_Kim.png)
Pyrimidine Derivatives as Selective COX-2 Inhibitors with Anti-Inflammatory and Antioxidant Properties

 ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. In Vitro Cyclooxygenase Inhibition Assay
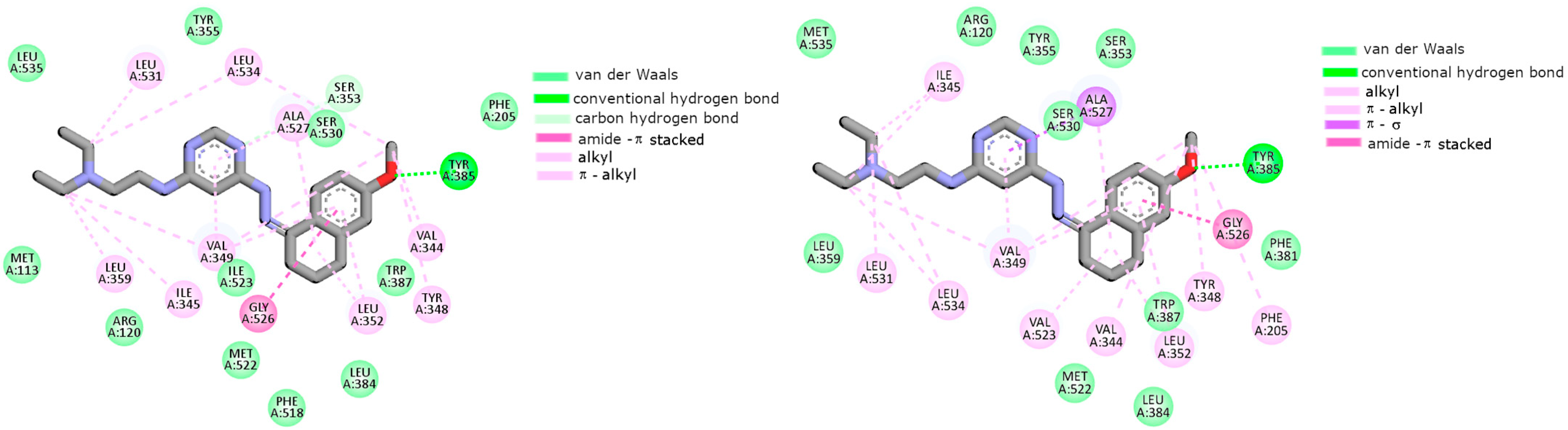
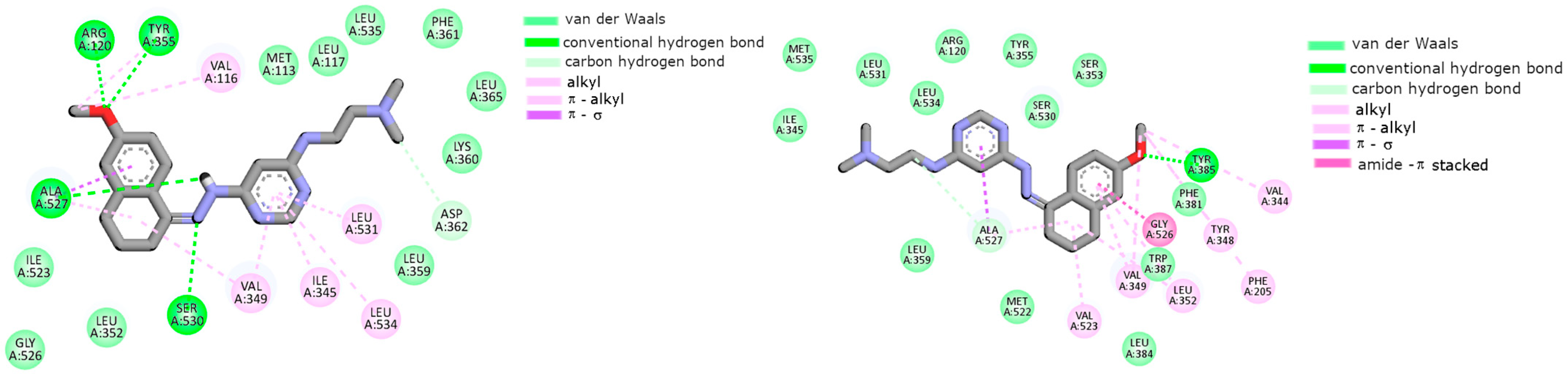
2.2. Cyclooxygenase Molecular Docking Study
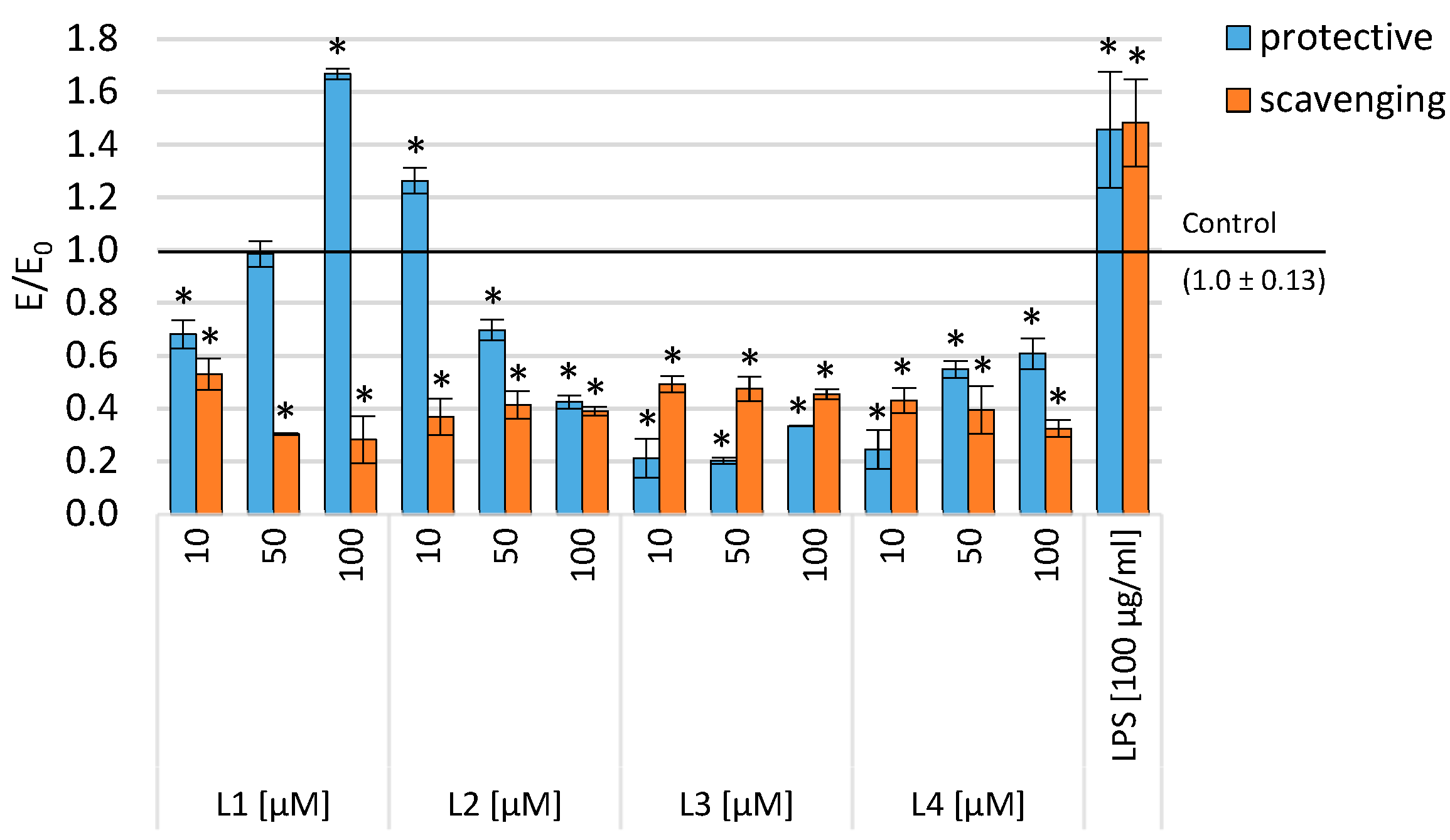
2.3. Evaluation of Viability in Inflammatory Model
2.4. Level of Intracellular ROS
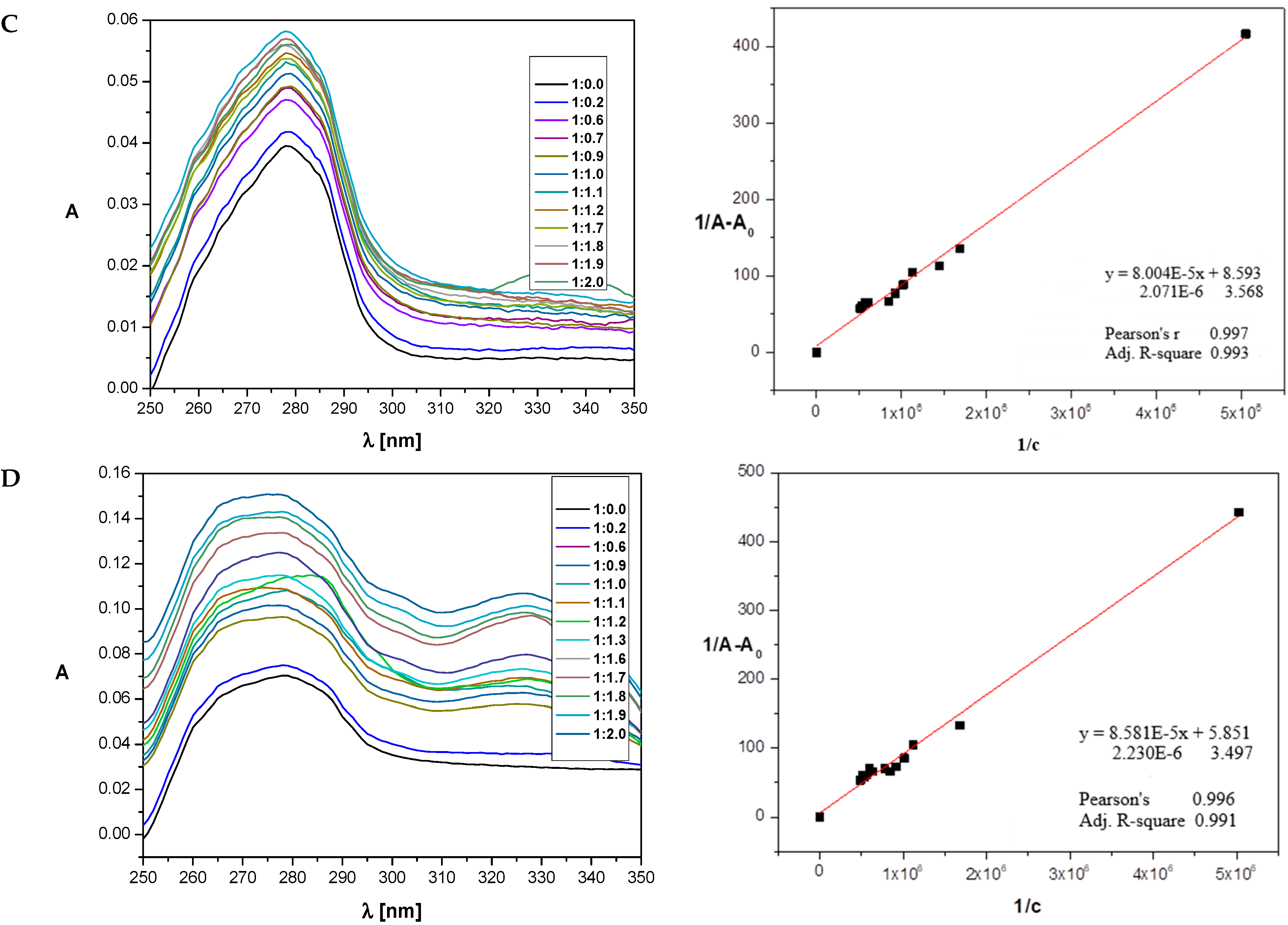
2.5. Determination of Binding Constants
3. Materials and Methods
3.1. Cyclooxygenase Inhibition Assay
3.2. Molecular Modeling
3.3. Biological Evaluation
3.3.1. Cell Line and Culture Conditions
3.3.2. Tested Compounds
3.3.3. Experimental Design
3.3.4. SRB Assay
3.3.5. DCF-DA Assay
3.3.6. Statistical Analysis
3.4. Spectroscopic Studies
3.4.1. UV–Vis Spectroscopy
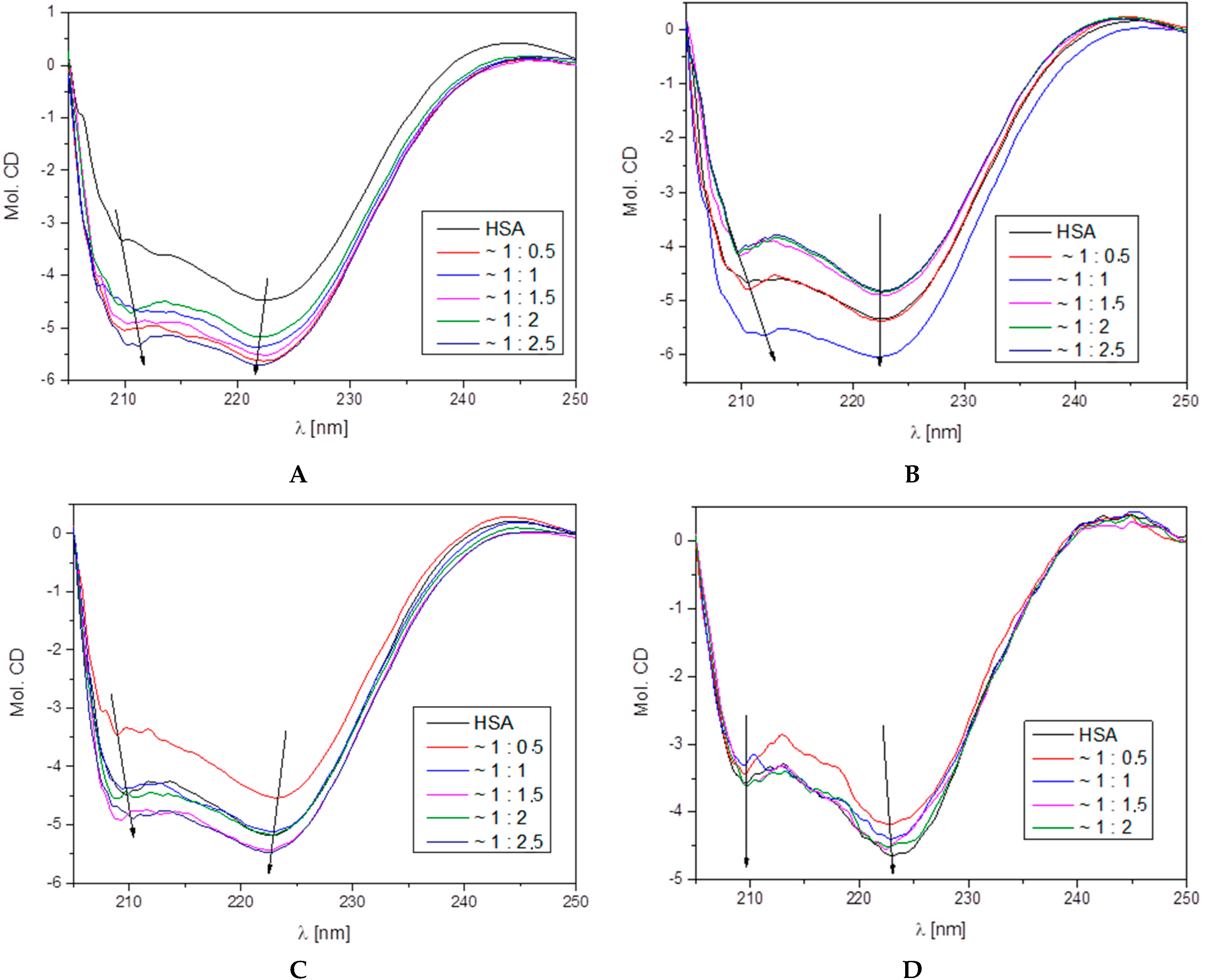
3.4.2. Circular Dichroism
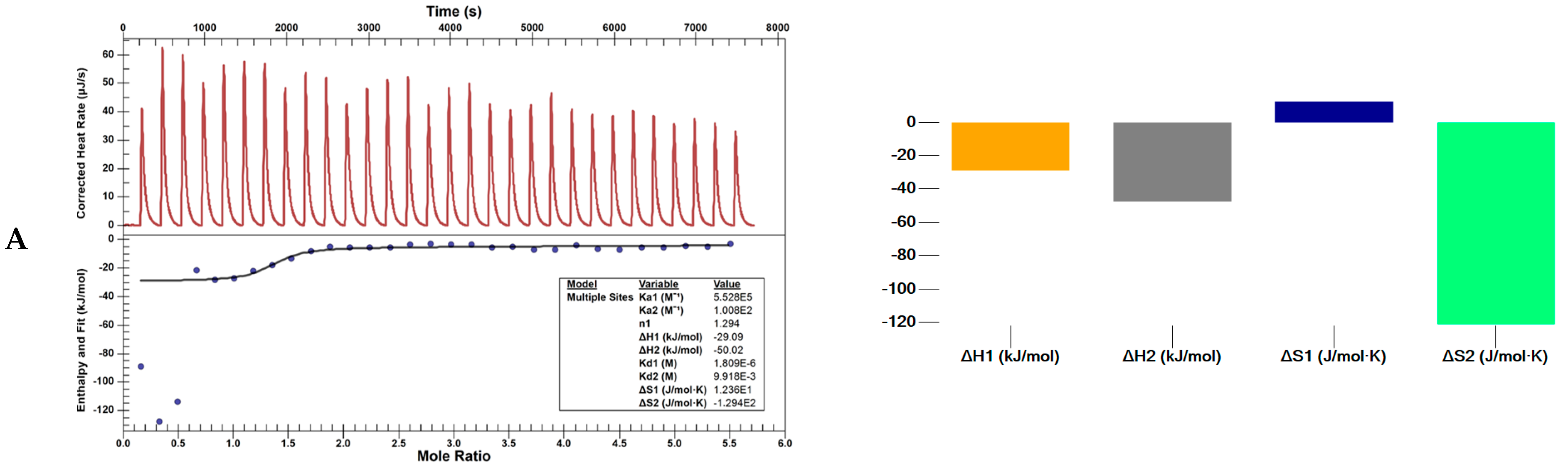
3.4.3. Nano ITC Calorimeter
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global Burden of Colorectal Cancer in 2020 and 2040: Incidence and Mortality Estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Didkowska, J.; Barańska, K.; Miklewska, M.J.; Wojciechowska, U. Cancer Incidence and Mortality in Poland in 2023. Nowotwory J. Oncol. 2024, 74, 75–93. [Google Scholar] [CrossRef]
- Chen, W.S.; Wei, S.J.; Liu, M.J.; Hsiao, M.; Kou-Lin, J.; Yang, W.K. Tumor Invasiveness and Liver Metastasis of Colon Cancer Cells Correlated with Cyclooxygenase-2 (COX-2) Expression and Inhibited by a COX-2-Selective Inhibitor, Etodolac—PubMed. Int. J. Cancer 2001, 15, 894–899. [Google Scholar] [CrossRef]
- El-Husseiny, W.M.; El-Sayed, M.A.A.; Abdel-Aziz, N.I.; El-Azab, A.S.; Asiri, Y.A.; Abdel-Aziz, A.A.M. Structural Alterations Based on Naproxen Scaffold: Synthesis, Evaluation of Antitumor Activity and COX-2 Inhibition, and Molecular Docking. Eur. J. Med. Chem. 2018, 158, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655. [Google Scholar]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Charlier, C.; Michaux, C. Dual Inhibition of Cyclooxygenase-2 (COX-2) and 5-Lipoxygenase (5-LOX) as a New Strategy to Provide Safer Non-Steroidal Anti-Inflammatory Drugs. Eur. J. Med. Chem. 2003, 38, 645–659. [Google Scholar] [CrossRef]
- Tazawa, R.; Xu, X.M.; Wu, K.K.; Wang, L.H. Characterization of the Genomic Structure, Chromosomal Location and Promoter of Human Prostaglandin H Synthase-2 Gene. Biochem. Biophys. Res. Commun. 1994, 203, 190–199. [Google Scholar] [CrossRef]
- Miller, S.B. Prostaglandins in Health and Disease: An Overview. Semin. Arthritis Rheum. 2006, 36, 37–49. [Google Scholar] [CrossRef]
- Schneider, C.; Pozzi, A. Cyclooxygenases and Lipoxygenases in Cancer. Cancer Metastasis Rev. 2011, 30, 277. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Chaki, R.; Mandal, V.; Mandal, S.C. COX-2 as a Target for Cancer Chemotherapy. Pharmacol. Rep. 2010, 62, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, I.; Goh, K.L. Chemoprevention of Colorectal Cancer with Nonsteroidal Anti-Inflammatory Drugs. Chin. J. Dig. Dis. 2006, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Bangali, H.; Hammoud, A.; Mustafa, Y.F.; Al-Hetty, H.R.A.K.; Alkhafaji, A.T.; Deorari, M.M.; Al-Taee, M.M.; Zabibah, R.S.; Alsalamy, A. COX 2-Inhibitors; a Thorough and Updated Survey into Combinational Therapies in Cancers. Med. Oncol. 2024, 41, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, M.; Wang, L.; Yu, S. Combined Chemotherapy with Cyclooxygenase-2 (COX-2) Inhibitors in Treating Human Cancers: Recent Advancement. Biomed. Pharmacother. 2020, 129, 110389. [Google Scholar] [CrossRef]
- Keche, A.P.; Hatnapure, G.D.; Tale, R.H.; Rodge, A.H.; Birajdar, S.S.; Kamble, V.M. A Novel Pyrimidine Derivatives with Aryl Urea, Thiourea and Sulfonamide Moieties: Synthesis, Anti-Inflammatory and Antimicrobial Evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 3445–3448. [Google Scholar] [CrossRef]
- Kumar, N.; Chauhan, A.; Drabu, S. Synthesis of Cyanopyridine and Pyrimidine Analogues as New Anti-Inflammatory and Antimicrobial Agents. Biomed. Pharmacother. 2011, 65, 375–380. [Google Scholar] [CrossRef]
- Giles, D.; Roopa, K.; Sheeba, F.R.; Gurubasavarajaswamy, P.M.; Divakar, G.; Vidhya, T. Synthesis Pharmacological Evaluation and Docking Studies of Pyrimidine Derivatives. Eur. J. Med. Chem. 2012, 58, 478–484. [Google Scholar] [CrossRef]
- Stolarczyk, M.; Wolska, A.; Mikołajczyk, A.; Bryndal, I.; Cieplik, J.; Lis, T.; Matera-Witkiewicz, A. A New Pyrimidine Schiff Base with Selective Activities against Enterococcus Faecalis and Gastric Adenocarcinoma. Molecules 2021, 26, 2296. [Google Scholar] [CrossRef]
- Bassyouni, F.; Tarek, M.; Salama, A.; Ibrahim, B.; Dine, S.S.E.; Yassin, N.; Hassanein, A.; Moharam, M.; Abdel-Rehim, M. Promising Antidiabetic and Antimicrobial Agents Based on Fused Pyrimidine Derivatives: Molecular Modeling and Biological Evaluation with Histopathological Effect. Molecules 2021, 26, 2370. [Google Scholar] [CrossRef]
- Fu, Y.; Leng, C.; Fan, Y.; Ma, X.; Li, X.; Wang, X.; Guo, Z.; Wang, X.; Shang, R. In Vitro and In Vivo Activity of 14-O-[(4,6-Diamino-Pyrimidine-2-Yl) Thioacetyl] Mutilin against Methicillin-Resistant Staphylococcus Aureus. Molecules 2021, 26, 3277. [Google Scholar] [CrossRef] [PubMed]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Enaminonitrile in Heterocyclic Synthesis: Synthesis and Antimicrobial Evaluation of Some New Pyrazole, Isoxazole and Pyrimidine Derivatives Incorporating a Benzothiazole Moiety. Eur. J. Med. Chem. 2009, 44, 4813–4818. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014, 2014, 202784. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Nakajima, T.; Ueda, Y.; Fujita, H.; Kawakami, H. Novel Piperidinylpyrimidine Derivatives as Inhibitors of HIV-1 LTR Activation. Bioorg. Med. Chem. 2008, 16, 9804–9816. [Google Scholar] [CrossRef] [PubMed]
- Rathwa, S.; Bhoi, M.; Borad, M.; Patel, K.; Rajani, D.; Rajani, S.; Patel, H. Microwave Assisted Synthesis, Biological Characterization and Docking Studies of Pyrimidine Derivatives. Curr. Microw. Chem. 2016, 3, 178–186. [Google Scholar] [CrossRef]
- Phuangsawai, O.; Beswick, P.; Ratanabunyong, S.; Tabtimmai, L.; Suphakun, P.; Obounchoey, P.; Srisook, P.; Horata, N.; Chuckowree, I.; Hannongbua, S.; et al. Evaluation of the Anti-Malarial Activity and Cytotoxicity of 2,4-Diamino-Pyrimidine-Based Kinase Inhibitors. Eur. J. Med. Chem. 2016, 124, 896–905. [Google Scholar] [CrossRef]
- Xie, F.; Zhao, H.; Zhao, L.; Lou, L.; Hu, Y. Synthesis and Biological Evaluation of Novel 2,4,5-Substituted Pyrimidine Derivatives for Anticancer Activity. Bioorg. Med. Chem. Lett. 2009, 19, 275–278. [Google Scholar] [CrossRef]
- Simonetti, G.; Boga, C.; Durante, J.; Micheletti, G.; Telese, D.; Caruana, P.; Di Rorà, A.G.L.; Mantellini, F.; Bruno, S.; Martinelli, G.; et al. Synthesis of Novel Tryptamine Derivatives and Their Biological Activity as Antitumor Agents. Molecules 2021, 26, 683. [Google Scholar] [CrossRef]
- Madia, V.N.; Nicolai, A.; Messore, A.; De Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; De Vita, D.; Scipione, L.; Artico, M.; et al. Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents. Molecules 2021, 26, 771. [Google Scholar] [CrossRef]
- Tylińska, B.; Wiatrak, B.; Czyżnikowska, Ż.; Cieśla-Niechwiadowicz, A.; Gębarowska, E.; Janicka-Kłos, A. Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 3825. [Google Scholar] [CrossRef]
- Mohamed, T.; Yeung, J.C.K.; Vasefi, M.S.; Beazely, M.A.; Rao, P.P.N. Development and Evaluation of Multifunctional Agents for Potential Treatment of Alzheimer’s Disease: Application to a Pyrimidine-2,4-Diamine Template. Bioorg. Med. Chem. Lett. 2012, 22, 4707–4712. [Google Scholar] [CrossRef] [PubMed]
- Rehman, T.U.; Khan, I.U.; Ashraf, M.; Tarazi, H.; Riaz, S.; Yar, M. An Efficient Synthesis of Bi-Aryl Pyrimidine Heterocycles: Potential New Drug Candidates to Treat Alzheimer’s Disease. Arch. Der Pharm. 2017, 350, 1600304. [Google Scholar] [CrossRef] [PubMed]
- Rivkin, A.; Ahearn, S.P.; Chichetti, S.M.; Kim, Y.R.; Li, C.; Rosenau, A.; Kattar, S.D.; Jung, J.; Shah, S.; Hughes, B.L.; et al. Piperazinyl Pyrimidine Derivatives as Potent Gamma-Secretase Modulators. Bioorg. Med. Chem. Lett. 2010, 20, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Prajapati, S.K.; Majumdar, S.; Raza, K.; Gabr, M.T.; Kumar, S.; Pal, K.; Rashid, H.; Kumar, S.; Krishnamurthy, S.; et al. Discovery of New Phenyl Sulfonyl-Pyrimidine Carboxylate Derivatives as the Potential Multi-Target Drugs with Effective Anti-Alzheimer’s Action: Design, Synthesis, Crystal Structure and In-Vitro Biological Evaluation. Eur. J. Med. Chem. 2021, 215, 113224. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.E.; Awadallah, F.M.; Ibrahin, N.A.; Said, E.G.; Kamel, G.M. New Quinazolinone–Pyrimidine Hybrids: Synthesis, Anti-Inflammatory, and Ulcerogenicity Studies. Eur. J. Med. Chem. 2012, 53, 141–149. [Google Scholar] [CrossRef]
- Nofal, Z.M.; Fahmy, H.H.; Zarea, E.S.; El-Eraky, W. Synthesis of New Pyrimidine Derivatives with Evaluation of Their Anti-Inflammatory and Analgesic Activities. Acta Pol. Pharm. 2011, 68, 507–517. [Google Scholar]
- Gupta, J.K.; Sharma, P.K.; Dudhe, R.; Chaudhary, A.; Singh, A.; Verma, P.K.; Mondal, S.C.; Yadav, R.K.; Kashyap, S. Analgesic Study of Novel Pyrimidine Derivatives Linked with Coumarin Moiety. Med. Chem. Res. 2012, 21, 1625–1632. [Google Scholar] [CrossRef]
- Abouzid, K.; Bekhit, S.A. Novel Anti-Inflammatory Agents Based on Pyridazinone Scaffold; Design, Synthesis and In Vivo Activity. Bioorg. Med. Chem. 2008, 16, 5547–5556. [Google Scholar] [CrossRef]
- Mogilski, S.; Kubacka, M.; Redzicka, A.; Kazek, G.; Dudek, M.; Malinka, W.; Filipek, B. Antinociceptive, Anti-Inflammatory and Smooth Muscle Relaxant Activities of the Pyrrolo [3,4-d]Pyridazinone Derivatives: Possible Mechanisms of Action. Pharmacol. Biochem. Behav. 2015, 133, 99–110. [Google Scholar] [CrossRef]
- Singh, J.; Saini, V.; Kumar, A.; Bansal, R. Synthesis, Molecular Docking and Biological Evaluation of Some Newer 2-Substituted-4-(Benzo[d][16,18]Dioxol-5-Yl)-6-Phenylpyridazin-3(2H)-Ones as Potential Anti-Inflammatory and Analgesic Agents. Bioorg. Chem. 2017, 71, 201–210. [Google Scholar] [CrossRef]
- Boukharsa, Y.; Lakhlili, W.; El harti, J.; Meddah, B.; Tiendrebeogo, R.Y.; Taoufik, J.; El Abbes Faouzi, M.; Ibrahimi, A.; Ansar, M. Synthesis, Anti-Inflammatory Evaluation In Vivo and Docking Studies of Some New 5-(Benzo[b]Furan-2-Ylmethyl)-6-Methyl-Pyridazin-3(2H)-One Derivatives. J. Mol. Struct. 2018, 1153, 119–127. [Google Scholar] [CrossRef]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 Inhibition Activities and Molecular Docking Study of Isothiazolopyridine Derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site via a Two-Water-Mediated h-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Meade, E.; Smith, W.; Chemistry, D.D.-J. Differential Inhibition of Prostaglandin Endoperoxide Synthase (Cyclooxygenase) Isozymes by Aspirin and Other Non-Steroidal Anti-Inflammatory Drugs. J. Biol. Chem. 1993, 268, 6610–6614. [Google Scholar] [CrossRef]
- Llorens, O.; Perez, J.; Palomer, A.; Mauleon, D. Structural Basis of the Dynamic Mechanism of Ligand Binding to Cyclooxygenase. Bioorg. Med. Chem. Lett. 1999, 9, 2779–2784. [Google Scholar] [CrossRef]
- Balon, K.; Wiatrak, B. PC12 and THP-1 Cell Lines as Neuronal and Microglia Model in Neurobiological Research. Appl. Sci. 2021, 11, 3729. [Google Scholar] [CrossRef]
- Feng, L.; Yasmeen, R.; Schoene, N.W.; Lei, K.Y.; Wang, T.T.Y. Resveratrol Differentially Modulates Immune Responses in Human THP-1 Monocytes and Macrophages. Nutr. Res. 2019, 72, 57–69. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. THP-1 Cells as a Model for Human Monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, Oxidative Stress and Inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Seal, B.K.; Sil, H.; Mukherjee, D.C. Independent Determination of Equilibrium Constant and Molar Extinction Coefficient of Molecular Complexes from Spectrophotometric Data by a Graphical Method. Spectrochim. Acta A 1982, 38, 289–292. [Google Scholar] [CrossRef]
- Yao, H.; Wynendaele, E.; Xu, X.; Kosgei, A.; De Spiegeleer, B. Circular Dichroism in Functional Quality Evaluation of Medicines. J. Pharm. Biomed. Anal. 2018, 147, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Maciążek-Jurczyk, M.; Morak-Młodawska, B.; Jeleń, M.; Kopeć, W.; Szkudlarek, A.; Owczarzy, A.; Kulig, K.; Rogóż, W.; Pożycka, J. The Influence of Oxidative Stress on Serum Albumin Structure as a Carrier of Selected Diazaphenothiazine with Potential Anticancer Activity. Pharmaceuticals 2021, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- Saboury, A.A. A Review on the Ligand Binding Studies by Isothermal Titration Calorimetry. J. Iran. Chem. Soc. 2006, 3, 1–21. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM Ab Initio Level. J. Mol. Struct. Theochem. 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef]
- Redzicka, A.; Wiatrak, B.; Jęśkowiak-Kossakowska, I.; Kochel, A.; Płaczek, R.; Czyżnikowska, Ż. Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-Aryl/Alkyl-2-[2-Hydroxy-3-(4-Substituted-1-Piperazinyl)Propyl]Pyrrolo [3,4-c]Pyrrole-1,3(2H,5H)-Diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX. Pharmaceuticals 2023, 16, 804. [Google Scholar] [CrossRef] [PubMed]
- Peregrym, K.; Szczukowski, Ł.; Wiatrak, B.; Potyrak, K.; Czyżnikowska, Ż.; Świątek, P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]Pyridazinone as Promising Cyclooxygenase Inhibitors. Int. J. Mol. Sci. 2021, 22, 9130. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cyclooxygenase Inhibition Assay IC50 [µM] | COX Selectivity Ratio IC50(COX-2)/IC50(COX-1) | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| L1 | 105.8 ± 22.39 | 74.6 ± 3.03 ^ | 0.70 |
| L2 | 91.0 ± 3.40 * | 76.8 ± 1.20 ^ | 0.84 |
| L3 | N/C | N/C | - |
| L4 | N/C | N/C | - |
| Piroxicam | 87.4 ± 5.10 | 80.1 ± 1.54 | 0.92 |
| Meloxicam | 128.8 ± 5.78 | 76.4 ± 7.91 | 0.59 |
| Ligand | L1 | L1 | L3 | L4 |
|---|---|---|---|---|
| Ka [dm3/mol] | 9.92 × 104 | 4.54 × 105 | 1.07 × 105 | 6.82 × 104 |
| ∆G [J/mol] | −2.85 × 104 | −3.23 × 105 | −2.87 × 104 | −2.76 × 104 |
| HSA–L1 Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
| 1:0.0 | 69.4 | 5.8 | 8.5 | 16.3 |
| 1:0.5 | 66.7 | 10.1 | 8.2 | 15.0 |
| 1:1.0 | 65.8 | 9.0 | 8.5 | 16.7 |
| 1:1.5 | 66.2 | 12.2 | 8.0 | 13.6 |
| 1:2.0 | 65.6 | 10.7 | 8.3 | 15.4 |
| 1:2.5 | 65.5 | 11.9 | 8.1 | 14.5 |
| HSA–L2 Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
| 1:0.0 | 68.2 | 8.6 | 8.2 | 15.0 |
| 1:0.5 | 68.6 | 7,3 | 8.3 | 15.8 |
| 1:1.0 | 67.7 | 6.2 | 8.6 | 17.5 |
| 1:1.5 | 68.2 | 11.7 | 7.7 | 12.4 |
| 1:2.0 | 67.1 | 7.3 | 8.5 | 17.1 |
| 1:2.5 | 66.5 | 7.7 | 8.6 | 17.2 |
| HSA–L3 Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
| 1:0.0 | 69.6 | 5.5 | 8.4 | 16.5 |
| 1:0.5 | 64.6 | 6.9 | 9.0 | 19.5 |
| 1:1.0 | 64.9 | 9.8 | 8.5 | 16.8 |
| 1:1.5 | 65.0 | 10.0 | 8.4 | 16.6 |
| 1:2.0 | 67.1 | 9.4 | 8.7 | 14.8 |
| 1:2.5 | 66.5 | 9.6 | 8.6 | 14.3 |
| HSA–L4 Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
| 1:0.0 | 65.4 | 3.5 | 8.9 | 22.2 |
| 1:0.5 | 55.5 | 7.5 | 9.7 | 27.3 |
| 1:1.0 | 66.2 | 5.2 | 9,1 | 19.5 |
| 1:1.5 | 60.2 | 9.0 | 8.9 | 21.9 |
| 1:2.0 | 64.2 | 6.4 | 9.1 | 20.3 |
| Ka (M−1) | n | ΔH (kJ/mol) | Kd1 (M) | ΔS (J/mol·K) | ΔG (J/mol) | ||
|---|---|---|---|---|---|---|---|
| L1 | 1 | 5.528 × 105 | 1.29 | −29.1 | 1.809 × 10−6 | 1.236 × 101 | −3.278 × 104 |
| 2 | 1.008 × 102 | 3.61 | −50.1 | 9.918 × 10−3 | −1.294 × 102 | −1.154 × 104 | |
| L4 | 1 | 5.724 × 108 | 2.56 | −41.9 | 1.747 × 10−9 | 2.715 × 101 | −4.999 × 104 |
| 2 | 2.093 × 108 | 1.06 | 99.1 | 4.779 × 10−9 | 4.917 × 102 | −4.742 × 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tylińska, B.; Janicka-Kłos, A.; Gębarowski, T.; Nowotarska, P.; Plińska, S.; Wiatrak, B. Pyrimidine Derivatives as Selective COX-2 Inhibitors with Anti-Inflammatory and Antioxidant Properties. Int. J. Mol. Sci. 2024, 25, 11011. https://doi.org/10.3390/ijms252011011
Tylińska B, Janicka-Kłos A, Gębarowski T, Nowotarska P, Plińska S, Wiatrak B. Pyrimidine Derivatives as Selective COX-2 Inhibitors with Anti-Inflammatory and Antioxidant Properties. International Journal of Molecular Sciences. 2024; 25(20):11011. https://doi.org/10.3390/ijms252011011
Chicago/Turabian StyleTylińska, Beata, Anna Janicka-Kłos, Tomasz Gębarowski, Paulina Nowotarska, Stanisława Plińska, and Benita Wiatrak. 2024. "Pyrimidine Derivatives as Selective COX-2 Inhibitors with Anti-Inflammatory and Antioxidant Properties" International Journal of Molecular Sciences 25, no. 20: 11011. https://doi.org/10.3390/ijms252011011
APA StyleTylińska, B., Janicka-Kłos, A., Gębarowski, T., Nowotarska, P., Plińska, S., & Wiatrak, B. (2024). Pyrimidine Derivatives as Selective COX-2 Inhibitors with Anti-Inflammatory and Antioxidant Properties. International Journal of Molecular Sciences, 25(20), 11011. https://doi.org/10.3390/ijms252011011