
The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies

 ,
,  , and
, and {kind=link}
{kind=link}
Abstract
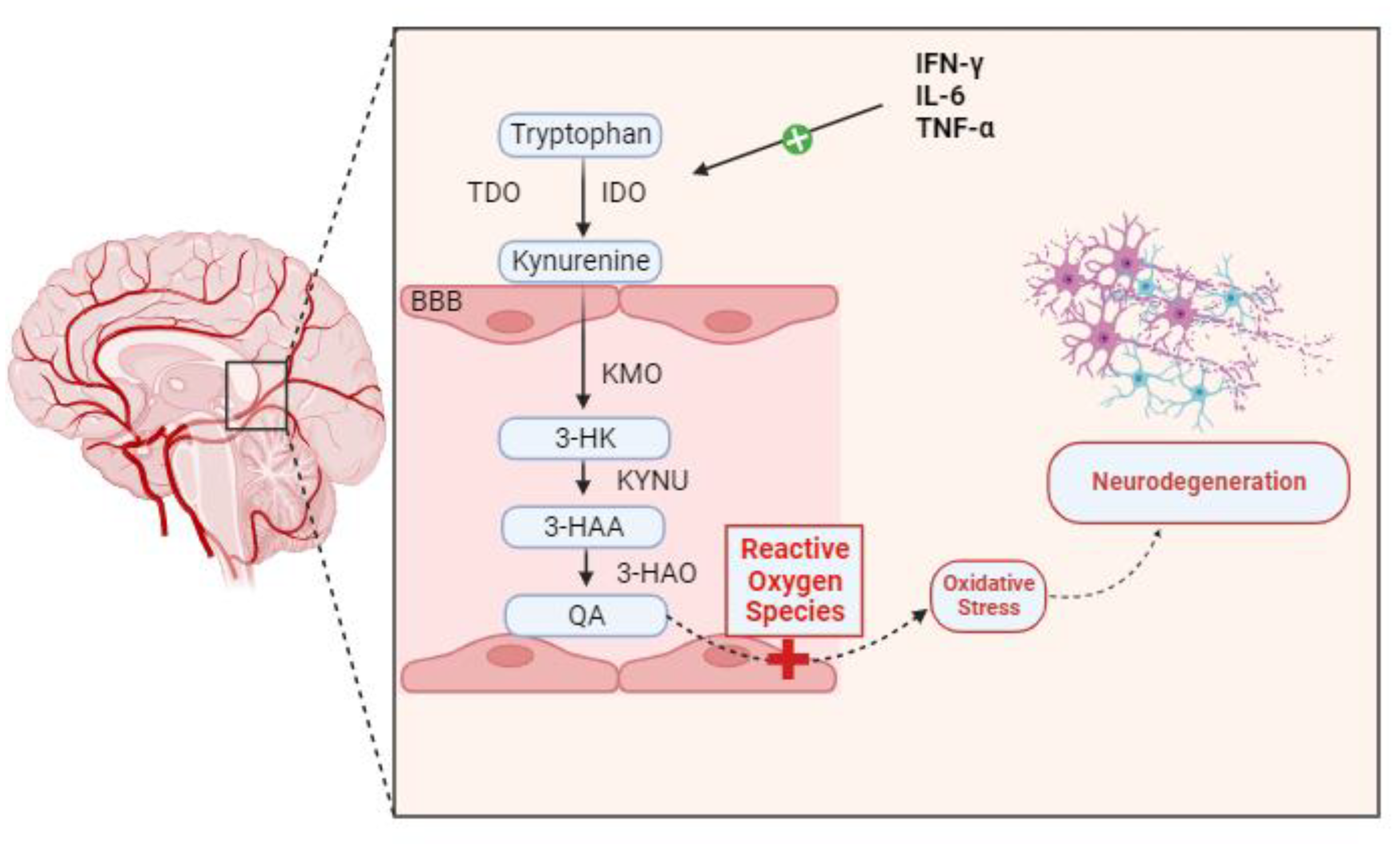
1. Kynurenine Pathway and Tryptophan Metabolism
2. Kynurenine Pathway and Inflammation
3. Kynurenine Pathway Dysfunction in Alzheimer’s Disease (AD)
4. Kynurenine Pathway Dysfunction in Parkinson’s Disease (PD)
5. Kynurenine Pathway Dysfunction in Other Neurodegenerative Diseases
6. Therapeutic Targets Modulate the Kynurenine Pathway
7. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Tryptophan Metabolism: A Versatile Area Providing Multiple Targets for Pharmacological Intervention. Egypt. J. Basic Clin. Pharmacol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xie, S.; He, Y.; Xu, M.; Qiao, X.; Zhu, Y.; Wu, W. Kynurenine Pathway Metabolites as Biomarkers in Alzheimer’s Disease. Dis Markers. 2022, 2022, 9484217. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother. 2015, 15, 719–721. [Google Scholar] [CrossRef]
- Garcez, M.L.; Jacobs, K.R.; Guillemin, G.J. Microbiota Alterations in Alzheimer’s Disease: Involvement of the Kynurenine Pathway and Inflammation. Neurotox. Res. 2019, 36, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Singh, T.G.; Prabhakar, N.K.; Mannan, A. Kynurenine Metabolism and Alzheimer’s Disease: The Potential Targets and Approaches. Neurochem. Res. 2022, 47, 1459–1476. [Google Scholar] [CrossRef]
- Bakker, L.; Choe, K.; Eussen, S.J.P.M.; Ramakers, I.H.G.B.; van den Hove, D.L.A.; Kenis, G.; Rutten, B.P.F.; Verhey, F.R.J.; Köhler, S. Relation of the kynurenine pathway with normal age: A systematic review. Mech. Ageing Dev. 2024, 217, 111890. [Google Scholar] [CrossRef]
- Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’Connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. Landmark Ed. 2015, 20, 1116–1143. [Google Scholar] [PubMed]
- Mingoti, M.; Bertollo, A.; Simões, J.; Francisco, G.; Bagatini, M.; Ignácio, Z.M. COVID-19, Oxidative Stress and Neuroinflammation in the Depression Route. J. Mol. Neurosci. 2022, 72, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Qin, Z.S.; Zheng, Y.; Xie, J.Y.; Liang, S.S.; Zhang, J.L.; Feng, Y.B.; Zhang, Z.J. Minocycline, a classic antibiotic, exerts psychotropic effects by normalizing microglial neuroinflammation-evoked tryptophan-kynurenine pathway dysregulation in chronically stressed male mice. Brain Behav. Immun. 2023, 107, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Gissen, P.; Smith, M.; Heales, S.J.R.; Clayton, P.T. The monoamine neurotransmitter disorders: An expanding range of neurological syndromes. Lancet Neurol. 2011, 10, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Endogenous neurotoxins from Tryptophan. Toxicon 2001, 39, 61–73. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxidative Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef]
- van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef]
- Hu, L.J.; Li, X.F.; Hu, J.Q.; Ni, X.J.; Lu, H.Y.; Wang, J.J.; Huang, X.N.; Lin, C.X.; Shang, D.W.; Wen, Y.G. A Simple HPLC-MS/MS Method for Determination of Tryptophan, Kynurenine and Kynurenic Acid in Human Serum and its Potential for Monitoring Antidepressant Therapy. J. Anal. Toxicol. 2017, 41, 37–44. [Google Scholar] [CrossRef]
- Grifka-Walk, H.M.; Jenkins, B.R.; Kominsky, D.J. Amino Acid Trp: The Far Out Impacts of Host and Commensal Tryptophan Metabolism. Front. Immunol. 2021, 12, 653208. [Google Scholar] [CrossRef]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major Developments in the Design of Inhibitors along the Kynurenine Pathway. Curr. Med. Chem. 2017, 24, 2471–2495. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, L.; Olah, M.; Smith, R.; Oatman, S.R.; Allen, M.; Pishva, E.; Zhang, B.; Menon, V.; Ertekin-Taner, N.; et al. Epigenomic features related to microglia are associated with attenuated effect of APOE ε4 on Alzheimer’s disease risk in humans. Alzheimers Dement. 2022, 18, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Katzourou, I.; Leonenko, G.; Ivanov, D.; Meggy, A.; Marshall, R.; Sims, R.; Williams, J.; Holmans, P.; Escott-Price, V. Cognitive Decline in Alzheimer’s Disease Is Not Associated with APOE. J. Alzheimers Dis. 2021, 84, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Zhang, L.; Hu, J.; Gao, H.; Zhang, B. Correlation of early cognitive dysfunction with inflammatory factors and metabolic indicators in patients with Alzheimer’s disease. Am. J. Transl. Res. 2021, 13, 9208–9215. [Google Scholar] [PubMed]
- de Leeuw, F.A.; Peeters, C.F.W.; Kester, M.I.; Harms, A.C.; Struys, E.A.; Hankemeier, T.; van Vlijmen, H.W.T.; van der Lee, S.J.; van Duijn, C.M.; Scheltens, P.; et al. Blood-based metabolic signatures in Alzheimer’s disease. Alzheimers Dement. 2017, 8, 196–207. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Lim, C.K.; Blennow, K.; Zetterberg, H.; Chatterjee, P.; Martins, R.N.; Brew, B.J.; Guillemin, G.J.; Lovejoy, D.B. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid-β and tau. Neurobiol. Aging 2019, 80, 11–20. [Google Scholar] [CrossRef]
- Willette, A.A.; Pappas, C.; Hoth, N.; Wang, Q.; Klinedinst, B.; Willette, S.A.; Larsen, B.; Pollpeter, A.; Li, T.; Le, S.; et al. Inflammation, negative affect, and amyloid burden in Alzheimer’s disease: Insights from the kynurenine pathway. Brain Behav. Immun. 2021, 95, 216–225. [Google Scholar] [CrossRef]
- Giil, L.M.; Midttun, Ø.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef]
- Gracia-Ramos, A.E.; Cruz-Domínguez, M.P.; Madrigal-Santillán, E.O. Incretin-based therapy for glycemic control of hospitalized patients with type 2 diabetes: A systematic review. Rev. Clín. Esp. Engl. Ed. 2022, 222, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Grant, R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen. Res. 2017, 12, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Vermeiren, Y.; Van Faassen, M.; van der Ley, C.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Bendheim, P.E.; Poeggeler, B.; Neria, E.; Ziv, V.; Pappolla, M.A.; Chain, D.G. Development of indole-3-propionic acid (OXIGON) for Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 213–217. [Google Scholar] [CrossRef]
- Wu, W.; Nicolazzo, J.A.; Wen, L.; Chung, R.; Stankovic, R.; Bao, S.S.; Lim, C.K.; Brew, B.J.; Cullen, K.M.; Guillemin, G.J. Expression of tryptophan 2,3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer’s disease brain. PLoS ONE 2013, 8, e59749. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Progress Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Laich, A.; Sperner-Unterweger, B.; Ledochowski, M.; Fuchs, D. Neopterin production, tryptophan degradation, and mental depression—What is the link? Brain Behav. Immun. 2002, 16, 590–595. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, L.; Liu, H.; Yang, J.; Yu, F.; Cui, C.; Huang, D. A Diagnostic Model for Parkinson’s Disease Based on Anoikis-Related Genes. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. TRYCAT pathways link peripheral inflammation, nicotine, somatization and depression in the etiology and course of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 137–149. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Kubesova, A.; Tejkalova, H.; Syslova, K.; Kacer, P.; Vondrousova, J.; Tyls, F.; Fujakova, M.; Palenicek, T.; Horacek, J. Biochemical, histopathological and morphological profiling of a rat model of early immune stimulation: Relation to psychopathology. PLoS ONE 2015, 10, e0115439. [Google Scholar] [CrossRef]
- Zhang, T.M.; Yu, S.Y.; Guo, P.; Du, Y.; Hu, Y.; Piao, Y.S.; Zuo, L.J.; Lian, T.H.; Wang, R.D.; Yu, Q.J.; et al. Nonmotor symptoms in patients with Parkinson disease: A cross-sectional observational study. Medicine 2016, 95, e5400. [Google Scholar] [CrossRef]
- Bai, M.Y.; Lovejoy, D.B.; Guillemin, G.J.; Kozak, R.; Stone, T.W.; Koola, M.M. Galantamine-Memantine Combination and Kynurenine Pathway Enzyme Inhibitors in the Treatment of Neuropsychiatric Disorders. Complex Psychiatry 2021, 7, 19–33. [Google Scholar] [CrossRef]
- Sun, C.; Armstrong, M.J. Treatment of Parkinson’s Disease with Cognitive Impairment: Current Approaches and Future Directions. Behav. Sci. 2021, 11, 54. [Google Scholar] [CrossRef]
- Wu, H.Q.; Rassoulpour, A.; Schwarcz, R. Kynurenic acid leads, dopamine follows: A new case of volume transmission in the brain? J. Neural Transm. 2007, 114, 33–41. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Obara-Michlewska, M.; Tuszyńska, P.; Albrecht, J. Ammonia upregulates KKynurenine aminotransferase II mRNA expression in rat brain: A role for astrocytic NMDA receptors? Metab. Brain Dis. 2013, 28, 161–165. [Google Scholar] [CrossRef]
- Yoon, J.H.; Maddock, R.J.; DongBo Cui, E.; Minzenberg, M.J.; Niendam, T.A.; Lesh, T.; Solomon, M.; Ragland, J.D.; Carter, C. Reduced in vivo visual cortex GABA in schizophrenia, a replication in a recent onset sample. Schizophr. Res. 2020, 215, 217–222. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Lowe, A.J.; Tortelli, R.; Heins, M.; Flik, G.; Johnson, E.B.; De Vita, E.; Scahill, R.I.; Giorgini, F.; et al. Kynurenine pathway metabolites in cerebrospinal fluid and blood as potential biomarkers in Huntington’s disease. J. Neurochem. 2021, 158, 539–553. [Google Scholar] [CrossRef]
- Byrne, L.M.; Wild, E.J. Cerebrospinal Fluid Biomarkers for Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Tavasol, A.; Jazi, K.; Hajibeygi, R.; Shool, S.; Sodeifian, F.; Klegeris, A.; McElhinney, A.; et al. Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A systematic Review and meta-analysis. Front. Immunol. 2022, 13, 997240. [Google Scholar] [CrossRef]
- Thevandavakkam, M.A.; Schwarcz, R.; Muchowski, P.J.; Giorgini, F. Targeting kynurenine 3-monooxygenase (KMO): Implications for therapy in Huntington’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 791–800. [Google Scholar] [CrossRef]
- NIH National Institute on Aging (NIA). What Is Lewy Body Dementia? Causes, Symptoms, and Treatments. 2021. Available online: https://www.nia.nih.gov/health/lewy-body-dementia/what-lewy-body-dementia-causes-symptoms-and-treatments (accessed on 29 July 2021).
- Solvang, S.H.; Nordrehaug, J.E.; Aarsland, D.; Lange, J.; Ueland, P.M.; McCann, A.; Midttun, Ø.; Tell, G.S.; Giil, L.M. Kynurenines, Neuropsychiatric Symptoms, and Cognitive Prognosis in Patients with Mild Dementia. Int. J. Tryptophan Res. 2019, 12, 1178646919877883. [Google Scholar]
- Jones, S.P.; Franco, N.F.; Varney, B.; Sundaram, G.; Brown, D.A.; de Bie, J.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Expression of the Kynurenine Pathway in Human Peripheral Blood Mononuclear Cells: Implications for Inflammatory and Neurodegenerative Disease. PLoS ONE 2015, 10, e0131389. [Google Scholar] [CrossRef]
- Braesch-Andersen, S.; Paulie, S.; Smedman, C.; Mia, S.; Kumagai-Braesch, M. ApoE production in human monocytes and its regulation by inflammatory cytokines. PLoS ONE 2013, 8, e79908. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef]
- Heylen, A.; Vermeiren, Y.; Kema, I.P.; van Faassen, M.; van der Ley, C.; Van Dam, D.; De Deyn, P.P. Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD. Pharmaceuticals 2023, 16, 615. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134 Pt 9, 2456–2477. [Google Scholar] [CrossRef]
- González-Sánchez, M.; Jiménez, J.; Narváez, A.; Antequera, D.; Llamas-Velasco, S.; Martín, A.H.; Arjona, J.A.M.; Munain, A.L.; Bisa, A.L.; Marco, M.P.; et al. Kynurenic Acid Levels are Increased in the CSF of Alzheimer’s Disease Patients. Biomolecules 2020, 10, 571. [Google Scholar] [CrossRef]
- Lee, J.-M.; Tan, V.; Lovejoy, D.; Braidy, N.; Rowe, D.B.; Brew, B.J.; Guillemin, G.J. Involvement of quinolinic acid in the neuropathogenesis of amyotrophic lateral sclerosis. Neuropharmacology 2017, 112, 346–364. [Google Scholar]
- Alarcan, H.; Chaumond, R.; Emond, P.; Benz-De Bretagne, I.; Lefèvre, A.; Bakkouche, S.E.; Veyrat-Durebex, C.; Vourc’h, P.; Andres, C.; Corcia, P.; et al. Some CSF Kynurenine Pathway Intermediates Associated with Disease Evolution in Amyotrophic Lateral Sclerosis. Biomolecules 2021, 11, 691. [Google Scholar] [CrossRef]
- Poewe, W.; Stankovic, I.; Halliday, G.; Meissner, W.G.; Wenning, G.K.; Pellecchia, M.T.; Seppi, K.; Palma, J.A.; Kaufmann, H. Multiple system atrophy. Nat. Rev. Dis. Primers 2022, 8, 56. [Google Scholar]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Tavasol, A.; Jazi, K.; Mohamadkhani, A.; Klegeris, A.; McElhinney, A.; Mafi, Z.; Hajiesmaeili, M.; et al. Dynamic changes in kynurenine pathway metabolites in multiple sclerosis: A systematic review. Front. Immunol. 2022, 13, 1013784. [Google Scholar] [CrossRef]
- Tafti, D.; Ehsan, M.; Xixis, K.L. Multiple Sclerosis; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tan, V.X.; Guillemin, G.J. Kynurenine Pathway Metabolites as Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurosci. 2019, 13, 1013. [Google Scholar]
- Hanson, J. Novel drug targets and therapeutic perspectives. Towards a precision medicine. Rev. Med. Liege 2020, 75, 460–465. [Google Scholar]
- Therapeutic Target: National Center for Advancing Translational Sciences (NCATS). 2017. Available online: https://registries.ncats.nih.gov/glossary/therapeutic-target/ (accessed on 7 January 2024).
- Solvang, S.H.; Hodge, A.; Watne, L.O.; Cabral-Marques, O.; Nordrehaug, J.E.; Giles, G.G.; Dugué, P.A.; Nygård, O.; Ueland, P.M.; McCann, A.; et al. Kynurenine Pathway Metabolites in the Blood and Cerebrospinal Fluid Are Associated with Human Aging. Oxidative Med. Cell. Longev. 2022, 2022, 5019752. [Google Scholar] [CrossRef]
- Wuebben, Y.; Winterer, G. Hypofrontality—A risk-marker related to schizophrenia? Schizophr. Res. 2001, 48, 207–217. [Google Scholar]
- Stone, T.W.; Darlington, L.G. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br. J. Pharmacol. 2013, 169, 1211–1227. [Google Scholar] [CrossRef]
- Klinkenberg, S.; van den Borne, C.J.; Aalbers, M.W.; Verschuure, P.; Kessels, A.G.; Leenen, L.; Rijkers, K.; Aldenkamp, A.P.; Vles, J.S.; Majoie, H.J. The effects of vagus nerve stimulation on tryptophan metabolites in children with intractable epilepsy. Epilepsy Behav. 2014, 37, 133–138. [Google Scholar]
- Tohgi, H.; Abe, T.; Takahashi, S.; Saheki, M.; Kimura, M. Indoleamine concentrations in cerebrospinal fluid from patients with Alzheimer type and Binswanger type dementias before and after administration of citalopram, a synthetic serotonin uptake inhibitor. J. Neural Transm. Park. Dis. Dement. Sect. 1995, 9, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Lunn, B.S.; Walker, L.L.; Gray, J.M.; Ballard, C.G.; O’Brien, J.T. Cognitive deficit induced by acute tryptophan depletion in patients with Alzheimer’s disease. Am. J. Psychiatry 2000, 157, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [PubMed]
- Krause, D.; Suh, H.S.; Tarassishin, L.; Cui, Q.L.; Durafourt, B.A.; Choi, N.; Bauman, A.; Cosenza-Nashat, M.; Antel, J.P.; Zhao, M.L.; et al. The tryptophan metabolite 3-hydroxyanthranilic acid plays anti-inflammatory and neuroprotective roles during inflammation: Role of hemeoxygenase-1. Am. J. Pathol. 2011, 179, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.S.; Majewska, A.K. An overview of microglia ontogeny and maturation in the homeostatic and pathological brain. Eur. J. Neurosci. 2021, 53, 3525–3547. [Google Scholar] [CrossRef]
- Kim, C.S.; Jung, S.; Hwang, G.S.; Shin, D.M. Gut microbiota indole-3-propionic acid mediates neuroprotective effect of probiotic consumption in healthy elderly: A randomized, double-blind, placebo-controlled, multicenter trial and in vitro study. Clin. Nutr. 2023, 42, 1025–1033. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, S.; Nadar, R.; Kim, S.; Liu, K.; Govindarajulu, M.; Cook, P.; Watts Alexander, C.S.; Dhanasekaran, M.; Moore, T. The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies. Int. J. Mol. Sci. 2024, 25, 853. https://doi.org/10.3390/ijms25020853
Pathak S, Nadar R, Kim S, Liu K, Govindarajulu M, Cook P, Watts Alexander CS, Dhanasekaran M, Moore T. The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies. International Journal of Molecular Sciences. 2024; 25(2):853. https://doi.org/10.3390/ijms25020853
Chicago/Turabian StylePathak, Suhrud, Rishi Nadar, Shannon Kim, Keyi Liu, Manoj Govindarajulu, Preston Cook, Courtney S. Watts Alexander, Muralikrishnan Dhanasekaran, and Timothy Moore. 2024. "The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies" International Journal of Molecular Sciences 25, no. 2: 853. https://doi.org/10.3390/ijms25020853
APA StylePathak, S., Nadar, R., Kim, S., Liu, K., Govindarajulu, M., Cook, P., Watts Alexander, C. S., Dhanasekaran, M., & Moore, T. (2024). The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies. International Journal of Molecular Sciences, 25(2), 853. https://doi.org/10.3390/ijms25020853