
Balancing Tumor Immunotherapy and Immune-Related Adverse Events: Unveiling the Key Regulators

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tumor Immunotherapy
2.1. Origin of Tumor Immunotherapy
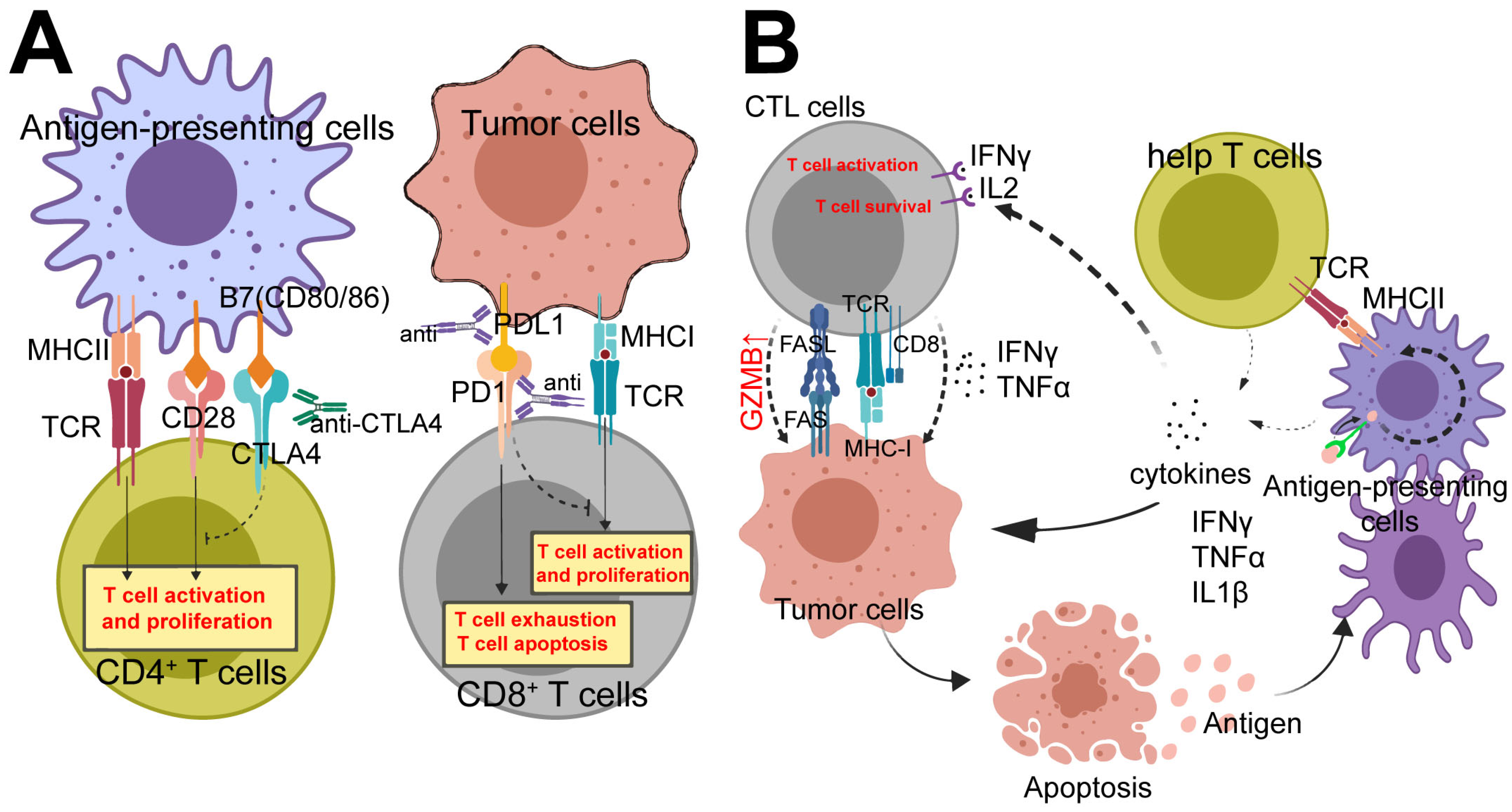
2.2. Current Strategies for Tumor Immunotherapy
3. Immunotherapy Target Cells
3.1. CD8+ T Cells
3.2. CD4+ T Cells
3.3. M1 Type Macrophages
3.4. NK Cells
3.5. Other Cells
3.5.1. DCs
3.5.2. γδT Cells
3.5.3. B Cells
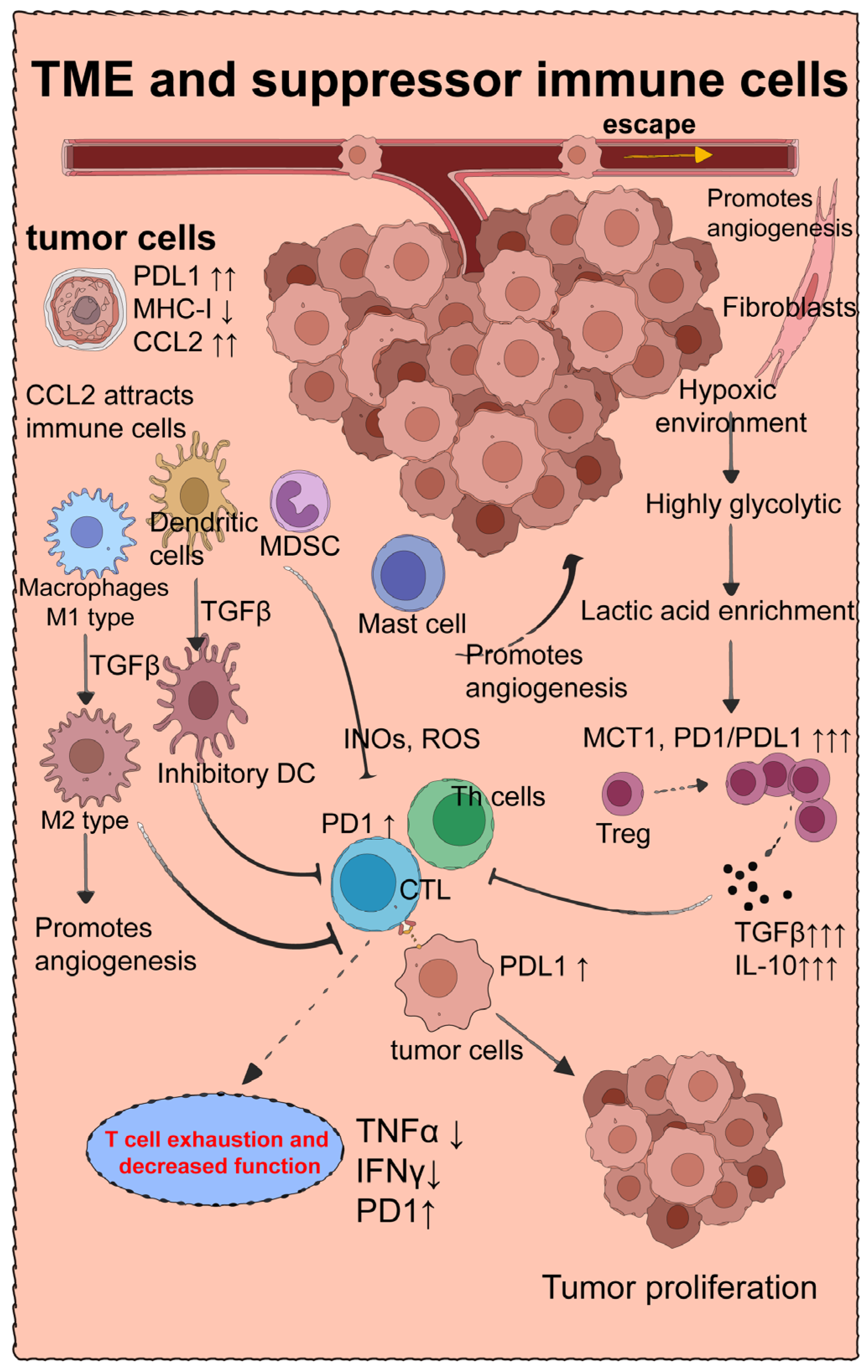
4. Suppressive Immune Cells That Downregulate Tumor Immune Efficacy
4.1. Tumor Microenvironment and Treg Cells
4.2. M2-Type Macrophages
4.3. Myeloid-Derived Suppressor Cells
4.4. Mast Cells
5. Tumor Immunotherapy and irAEs
5.1. The Epidemiology of irAEs
5.2. Characteristics of Intestine-Related irAES
6. Strategies to Mitigate Adverse Effects of Tumor Immunotherapy
6.1. Conventional Remission Therapy for irAEs
6.2. Infliximab
6.3. Targets for Mitigating irAEs and Enhancing Tumor Immunotherapy
7. Targeting Epithelial Cells May Represent a Crucial Approach for Modulating the Delicate Equilibrium between Tumor Immunotherapy and irAEs
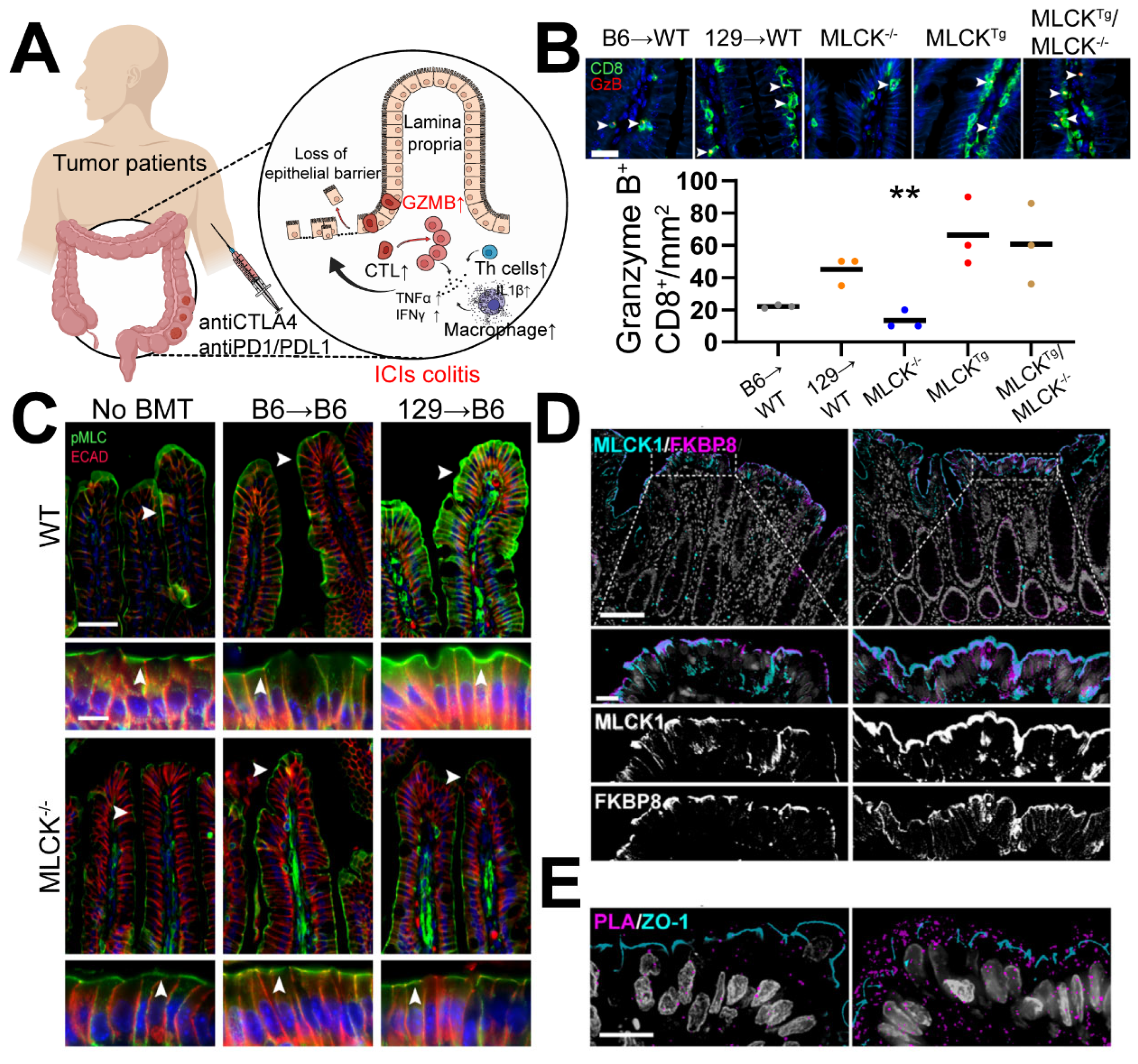
7.1. Immune Dysregulation Stands as a Significant Contributing Factor to ICI Colitis
7.2. Similarities between ICIs-Related IBD and GVHD-Related IBD
7.3. MLCK Targeting for GVHD-Related IBD and ICIs-Related IBD: Promising but Imperfect
7.4. Promising Regimen for Remission of ICIs Colitis
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2018, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Edward, B.G.; Naiyer, A.R.; Rina, H.; Natasha, L.; Ani, S.B.; Joseph, P.E.; Amita, P.; Charu, A.; Matthew, G.; Leora, H.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Robert, J.M.; Nizar, M.T.; David, F.M.; Osvaldo, A.F.; Bohuslav, M.; Toni, K.C.; Elizabeth, R.P.; Philippe, B.; Camillo, P.; Saby, G.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Suzanne, L.T.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 372, 2443–2454. [Google Scholar] [CrossRef]
- Alice, T.; Gilles, Q.; Laurent, M.; Elisa, F.-B.; François-Xavier, D.; Emilie, R.; Caroline, R.; Yohann, L.; Olivier, L.; Bertille, B.; et al. Safety and Efficacy of Immune Checkpoint Inhibitors in Patients with Cancer and Preexisting Autoimmune Disease: A Nationwide, Multicenter Cohort Study. Arthritis Rheumatol. 2019, 71, 2100–2111. [Google Scholar] [CrossRef]
- Nan-Chun, W.; Yin-Hsun, F.; Yu-Hsuan, K.; Wei-Yu, C.; Hong-Chang, W.; Chien-Tai, H.; Wen-Ching, W.; Nai-Wen, K.; Jhih-Yuan, S.; Zhih-Cherng, C.; et al. Clinical Features and Outcomes of Immune Checkpoint Inhibitor-Associated Cardiovascular Toxicities. Acta Cardiol. Sin. 2022, 38, 39–46. [Google Scholar] [CrossRef]
- Rongmao, G.; Fuxun, Y.; Chen, Y.; Zhao, Z.; Mingzong, L.; Chunlin, X.; Huan, H.; Xiaoxiu, L.; Jiajia, L.; Rongan, L. A case report and literature review of immune checkpoint inhibitor-associated pneumonia caused by penpulimab. Front. Immunol. 2023, 14, 1114994. [Google Scholar] [CrossRef]
- Virginia, H.S.; Julius, C.H.; Ibrahim, H.; Vineet, K.R.; Chia-Yun, W.; Leyre, Z.; Azin, G.; Nicole, R.L.; Osama, A.-S.; Kenneth, L.K.; et al. Enhancing Precision in Detecting Severe Immune-Related Adverse Events: Comparative Analysis of Large Language Models and International Classification of Disease Codes in Patient Records. J. Clin. Oncol. 2024, JCO2400326. [Google Scholar] [CrossRef]
- Siwicki, M.; Gort-Freitas, N.A.; Messemaker, M.; Bill, R.; Gungabeesoon, J.; Engblom, C.; Zilionis, R.; Garris, C.; Gerhard, G.M.; Kohl, A.; et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci. Immunol. 2021, 6, eabi7083. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, E.; Minute, L.; Otano, I.; Alvarez, M.; Ochoa, M.C.; Belsue, V.; de Andrea, C.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Marquez-Rodas, I.; et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 2019, 569, 428–432. [Google Scholar] [CrossRef]
- Postow, M.A.; Longo, D.L.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Coley, W.B. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- Brunet, J.; Denizot, F.; Luciani, M.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Brahmer, J.; Tykodi, S.; Chow, L.; Hwu, W.; Topalian, S.; Hwu, P.; Drake, C.; Camacho, L.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Du, Y.; Liu, Y.; Wang, D.; Bai, H.; Wang, Z.; He, X.; Zhang, P.; Tian, J.; Wang, J. Peptidic microarchitecture-trapped tumor vaccine combined with immune checkpoint inhibitor or PI3Kγ inhibitor can enhance immunogenicity and eradicate tumors. J. Immunother. Cancer 2022, 10, e003564. [Google Scholar] [CrossRef]
- Sakemura, R.; Hefazi, M.; Siegler, E.; Cox, M.; Larson, D.; Hansen, M.; Manriquez Roman, C.; Schick, K.; Can, I.; Tapper, E.; et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022, 139, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Basar, R.; Wang, G.; Liu, E.; Moyes, J.; Li, L.; Kerbauy, L.; Uprety, N.; Fathi, M.; Rezvan, A.; et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med. 2022, 28, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Q.; Qiu, Z.; Kang, Y.; Liu, J.; Ning, S.; Yin, Y.; Pang, D.; Xu, S. Noncoding RNAs: The shot callers in tumor immune escape. Signal Transduct. Target. Ther. 2020, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magaña, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef]
- McLane, L.; Abdel-Hakeem, M.; Wherry, E. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Daassi, D.; Mahoney, K.; Freeman, G. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef]
- Borst, L.; van der Burg, S.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Kokka, F.; Bryant, A.; Olaitan, A.; Brockbank, E.; Powell, M.; Oram, D. Hysterectomy with radiotherapy or chemotherapy or both for women with locally advanced cervical cancer. Cochrane Database Syst. Rev. 2022, 8, CD010260. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.; Kuhns, M.; Egen, J.; Allison, J. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Kuzel, T.; Zhang, Y.; Zhang, B. The Discovery of Biomarkers in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 484–497. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Matias, E.V. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Toni, K.C.; Thomas, P.; Laurence, A.; Mauricio, B.; Cezary, S.; Bogdan, Z.; Eduardo, Y.R.; Marco, M.; Alberto, S.Z.; Luis, E.F.; et al. Cabozantinib plus Nivolumab and Ipilimumab in Renal-Cell Carcinoma. N. Engl. J. Med. 2023, 388, 1767–1778. [Google Scholar] [CrossRef]
- Melissa, B.; Marcello, T.; Giuseppe, L.B. Nivolumab plus Ipilimumab in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 382, 2020–2031. [Google Scholar] [CrossRef]
- James, L.; Vanna, C.-S.; Rene, G.; Jean-Jacques, G.; Piotr, R.; Christopher, D.L.; Lance, C.C.; Dirk, S.; John, W.; Reinhard, D.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Perea, F.; Sánchez-Palencia, A.; Gómez-Morales, M.; Bernal, M.; Concha, Á.; García, M.; González-Ramírez, A.; Kerick, M.; Martin, J.; Garrido, F.; et al. HLA class I loss and PD-L1 expression in lung cancer: Impact on T-cell infiltration and immune escape. Oncotarget 2018, 9, 4120–4133. [Google Scholar] [CrossRef]
- Luoma, A.; Suo, S.; Williams, H.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.; Rahma, O.; Sullivan, R.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, N.H. Lymphotoxin and TNF: How it all began—A tribute to the travelers. Cytokine Growth Factor Rev. 2014, 25, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yu, L.; Dongge, X.; Dan, Z.; Yang, Y.; Yuqing, M.; Susu, H.; Qing, X.; Erguang, L. An engineered TNFR1-selective human lymphotoxin-alpha mutant delivered by an oncolytic adenovirus for tumor immunotherapy. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167122. [Google Scholar] [CrossRef]
- Cheroutre, H.; Husain, M. CD4 CTL: Living up to the challenge. Semin. Immunol. 2013, 25, 273–281. [Google Scholar] [CrossRef]
- Eilon, W.; Cuiying, X.; Ofer, F.; Joseph, L.; Dalia, R.; Varda, N.; Yael, B.; Dalia, G.; Ori, B.; Gideon, B.; et al. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7731–7736. [Google Scholar] [CrossRef]
- Ichiro, T.; Motomi, O.; Takeshi, E.; Mary Jean, S.; Suk Chul, B.; Toshihisa, K.; Yoshiaki, I.; Dan, R.L. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002, 111, 621–633. [Google Scholar] [CrossRef]
- Brettschneider, E.E.S.; Terabe, M. The Role of NKT Cells in Glioblastoma. Cells 2021, 10, 1641. [Google Scholar] [CrossRef]
- Bauché, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.; Blumenschein, W.; Ganal-Vonarburg, S.; Wilson, D.; McClanahan, T.; Malefyt, R.; et al. LAG3 Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1 Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity 2018, 49, 342–352.e5. [Google Scholar] [CrossRef]
- Han, S.; Zhuang, H.; Lee, P.Y.; Li, M.; Yang, L.; Nigrovic, P.A.; Reeves, W.H. Differential Responsiveness of Monocyte and Macrophage Subsets to Interferon. Arthritis Rheumatol. 2020, 72, 100–113. [Google Scholar] [CrossRef]
- Wang, C.; Cui, A.; Bukenya, M.; Aung, A.; Pradhan, D.; Whittaker, C.A.; Agarwal, Y.; Thomas, A.; Liang, S.; Amlashi, P.; et al. Reprogramming NK cells and macrophages via combined antibody and cytokine therapy primes tumors for elimination by checkpoint blockade. Cell Rep. 2021, 37, 110021. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Jung, I.; Moon, S.; Jeon, S.; Jeong, S.; Sonn, S.; Seo, S.; Lee, M.; Song, E.; Kweon, H.; et al. CD137 Signaling Regulates Acute Colitis via RALDH2-Expressing CD11bCD103 DCs. Cell Rep. 2020, 30, 4124–4136.e5. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Chiang, Y.-H.; Hsu, K.; Chuang, C.-K.; Kao, C.-W.; Chang, Y.-F.; Chang, M.-C.; Lim, K.-H.; Cheng, H.-I.; Hsu, Y.-N.; et al. Activated naïve γδ T cells accelerate deep molecular response to BCR-ABL inhibitors in patients with chronic myeloid leukemia. Blood Cancer J. 2021, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.-P. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 2017, 8, 764. [Google Scholar] [CrossRef]
- Cui, C.; Wang, J.; Fagerberg, E.; Chen, P.M.; Connolly, K.A.; Damo, M.; Cheung, J.F.; Mao, T.; Askari, A.S.; Chen, S.; et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell 2021, 184, 6101–6118.e13. [Google Scholar] [CrossRef]
- Kato, T.; Rossetti, R.A.M.; Lorenzi, N.P.C.; Yokochi, K.; Rosa, M.B.S.D.F.; Benevides, L.; Margarido, P.F.R.; Baracat, E.C.; Carvalho, J.P.; Villa, L.L.; et al. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 2018, 13, e0199034. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Kumagai, S. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.e9. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Lainé, A.; Labiad, O.; Hernandez-Vargas, H.; This, S.; Sanlaville, A.; Léon, S.; Dalle, S.; Sheppard, D.; Travis, M.A.; Paidassi, H.; et al. Regulatory T cells promote cancer immune-escape through integrin αvβ8-mediated TGF-β activation. Nat. Commun. 2021, 12, 6228. [Google Scholar] [CrossRef] [PubMed]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Rey-Giraud, F.; Hafner, M.; Ries, C.H. In Vitro Generation of Monocyte-Derived Macrophages under Serum-Free Conditions Improves Their Tumor Promoting Functions. PLoS ONE 2012, 7, e42656. [Google Scholar] [CrossRef]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Lu, L.; Deng, S.; Meng, J.; Wan, C.; Huang, J.; Sun, Y.; Hu, Y.; Wu, B.; Wu, G.; et al. USP7 targeting modulates anti-tumor immune response by reprogramming Tumor-associated Macrophages in Lung Cancer. Theranostics 2020, 10, 9332–9347. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. OncoImmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.; Almeida, G.; Souza, I.; Borges, D.; de Lima, W.; Prazeres, P.; Birbrair, A.; Arantes, R.; Mosser, D.; Goncalves, R. High-Density-Immune-Complex Regulatory Macrophages Promote Recovery of Experimental Colitis in Mice. Inflammation 2021, 44, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Lyon, C.J.; Fletcher, J.K.; Tang, W.; Wan, M.; Hu, T.Y. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses. Acta Pharm. Sin. B 2021, 11, 1493–1512. [Google Scholar] [CrossRef]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef]
- Giri, J.; Das, R.; Nylen, E.; Chinnadurai, R.; Galipeau, J. CCL2 and CXCL12 Derived from Mesenchymal Stromal Cells Cooperatively Polarize IL-10+ Tissue Macrophages to Mitigate Gut Injury. Cell Rep. 2020, 30, 1923–1934.e4. [Google Scholar] [CrossRef] [PubMed]
- Cédile, O.; Løbner, M.; Toft-Hansen, H.; Frank, I.; Wlodarczyk, A.; Irla, M.; Owens, T. Thymic CCL2 influences induction of T-cell tolerance. J. Autoimmun. 2014, 55, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Patel, R.S.; Cook, D.R.; Choi, M.Y.; Patil, A.; Liang, A.C.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 2021, 373, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Bawazeer, M.A.; Theoharides, T.C. IL-33 stimulates human mast cell release of CCL5 and CCL2 via MAPK and NF-κB, inhibited by methoxyluteolin. Eur. J. Pharmacol. 2019, 865, 172760. [Google Scholar] [CrossRef]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.-L.; Rovère, C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef]
- Fujita, Y.; Khateb, A.; Li, Y.; Tinoco, R.; Zhang, T.; Bar-Yoseph, H.; Tam, M.A.; Chowers, Y.; Sabo, E.; Gerassy-Vainberg, S.; et al. Regulation of S100A8 Stability by RNF5 in Intestinal Epithelial Cells Determines Intestinal Inflammation and Severity of Colitis. Cell Rep. 2018, 24, 3296–3311.e6. [Google Scholar] [CrossRef]
- Zheng, W.; Song, H.; Luo, Z.; Wu, H.; Chen, L.; Wang, Y.; Cui, H.; Zhang, Y.; Wang, B.; Li, W.; et al. Acetylcholine ameliorates colitis by promoting IL-10 secretion of monocytic myeloid-derived suppressor cells through the nAChR/ERK pathway. Proc. Natl. Acad. Sci. USA 2021, 118, e2017762118. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. Mast cells, angiogenesis, and tumour growth. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 2–8. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2019, 58, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Herlyn, M. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, J.; Xu, L.; Yang, H.; Liang, N.; Zhang, L.; Zhang, F.; Zhang, X. Safety and Efficacy of the Rechallenge of Immune Checkpoint Inhibitors After Immune-Related Adverse Events in Patients With Cancer: A Systemic Review and Meta-Analysis. Front. Immunol. 2021, 12, 730320. [Google Scholar] [CrossRef]
- Hountondji, L.; Faure, S.; Palassin, P.; Viel, P.; Dupuy, M.; Larrey, D.; Lamoureux, A.; Coustal, C.; Pureur, D.; Lesage, C.; et al. Time to use the right classification to predict the severity of checkpoint inhibitor-induced liver injury, as assessed for causality using the updated RUCAM. Aliment. Pharmacol. Ther. 2024, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Netchiporouk, E.; Gerstein, W.; Gniadecki, R.; Litvinov, I.V. Cutaneous Immune-Related Adverse Events (irAEs) to Immune Checkpoint Inhibitors: A Dermatology Perspective on Management. J. Cutan. Med. Surg. 2021, 25, 59–76. [Google Scholar] [CrossRef]
- Wang, D.; Ye, F.; Zhao, S.; Johnson, D. Incidence of immune checkpoint inhibitor-related colitis in solid tumor patients: A systematic review and meta-analysis. OncoImmunology 2017, 6, e1344805. [Google Scholar] [CrossRef]
- Wang, D.; Salem, J.; Cohen, J.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Shivaji, U.N.; Jeffery, L.; Gui, X.; Smith, S.C.L.; Ahmad, O.F.; Akbar, A.; Ghosh, S.; Iacucci, M. Immune checkpoint inhibitor-associated gastrointestinal and hepatic adverse events and their management. Ther. Adv. Gastroenterol. 2019, 12, 175628481988419. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Autoimmune diseases surface after cancer treatment. Science 2017, 358, 852. [Google Scholar] [CrossRef]
- de Filette, J.; Pen, J.; Decoster, L.; Vissers, T.; Bravenboer, B.; Van der Auwera, B.; Gorus, F.; Roep, B.; Aspeslagh, S.; Neyns, B.; et al. Immune checkpoint inhibitors and type 1 diabetes mellitus: A case report and systematic review. Eur. J. Endocrinol. 2019, 181, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Günther, C.; Kremer, A.E.; Zaiss, M.M.; Rothhammer, V.; Neurath, M.F. Gut immune cell trafficking: Inter-organ communication and immune-mediated inflammation. Nat. Rev. Gastroenterol. Hepatol. 2022, 20, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut microbiome, liver immunology, and liver diseases. Cell. Mol. Immunol. 2020, 18, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Soularue, E.; Marthey, L.; Carbonnel, F. Management of Patients with Immune Checkpoint Inhibitor-Induced Enterocolitis: A Systematic Review. Clin. Gastroenterol. Hepatol. 2020, 18, 1393–1403.e1. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, G.; Je, Y.; Bossé, D.; Awad, M.; Ott, P.; Moreira, R.; Schutz, F.; Bellmunt, J.; Sonpavde, G.; Hodi, F.; et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol. Res. 2017, 5, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Komaki, Y.; Komaki, F.; Yamada, A.; Micic, D.; Ido, A.; Sakuraba, A. Meta-Analysis of the Risk of Immune-Related Adverse Events with Anticytotoxic T-Lymphocyte-Associated Antigen 4 and Antiprogrammed Death 1 Therapies. Clin. Pharmacol. Ther. 2018, 103, 318–331. [Google Scholar] [CrossRef]
- Tandon, P.; Bourassa-Blanchette, S.; Bishay, K.; Parlow, S.; Laurie, S.; McCurdy, J. The Risk of Diarrhea and Colitis in Patients with Advanced Melanoma Undergoing Immune Checkpoint Inhibitor Therapy: A Systematic Review and Meta-Analysis. J. Immunother. 2018, 41, 101–108. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Zhou, Y.; Guo, X.; Ge, J.; Fu, J. Immune-related adverse events from combination immunotherapy in cancer patients: A comprehensive meta-analysis of randomized controlled trials. Int. Immunopharmacol. 2018, 63, 292–298. [Google Scholar] [CrossRef]
- Eggermont, A.; Chiarion-Sileni, V.; Grob, J.; Dummer, R.; Wolchok, J.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.; Richards, J.; et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef]
- Masciullo, V.; Mainenti, S.; Lorusso, D.; Margariti, P.A.; Scambia, G. LethalClostridium difficileColitis Associated with Paclitaxel and Carboplatin Chemotherapy in Ovarian Carcinoma: Case Report and Review of the Literature. Obstet. Gynecol. Int. 2010, 2010, 1–3. [Google Scholar] [CrossRef]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Nosaka, T.; Takahashi, K.; Ofuji, K.; Matsuda, H.; Ohtani, M.; Hiramatsu, K.; Imamura, Y.; Ishizuka, T.; Nakamoto, Y. A case of immune checkpoint inhibitor-related colitis with a distinctive endoscopic finding of colonic pseudolipomatosis. Clin. J. Gastroenterol. 2021, 14, 1431–1436. [Google Scholar] [CrossRef]
- Lankes, K.; Hundorfean, G.; Harrer, T.; Pommer, A.J.; Agaimy, A.; Angelovska, I.; Tajmir-Riahi, A.; Göhl, J.; Schuler, G.; Neurath, M.F.; et al. Anti-TNF-refractory colitis after checkpoint inhibitor therapy: Possible role of CMV-mediated immunopathogenesis. OncoImmunology 2016, 5, e1128611. [Google Scholar] [CrossRef]
- Iborra, M.; Beltrán, B.; Bastida, G.; Aguas, M.; Nos, P. Infliximab and adalimumab-induced psoriasis in Crohn’s disease: A paradoxical side effect. J. Crohn’s Colitis 2011, 5, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shah, M.; Bansal, N.; Manocha, D. Infliximab-induced aseptic meningitis. Am. J. Emerg. Med. 2014, 32, 1560.e3–1560.e4. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lin, W.; Liao, Q.; Wang, H.; Lin, C.; Tang, L.; Lian, W.; Chen, Z.; Li, K.; Xu, L.; et al. Calcium Channel Blocker Nifedipine Suppresses Colorectal Cancer Progression and Immune Escape by Preventing NFAT2 Nuclear Translocation. Cell Rep. 2020, 33, 108327. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e13. [Google Scholar] [CrossRef]
- Zhao, Q.; Duck, L.W.; Huang, F.; Alexander, K.L.; Maynard, C.L.; Mannon, P.J.; Elson, C.O. CD4+ T cell activation and concomitant mTOR metabolic inhibition can ablate microbiota-specific memory cells and prevent colitis. Sci. Immunol. 2020, 5, eabc6373. [Google Scholar] [CrossRef]
- Yao, S.; Zhao, Z.; Wang, W.; Liu, X. Bifidobacterium Longum: Protection against Inflammatory Bowel Disease. J. Immunol. Res. 2021, 2021, 8030297. [Google Scholar] [CrossRef]
- Rizvi, Z.A.; Dalal, R.; Sadhu, S.; Kumar, Y.; Kumar, S.; Gupta, S.K.; Tripathy, M.R.; Rathore, D.K.; Awasthi, A. High-salt diet mediates interplay between NK cells and gut microbiota to induce potent tumor immunity. Sci. Adv. 2021, 7, eabg5016. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Wang, M.; Ajibade, A.A.; Tan, P.; Fu, C.; Chen, L.; Zhu, M.; Hao, Z.-Z.; Chu, J.; Yu, X.; et al. Microbiota regulate innate immune signaling and protective immunity against cancer. Cell Host Microbe 2021, 29, 959–974.e7. [Google Scholar] [CrossRef]
- Hanash, A.M.; Dudakov, J.A.; Hua, G.; O’Connor, M.H.; Young, L.F.; Singer, N.V.; West, M.L.; Jenq, R.R.; Holland, A.M.; Kappel, L.W.; et al. Interleukin-22 Protects Intestinal Stem Cells from Immune-Mediated Tissue Damage and Regulates Sensitivity to Graft versus Host Disease. Immunity 2012, 37, 339–350. [Google Scholar] [CrossRef]
- Nalle, S.; Zuo, L.; Ong, M.; Singh, G.; Worthylake, A.; Choi, W.; Manresa, M.; Southworth, A.; Edelblum, K.; Baker, G.; et al. Graft-versus-host disease propagation depends on increased intestinal epithelial tight junction permeability. J. Clin. Investig. 2019, 129, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Kuo, W.-T.; Cao, F.; Chanez-Paredes, S.D.; Zeve, D.; Mannam, P.; Jean-François, L.; Day, A.; Vallen Graham, W.; Sweat, Y.Y.; et al. Tacrolimus-binding protein FKBP8 directs myosin light chain kinase-dependent barrier regulation and is a potential therapeutic target in Crohn’s disease. Gut 2022, 72, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.; Taylor, J. Diagnosis and Treatment of Myelodysplastic Syndromes: A Review. JAMA 2022, 328, 872–880. [Google Scholar] [CrossRef]
- Abbasi, J. Reducing Graft-vs-Host Disease in Blood Stem Cell Transplants. JAMA 2022, 327, 517. [Google Scholar] [CrossRef] [PubMed]
- Clayburgh, D.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Investig. 2005, 115, 2702–2715. [Google Scholar] [CrossRef]
- Shen, L.; Black, E.D.; Witkowski, E.D.; Lencer, W.I.; Guerriero, V.; Schneeberger, E.E.; Turner, J.R. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J. Cell Sci. 2006, 119, 2095–2106. [Google Scholar] [CrossRef]
- Somlyo, A.V.; Wang, H.; Choudhury, N.; Khromov, A.S.; Majesky, M.; Owens, G.K.; Somlyo, A.P. Myosin light chain kinase knockout. J. Muscle Res. Cell Motil. 2004, 25, 241–242. [Google Scholar] [CrossRef]
- He, W.; Peng, Y.; Zhang, W.; Lv, N.; Tang, J.; Chen, C.; Zhang, C.; Gao, S.; Chen, H.; Zhi, G.; et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 2008, 135, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted Epithelial Tight Junction Dysfunction Causes Immune Activation and Contributes to Development of Experimental Colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Graham, W.V.; He, W.; Marchiando, A.M.; Zha, J.; Singh, G.; Li, H.-S.; Biswas, A.; Ong, M.L.D.M.; Jiang, Z.-H.; Choi, W.; et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat. Med. 2019, 25, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-T.; Zuo, L.; Odenwald, M.A.; Madha, S.; Singh, G.; Gurniak, C.B.; Abraham, C.; Turner, J.R. The Tight Junction Protein ZO-1 Is Dispensable for Barrier Function but Critical for Effective Mucosal Repair. Gastroenterology 2021, 161, 1924–1939. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Xiong, L.; Tang, S.; Zhao, J.; Zuo, L. Balancing Tumor Immunotherapy and Immune-Related Adverse Events: Unveiling the Key Regulators. Int. J. Mol. Sci. 2024, 25, 10919. https://doi.org/10.3390/ijms252010919
Huang J, Xiong L, Tang S, Zhao J, Zuo L. Balancing Tumor Immunotherapy and Immune-Related Adverse Events: Unveiling the Key Regulators. International Journal of Molecular Sciences. 2024; 25(20):10919. https://doi.org/10.3390/ijms252010919
Chicago/Turabian StyleHuang, Jianshang, Lei Xiong, Sainan Tang, Junhao Zhao, and Li Zuo. 2024. "Balancing Tumor Immunotherapy and Immune-Related Adverse Events: Unveiling the Key Regulators" International Journal of Molecular Sciences 25, no. 20: 10919. https://doi.org/10.3390/ijms252010919
APA StyleHuang, J., Xiong, L., Tang, S., Zhao, J., & Zuo, L. (2024). Balancing Tumor Immunotherapy and Immune-Related Adverse Events: Unveiling the Key Regulators. International Journal of Molecular Sciences, 25(20), 10919. https://doi.org/10.3390/ijms252010919