.6. Phylogenetic Analysis Based on Chloroplast Genome
4.7. Analysis of Genetic Diversity and Population Structure Based on SSR Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- My, S.; Kalu, M.; Syazana, A. Orchidantha sarawakensis sp. Nov. (Zingiberales: Lowiaceae), a new species endemic to east malaysia, borneo. J. Trop. For. Sci. 2019, 31, 371–376. [Google Scholar]
- Zou, P.; Cai, X.A.; Xu, K.; Cui, C.J.; Ye, Y.S.; Liao, J.P. Orchidantha crassinervia sp. Nov. (Lowiaceae) from Guangxi, China. Nord. J. Bot. 2019, 37, e02493. [Google Scholar] [CrossRef]
- Leong-Skornickova, J.; Binh Nguy, Q.; Sida, O. Orchidantha virosa Skornick. & Q.B.Nguyen, sp. Nov. (Lowiaceae), a new species endemic to northern vietnam. Adansonia 2014, 36, 237–243. [Google Scholar]
- ăng Trân, H.; Leong-škorničková, J. Orchidantha stercorea sp. Nov. (Lowiaceae) from vietnam. Nord. J. Bot. 2010, 28, 299–303. [Google Scholar] [CrossRef]
- Cai, L.; Dao, Z.; Sun, W. Discovery of a wild population of Orchidantha yunnanensis in south-east yunnan, China. Oryx 2019, 53, 412. [Google Scholar] [CrossRef]
- Johansen, L.B. Phylogeny of Orchidantha (Lowiaceae) and the Zingiberales based on six dna regions. Syst. Bot. 2005, 30, 106–117. [Google Scholar] [CrossRef]
- Niissalo, M.A.; Gardner, E.M.; Khew, G.S.; šída, O.; Poulsen, A.D.; Leong-škorničková, J. Whence came these plants most foul? Phylogenomics and biogeography of Lowiaceae (Zingiberales). Front. Ecol. Evol. 2022, 9, 794977. [Google Scholar] [CrossRef]
- Kress, W.J.; Prince, L.M.; Hahn, W.J.; Zimmer, E.A.; Olmstead, R.; Olmstead, R. Unraveling the evolutionary radiation of the families of the Zingiberales using morphological and molecular evidence. Syst. Biol. 2001, 50, 926–944. [Google Scholar] [CrossRef]
- Barrett, C.F.; Specht, C.D.; Leebens-Mack, J.; Stevenson, D.W.; Zomlefer, W.B.; Davis, J.I. Resolving ancient radiations: Can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Ann. Bot. 2014, 113, 119–133. [Google Scholar] [CrossRef]
- Sass, C.; Iles, W.J.D.; Barrett, C.F.; Smith, S.Y.; Specht, C.D. Revisiting the Zingiberales: Using multiplexed exon capture to resolve ancient and recent phylogenetic splits in a charismatic plant lineage. PeerJ 2016, 4, e1584. [Google Scholar] [CrossRef]
- Carlsen, M.M.; Fér, T.; Schmickl, R.; Leong-škorničková, J.; Newman, M.; Kress, W.J. Resolving the rapid plant radiation of early diverging lineages in the tropical Zingiberales: Pushing the limits of genomic data. Mol. Phylogenet. Evol. 2018, 128, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zeng, P.; Zhang, L.; Huang, W.; Wang, H.; Chen, G. Symbiosis theory-directed green synthesis of silver nanoparticles and their application in infected wound healing. Int. J. Nanomed. 2016, 11, 2757–2767. [Google Scholar]
- Luo, Y.; Chen, W.; Wen, L.; Zhou, L.; Kang, X.; Chen, G. A new hexanedioic acid analogue from the endophytic fungus penicillium sp. Oc-4 of Orchidantha chinensis. Chem. Nat. Compd. 2017, 53, 834–838. [Google Scholar] [CrossRef]
- Kirchoff, B.K.; Liu, H.; Liao, J. Inflorescence and flower development in Orchidantha chinensis T. L. Wu (Lowiaceae; Zingiberales): Similarities to inflorescence structure in the strelitziaceae. Int. J. Plant Sci. 2020, 181, 716–731. [Google Scholar] [CrossRef]
- Pedersen, L.B.; Johansen, B. Anatomy of the unusual stigma in Orchidantha (Lowiaceae). Am. J. Bot. 2004, 91, 299–305. [Google Scholar] [CrossRef]
- Song, J.; Liao, J.; Tang, Y.; Chen, Z. Chromosome numbers in Orchidantha (Lowiaceae) and their biogeographic and systematic implications. Ann. Bot. Fenn. 2004, 41, 429–433. [Google Scholar]
- Zou, P.; Ye, Y.S.; Cai, X.A.; Liao, J.P. Rediscovery and improved description of Orchidantha insularis (Lowiaceae), a rare species from hainan, china. Nord. J. Bot. 2014, 32, 819–823. [Google Scholar] [CrossRef]
- Li, R.; Tang, Y.; Liao, J. Study on genetic diversity of Orchidantha chinensis (Lowiaceae). J. Trop. Subtrop. Bot. 2005, 13, 253–258. (in Chinese). [Google Scholar]
- Li, R.; Zou, P.; Tang, Y.; Liao, J. Study on the population biology of endangered plant Orchidanha. J. Anhui Agric. Sci. 2008, 36, 4086–4088. (In Chinese) [Google Scholar]
- Taheri, S.; Lee Abdullah, T.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast Microsatellites: New Tools for Studies in Plant Ecology and Evolution; Elsevier Ltd.: Oxford, UK, 2001; Volume 16, pp. 142–147. [Google Scholar]
- Jakobsson, M.; Säll, T.; Lind-Hallden, C.; Hallden, C. Evolution of chloroplast mononucleotide microsatellites in Arabidopsis thaliana. Theor. Appl. Genet. 2007, 114, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and prospects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpssr discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Parthiban, S.; Govindaraj, P.; Senthilkumar, S. Comparison of relative efficiency of genomic SSR and EST-SSR markers in estimating genetic diversity in sugarcane. 3 Biotech 2018, 8, 112–144. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Hu, L.; Wang, J.; Wang, X.; Zhang, D.; Sun, Y.; Lu, T.; Shi, W. Development of SSR markers and evaluation of genetic diversity of endangered plant Saussurea involucrata. Biomolecules 2024, 14, 1010. [Google Scholar] [CrossRef]
- Szczecinska, M.; Sramko, G.; Wolosz, K.; Sawicki, J. Genetic diversity and population structure of the rare and endangered plant species Pulsatilla patens (L.) Mill in east central europe. PLoS ONE 2016, 11, e0151730. [Google Scholar] [CrossRef]
- Sakai, S.; Inoue, T. A new pollination system: Dung-beetle pollination discovered in Orchidantha inouei (Lowiaceae, Zingiberales) in sarawak, malaysia. Am. J. Bot. 1999, 86, 56–61. [Google Scholar] [CrossRef]
- Kramer, A.T.; Havens, K. Plant conservation genetics in a changing world. Trends Plant Sci. 2009, 14, 599–607. [Google Scholar] [CrossRef]
- Volis, S. How to conserve threatened chinese plant species with extremely small populations? Plant Divers. 2016, 38, 45–52. [Google Scholar] [CrossRef]
- Wauters, L.A.; Lens, L. Effects of food availability and density on red squirrel (Sciurus vulgaris) reproduction. Ecology 1995, 76, 2460–2469. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; Depamphilis, C.W.; Yi, T.; Li, D. Getorganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Qu, X.; Moore, M.J.; Li, D.; Yi, T. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Wu, M.; Yan, R.; Xu, X.; Gou, G.; Dai, Z. Characterization of the plastid genome of the vulnerable endemic Indosasa lipoensis and phylogenetic analysis. Diversity 2023, 15, 197. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M.; Valencia, A. Misa-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Zhou, Y.; Ye, Y.; Zhu, G.; Xu, Y.; Tan, J.; Liu, J. Diversity, classification, and est-ssr-based association analysis of caladium ornamental traits. Physiol. Plant. 2023, 175, e13841. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. Powermarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure harvester: A website and program for visualizing structure output and implementing the evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. Clumpp: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 21 August 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| Genes for photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI |
| Subunits of photosystem II | psbA, psbB, psbC, psbD (3), psbE, psbF, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, ycf3 | |
| Subunits of NADH dehydrogenase | ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| Subunits of photosystem I | psaA, psaB, psaI, psaJ | |
| Subunit of rubisco | rbcL | |
| Self-replication | Large subunit of ribosome | rpl14, rpl16, rpl20, rpl22, rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Small subunit of ribosome | rps11, rps12, rps14, rps15, rps16, rps19, rps2, rps4, rps7, rps8 | |
| Transfer RNA | trnQ-UUG, trnH-GUG (2), trnL-UAG, trnL-UAA, trnL-CAA, trnS-GCU, trnG-UCC (2), trnI-GAU, trnW-CCA, trnC-GCA, trnS-UGA, trnM-CAU (3), trnV-GAC, trnT-GGU, trnR-UCU, trnT-UGU, trnP-UGG, trnA-UGC, trnK-UUU, trnR-ACG, trnY-GUA, trnN-GUU, trnS-GGA (2), trnfM-CAU, trnV-UAC, trnF-GAA, trnE-UUC, trnD-GUC | |
| Ribosomal RNA | rrn23S, rrn4.5S, rrn16S, rrn5S | |
| Other genes | c-Type cytochrome synthesis gene | ccsA |
| Envelop membrane protein | cemA | |
| Translational initiation factor | infA | |
| Maturase | matK | |
| Conserved open reading frames | ycf4 |
| Item | Marker | Major Allele Frequency | Na * | GD * | PIC * |
|---|---|---|---|---|---|
| cpSSR | OccpSSR1 | 0.98 | 2 | 0.04 | 0.04 |
| OccpSSR8 | 0.89 | 2 | 0.2 | 0.18 | |
| OccpSSR12 | 0.88 | 2 | 0.22 | 0.19 | |
| OccpSSR19 | 0.76 | 2 | 0.36 | 0.30 | |
| OccpSSR40 | 0.77 | 5 | 0.37 | 0.33 | |
| OccpSSR44 | 0.72 | 5 | 0.46 | 0.43 | |
| OccpSSR45 | 0.52 | 4 | 0.59 | 0.52 | |
| Mean | 0.79 | 3.14 | 0.32 | 0.29 | |
| EST-SSR | OcESTSSR17 | 0.29 | 11 | 0.83 | 0.80 |
| OcESTSSR25 | 0.20 | 10 | 0.86 | 0.85 | |
| OcESTSSR34 | 0.46 | 4 | 0.58 | 0.50 | |
| OcESTSSR35 | 0.44 | 10 | 0.76 | 0.74 | |
| OcESTSSR39 | 0.27 | 29 | 0.89 | 0.89 | |
| OcESTSSR41 | 0.32 | 16 | 0.82 | 0.80 | |
| OcESTSSR62 | 0.49 | 10 | 0.66 | 0.60 | |
| OcESTSSR63 | 0.22 | 9 | 0.84 | 0.82 | |
| OcESTSSR69 | 0.29 | 10 | 0.83 | 0.81 | |
| OcESTSSR71 | 0.80 | 5 | 0.33 | 0.31 | |
| OcESTSSR75 | 0.43 | 6 | 0.74 | 0.70 | |
| OcESTSSR77 | 0.42 | 9 | 0.75 | 0.72 | |
| OcESTSSR78 | 0.43 | 7 | 0.70 | 0.65 | |
| OcESTSSR83 | 0.47 | 3 | 0.59 | 0.50 | |
| Mean | 0.39 | 9.93 | 0.73 | 0.69 |
| Populations | Sample Size | Major Allele Frequency | Na * | GD * | PIC * |
|---|---|---|---|---|---|
| G1 | 13 | 0.59 | 3.33 | 0.50 | 0.46 |
| G2 | 18 | 0.67 | 3.43 | 0.46 | 0.42 |
| G3 | 18 | 0.74 | 2.33 | 0.37 | 0.32 |
| G4 | 16 | 0.78 | 2.76 | 0.31 | 0.28 |
| G5 | 10 | 0.97 | 1.29 | 0.05 | 0.05 |
| G6 | 11 | 0.90 | 1.62 | 0.14 | 0.12 |
| G7 | 10 | 0.63 | 2.71 | 0.45 | 0.40 |
| Source | df | SS | MS | Est. Var. | Variation |
|---|---|---|---|---|---|
| Among Pops | 6 | 252.433 | 42.072 | 2.813 | 42% |
| Within Pops | 89 | 344.556 | 3.871 | 3.871 | 58% |
| Total | 95 | 596.990 | 6.684 | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Tan, J.; Huang, L.; Ye, Y.; Xu, Y. Assessing Genetic Diversity in Endangered Plant Orchidantha chinensis: Chloroplast Genome Assembly and Simple Sequence Repeat Marker-Based Evaluation. Int. J. Mol. Sci. 2024, 25, 11137. https://doi.org/10.3390/ijms252011137
Zhou Y, Tan J, Huang L, Ye Y, Xu Y. Assessing Genetic Diversity in Endangered Plant Orchidantha chinensis: Chloroplast Genome Assembly and Simple Sequence Repeat Marker-Based Evaluation. International Journal of Molecular Sciences. 2024; 25(20):11137. https://doi.org/10.3390/ijms252011137
Chicago/Turabian StyleZhou, Yiwei, Jianjun Tan, Lishan Huang, Yuanjun Ye, and Yechun Xu. 2024. "Assessing Genetic Diversity in Endangered Plant Orchidantha chinensis: Chloroplast Genome Assembly and Simple Sequence Repeat Marker-Based Evaluation" International Journal of Molecular Sciences 25, no. 20: 11137. https://doi.org/10.3390/ijms252011137
APA StyleZhou, Y., Tan, J., Huang, L., Ye, Y., & Xu, Y. (2024). Assessing Genetic Diversity in Endangered Plant Orchidantha chinensis: Chloroplast Genome Assembly and Simple Sequence Repeat Marker-Based Evaluation. International Journal of Molecular Sciences, 25(20), 11137. https://doi.org/10.3390/ijms252011137