
CRISPR/Cas9 as a Mutagenic Factor

 , , , ,
, , , ,
Abstract
1. Introduction
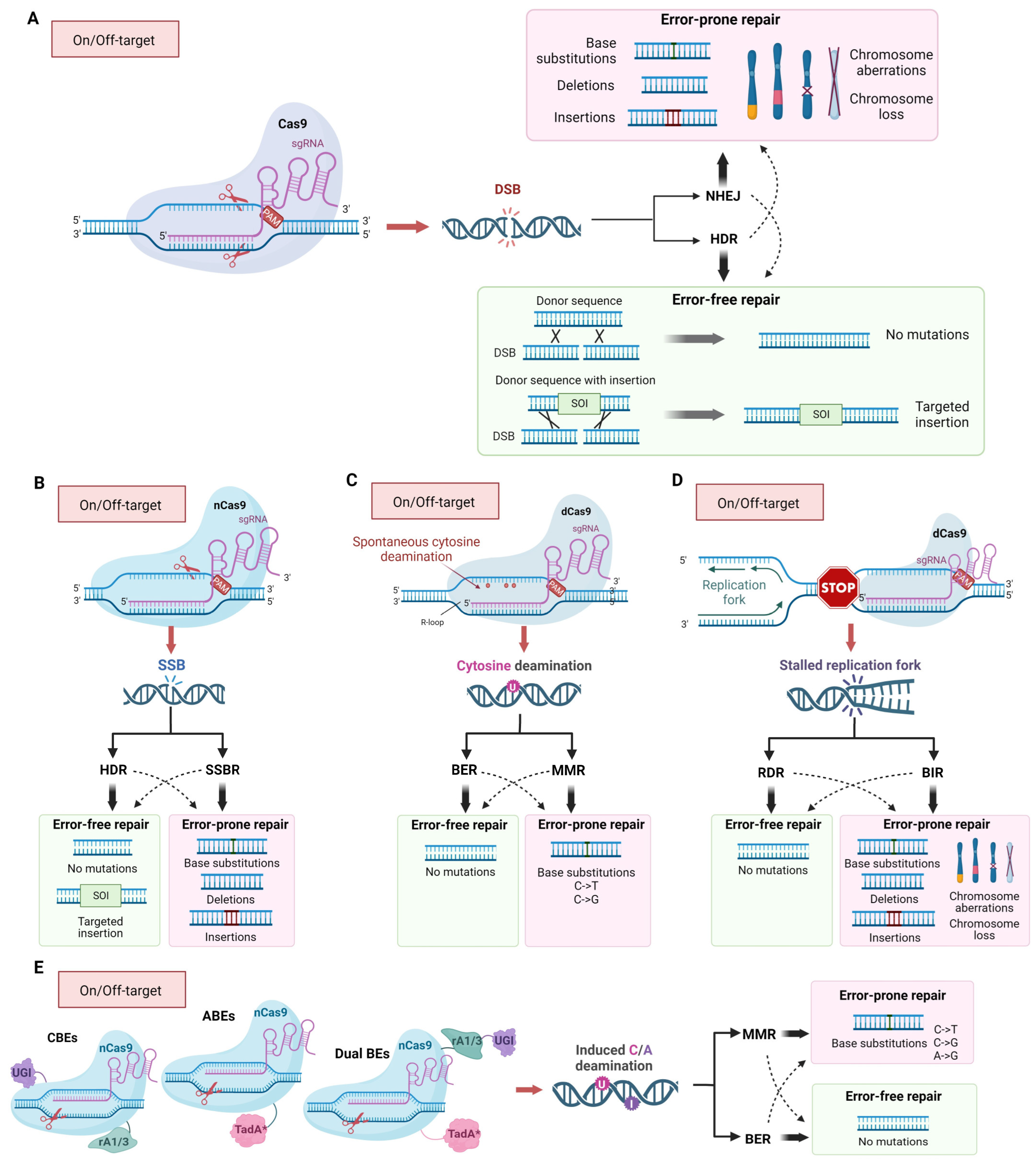
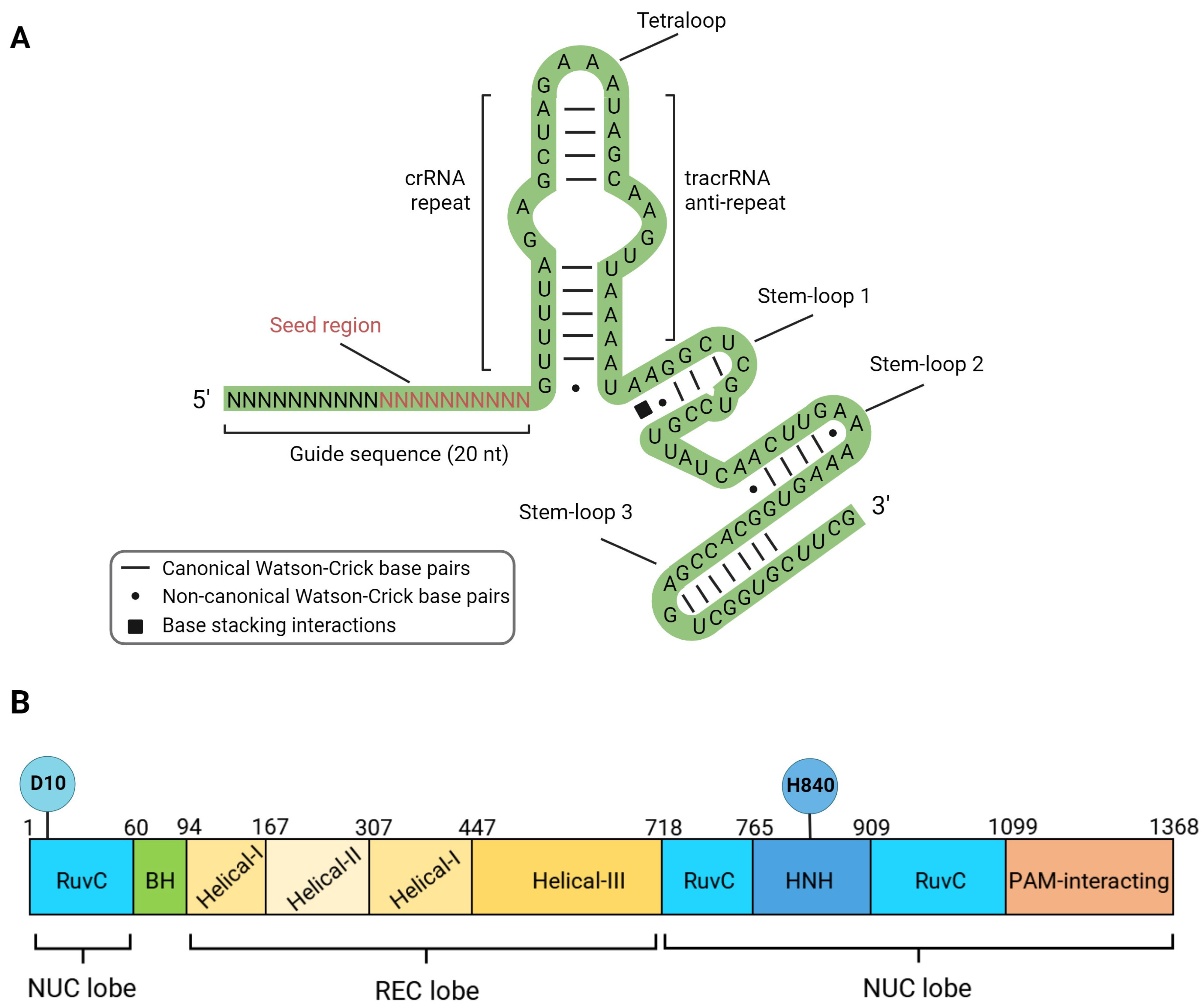
2. Structure, Mechanism of Action and DNA Lesions Caused by CRISPR/Cas9 Editing Tools
3. The Mechanism of Inaccurate Repair of Cas9-Induced DNA Lesions
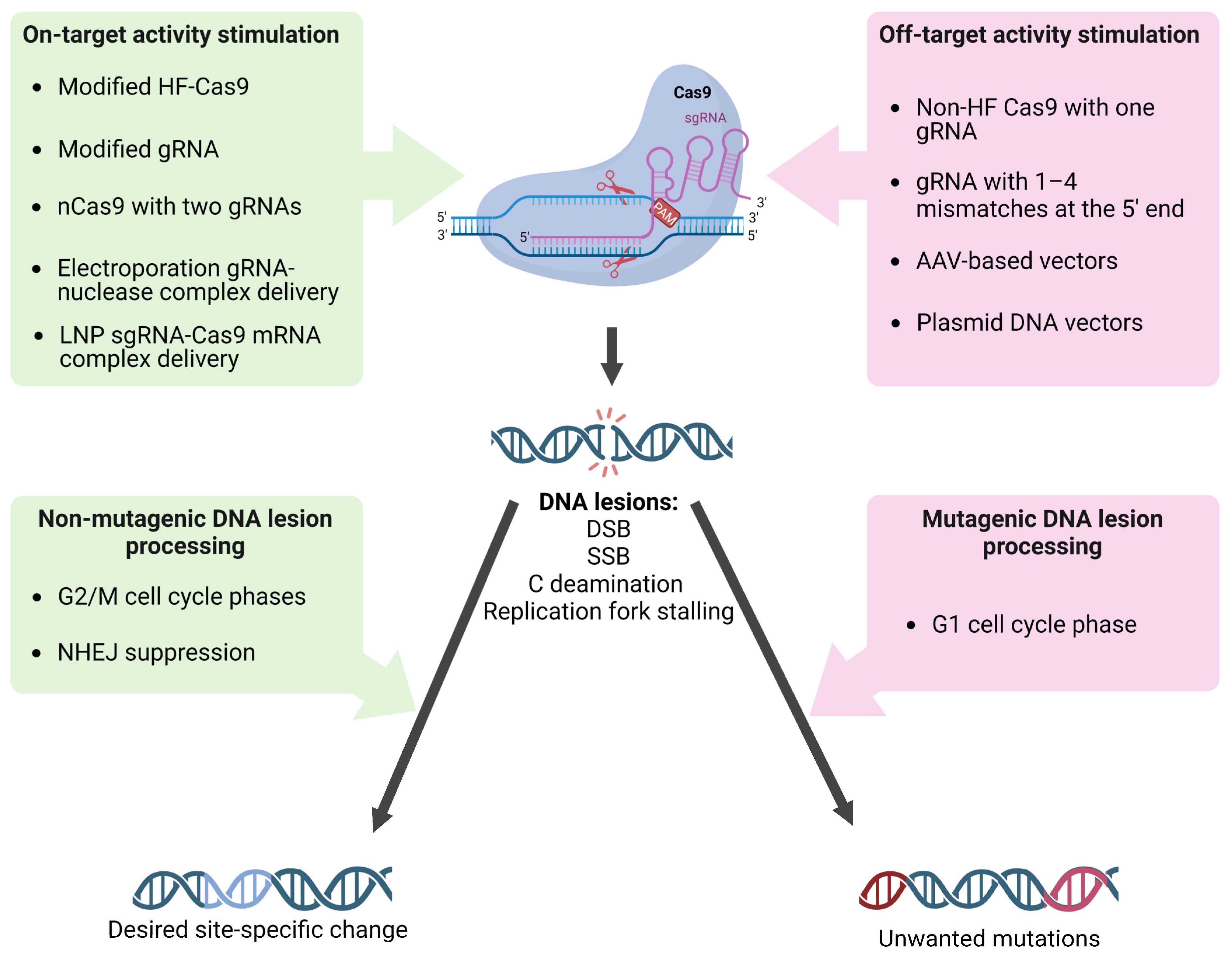
4. On-Target and Off-Target Activity: Hot Points of the sgRNA/Cas9-Dependent Mutagenesis
5. Types and Frequency of Mutations Caused by sgRNA/Cas9
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid Restriction Enzymes: Zinc Finger Fusions to Fok I Cleavage Domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Sirk, S.J.; Shui, S.L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the Genetic Editing Tool Kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L. Homing Endonucleases from Mobile Group I Introns: Discovery to Genome Engineering. Mob. DNA 2014, 5, 7. [Google Scholar] [CrossRef]
- Greig, J.A.; Breton, C.; Ashley, S.N.; Martins, K.M.; Gorsuch, C.; Chorazeczewski, J.K.; Furmanak, T.; Smith, M.K.; Zhu, Y.; Bell, P.; et al. Treating Transthyretin Amyloidosis via Adeno-Associated Virus Vector Delivery of Meganucleases. Hum. Gene Ther. 2022, 33, 1174–1186. [Google Scholar]
- Zeng, Y.; Li, J.; Li, G.; Huang, S.; Yu, W.; Zhang, Y.; Chen, D.; Chen, J.; Liu, J.; Huang, X. Correction of the Marfan Syndrome Pathogenic FBN1 Mutation by Base Editing in Human Cells and Heterozygous Embryos. Mol. Ther. 2018, 26, 2631–2637. [Google Scholar] [CrossRef]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Palmerola, K.L.; Smith, M.E.; et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell 2020, 183, 1650–1664.e15. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas Guides the Future of Genetic Engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- El-Mounadi, K.; Morales-Floriano, M.L.; Garcia-Ruiz, H. Principles, Applications, and Biosafety of Plant Genome Editing Using CRISPR-Cas9. Front. Plant Sci. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted Genome Engineering in Human Cells with the Cas9 RNA-Guided Endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wang, Q.; Mendez-Dorantes, C.; Burns, K.H.; Chiarle, R. Frequency and Mechanisms of LINE-1 Retrotransposon Insertions at CRISPR/Cas9 sites. Nat. Commun. 2022, 13, 3685. [Google Scholar] [CrossRef]
- Hussmann, J.A.; Ling, J.; Ravisankar, P.; Yan, J.; Cirincione, A.; Xu, A.; Simpson, D.; Yang, D.; Bothmer, A.; Cotta-Ramusino, C.; et al. Mapping the Genetic Landscape of DNA Double-Strand Break Repair. Cell 2021, 184, 5653–5669.e25. [Google Scholar] [CrossRef]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef]
- Alkan, F.; Wenzel, A.; Anthon, C.; Havgaard, J.H.; Gorodkin, J. CRISPR-Cas9 off-Targeting Assessment with Nucleic Acid Duplex Energy Parameters. Genome Biol. 2018, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Laughery, M.F.; Mayes, H.C.; Pedroza, I.K.; Wyrick, J.J. R-Loop Formation by dCas9 Is Mutagenic in Saccharomyces cerevisiae. Nucleic Acids Res. 2019, 47, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Doi, G.; Okada, S.; Yasukawa, T.; Sugiyama, Y.; Bala, S.; Miyazaki, S.; Kang, D.; Ito, T. Catalytically Inactive Cas9 Impairs DNA Replication Fork Progression to Induce Focal Genomic Instability. Nucleic Acids Res. 2021, 49, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pham, N.; Xia, B.; Papusha, A.; Wang, G.; Yan, Z.; Peng, G.; Chen, K.; Ira, G. Dna2 Nuclease Deficiency Results in large and Complex DNA Insertions at Chromosomal Breaks. Nature 2018, 564, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.L.; Lee, S.S.; Greenlees, K.J.; Tadesse, D.A.; Miller, M.F.; Lombardi, H.A. Template Plasmid Integration in Germline Genome-Edited Cattle. Nat. Biotechnol. 2020, 38, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Xie, H.; Tang, L.; He, X.; Liu, X.; Zhou, C.; Liu, J.; Ge, X.; Li, J.; Liu, C.; Zhao, J.; et al. SaCas9 Requires 5′-NNGRRT-3′ PAM for Sufficient Cleavage and Possesses Higher Cleavage Activity than SpCas9 or FnCpf1 in Human Cells. Biotechnol. J. 2018, 13, e1700561. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In Vivo Genome Editing Using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Heidrich, N.; Ampattu, B.J.; Gunderson, C.W.; Seifert, H.S.; Schoen, C.; Vogel, J.; Sontheimer, E.J. Processing-Independent CRISPR RNAs Limit Natural Transformation in Neisseria meningitidis. Mol. Cell 2013, 50, 488–503. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In Vivo Genome Editing with a Small Cas9 Orthologue Derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas Bacterial Immune System Cleaves Bacteriophage and Plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR-Cas Systems: A Burst of Class 2 and Derived Variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R. Diversity of CRISPR-Cas Immune Systems and Molecular Machines. Genome Biol. 2015, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of Functional Guide RNAs for the CRISPR/Cas9 System. Genome Biol. 2015, 16, 218. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef] [PubMed]
- Skrekas, C.; Limeta, A.; Siewers, V.; David, F. Targeted In Vivo Mutagenesis in Yeast Using CRISPR/Cas9 and Hyperactive Cytidine and Adenine Deaminases. ACS Synth. Biol. 2023, 12, 2278–2289. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, B.; Ru, G.; Meng, H.; Yan, Y.; Hong, M.; Zhang, D.; Luan, C.; Zhang, S.; Wu, H.; et al. Re-Engineering the Adenine Deaminase TadA-8e for Efficient and Specific CRISPR-Based Cytosine Base Editing. Nat. Biotechnol. 2023, 41, 663–672. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Wang, Z.; Wu, Z.; Wang, Y.; Tang, N.; Xu, X.; Zhao, S.; Chen, W.; Ji, Q. Programmable Adenine Deamination in Bacteria Using a Cas9-Adenine-Deaminase fusion. Chem. Sci. 2020, 11, 1657–1664. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, X.H.; Li, D.L. The “New Favorite” of Gene Editing Technology-Single Base Editors. Yi Chuan 2017, 39, 1115–1121. [Google Scholar]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A Dual-Deaminase CRISPR Base Editor Enables Concurrent Adenine and Cytosine Editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, M.; Liu, J.; Xue, X.; Zhong, J.; Lin, J.; Ye, B.; Chen, J.; Qiao, Y. Dual Guide RNA-Mediated Concurrent C&G-to-T&A and A&T-to-G&C Conversions Using CRISPR Base Editors. Comput. Struct. Biotechnol. J. 2023, 21, 856–868. [Google Scholar] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Marini, F.; Rawal, C.C.; Liberi, G.; Pellicioli, A. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front. Mol. Biosci. 2019, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA Break Repair by the Nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Liu, L.; Malkova, A. Break-Induced Replication: Unraveling Each Step. Trends Genet. 2022, 38, 752–765. [Google Scholar] [CrossRef]
- Zhuk, A.S.; Shiriaeva, A.A.; Andreychuk, Y.V.; Kochenova, O.V.; Tarakhovskaya, E.R.; Bure, V.M.; Pavlov, Y.I.; Inge-Vechtomov, S.G.; Stepchenkova, E.I. Detection of Primary DNA Lesions by Transient Changes in Mating Behavior in Yeast Saccharomyces cerevisiae Using the Alpha-Test. Int. J. Mol. Sci. 2023, 24, 12163. [Google Scholar] [CrossRef]
- Geisinger, J.M.; Turan, S.; Hernandez, S.; Spector, L.P.; Calos, M.P. In Vivo Blunt-End Cloning through CRISPR/Cas9-Facilitated Non-Homologous End-Joining. Nucleic Acids Res. 2016, 44, e76. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA Damage Response Revisited: The p53 Family and Its Regulators Provide Endless Cancer Therapy Opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Mathiasen, D.P.; Lisby, M. Cell Cycle Regulation of Homologous Recombination in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Homma, A.; Sayadi, J.; Yang, S.; Ohashi, J.; Takumi, T. Sequence Features Associated with the Cleavage Efficiency of CRISPR/Cas9 System. Sci. Rep. 2016, 6, 19675. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-Induced Precise Gene Editing in Mammalian Cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the Efficiency of Precise Genome Editing with CRISPR-Cas9 by Inhibition of Nonhomologous End Joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological Inhibition of DNA-PK Stimulates Cas9-Mediated Genome Editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear Domain ‘Knock-in’ Screen for the Evaluation and Identification of Small Molecule Enhancers of CRISPR-Based Genome Editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M Cell Cycle Phase in Human Pluripotent Stem Cells Enhances HDR-Mediated Gene Repair with Customizable Endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-Strand Break Repair and Genetic Disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Beal, P.A. Off-Target Editing by CRISPR-Guided DNA Base Editors. Biochemistry 2019, 58, 3727–3734. [Google Scholar] [CrossRef]
- Kouzminova, E.A.; Kuzminov, A. Fragmentation of Replicating Chromosomes Triggered by Uracil in DNA. J. Mol. Biol. 2006, 355, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Single-Strand Interruptions in Replicating Chromosomes Cause Double-Strand Breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Northam, M.R.; Robinson, H.A.; Kochenova, O.V.; Shcherbakova, P.V. Participation of DNA Polymerase Zeta in Replication of Undamaged DNA in Saccharomyces cerevisiae. Genetics 2010, 184, 27–42. [Google Scholar] [CrossRef]
- Northam, M.R.; Moore, E.A.; Mertz, T.M.; Binz, S.K.; Stith, C.M.; Stepchenkova, E.I.; Wendt, K.L.; Burgers, P.M.; Shcherbakova, P.V. DNA Polymerases Zeta and Rev1 Mediate Error-Prone bypass of Non-B DNA Structures. Nucleic Acids Res. 2014, 42, 290–306. [Google Scholar] [CrossRef]
- Stepchenkova, E.I.; Zadorsky, S.P.; Shumega, A.R.; Aksenova, A.Y. Practical Approaches for the Yeast Saccharomyces cerevisiae Genome Modification. Int. J. Mol. Sci. 2023, 24, 11960. [Google Scholar] [CrossRef]
- Wang, S.; Zong, Y.; Lin, Q.; Zhang, H.; Chai, Z.; Zhang, D.; Chen, K.; Qiu, J.L.; Gao, C. Precise, Predictable Multi-Nucleotide Deletions in Rice and Wheat Using APOBEC-Cas9. Nat. Biotechnol. 2020, 38, 1460–1465. [Google Scholar] [CrossRef]
- Corsi, G.I.; Qu, K.; Alkan, F.; Pan, X.; Luo, Y.; Gorodkin, J. CRISPR/Cas9 gRNA Activity Depends on Free Energy Changes and on the Target PAM Context. Nat. Commun. 2022, 13, 3006. [Google Scholar] [CrossRef]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Lin-Shiao, E.; Trinidad, M.; Saffari Doost, M.; Colognori, D.; Doudna, J.A. Decorating Chromatin for Enhanced Genome Editing Using CRISPR-Cas9. Proc. Natl. Acad. Sci. USA 2022, 119, e2204259119. [Google Scholar] [CrossRef] [PubMed]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, T.S.; Baudrier, L.; Billon, P.; Ciccia, A. CRISPR-Based Genome Editing through the Lens of DNA Repair. Mol. Cell 2022, 82, 348–388. [Google Scholar] [CrossRef] [PubMed]
- Lohia, A.; Sahel, D.K.; Salman, M.; Singh, V.; Mariappan, I.; Mittal, A.; Chitkara, D. Delivery Strategies for CRISPR/Cas Genome Editing Tool for Retinal Dystrophies: Challenges and Opportunities. Asian J. Pharm. Sci. 2022, 17, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.A.; Law, E.K.; Serebrenik, A.; Brown, W.L.; Harris, R.S. A Lentivirus-Based System for Cas9/gRNA Expression and Subsequent Removal by Cre-Mediated Recombination. Methods 2019, 156, 79–84. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene Disruption by Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid Nanoparticle-Mediated Codelivery of Cas9 mRNA and Single-Guide RNA Achieves Liver-Specific In Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef]
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas Tools for In Vivo Genome Editing Therapy: Trends and Challenges. J. Control. Release 2022, 342, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, F.; Begum, A.A.; Dai, C.C.; Toth, I.; Moyle, P.M. Recent Advances in the Delivery and Applications of Nonviral CRISPR/Cas9 Gene Editing. Drug Deliv. Transl. Res. 2023, 13, 1500–1519. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yang, Z.; Zhang, Y.; Xia, J.; Zhang, J.; Han, Q.; Yu, H.; Wu, C.; Xu, Y.; Xu, W.; et al. Lipid Nanoparticle Delivery of Chemically Modified NGF(R100W) mRNA Alleviates Peripheral Neuropathy. Adv. Healthc. Mater. 2023, 12, e2202127. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-Target Effects in CRISPR/Cas9 Gene Editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.W.; Du, W.; Yang, Q.; Kooijmans, S.A.A.; Vink, A.; van Steenbergen, M.; Vader, P.; de Jager, S.C.A.; Fuchs, S.A.; Mastrobattista, E.; et al. Delivery of Modified mRNA to Damaged Myocardium by Systemic Administration of Lipid Nanoparticles. J. Control. Release 2022, 343, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for Nonviral Nanoparticle-Based Delivery of CRISPR/Cas9 Therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1609. [Google Scholar] [CrossRef] [PubMed]
- Greig, J.A.; Martins, K.M.; Breton, C.; Lamontagne, R.J.; Zhu, Y.; He, Z.; White, J.; Zhu, J.X.; Chichester, J.A.; Zheng, Q.; et al. Integrated Vector Genomes May Contribute to Long-Term Expression in Primate Liver after AAV Administration. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High Levels of AAV Vector Integration into CRISPR-Induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Kim, D.; Luk, K.; Wolfe, S.A.; Kim, J.S. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu. Rev. Biochem. 2019, 88, 191–220. [Google Scholar] [CrossRef]
- Ghani, M.W.; Iqbal, A.; Ghani, H.; Bibi, S.; Wang, Z.; Pei, R. Recent Advances in Nanocomposite-Based Delivery Systems for Targeted CRISPR/Cas Delivery and Therapeutic Genetic Manipulation. J. Mater. Chem. B 2023, 11, 5251–5271. [Google Scholar] [CrossRef] [PubMed]
- Kulishova, L.M.; Vokhtantsev, I.P.; Kim, D.V.; Zharkov, D.O. Mechanisms of the Specificity of the CRISPR/Cas9 System in Genome Editing. Mol. Biol. 2023, 57, 269–284. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR-Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced Proofreading Governs CRISPR-Cas9 Targeting Accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.P.K.; Liu, M.S.; Hibshman, G.N.; Dangerfield, T.L.; Jung, K.; McCool, R.S.; Johnson, K.A.; Taylor, D.W. Structural Basis for Mismatch Surveillance by CRISPR-Cas9. Nature 2022, 603, 343–347. [Google Scholar] [CrossRef]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A Highly Specific SpCas9 Variant is Identified by In Vivo Screening in Yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed Evolution of CRISPR-Cas9 to Increase Its Specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A High-Fidelity Cas9 Mutant Delivered as a Ribonucleoprotein Complex Enables Efficient Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput Profiling of off-Target DNA Cleavage Reveals RNA-Programmed Cas9 Nuclease Specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural Basis of PAM-Dependent Target DNA Recognition by the Cas9 Endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-Target Effects of CRISPR/Cas-Derived RNA-Guided Endonucleases and Nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, M.; Suzuki, H.I.; Kimura, R.; Suzuki, A. Optimization of Cas9 Activity through the Addition of Cytosine Extensions to Single-Guide RNAs. Nat. Biomed. Eng. 2023, 7, 672–691. [Google Scholar] [CrossRef]
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015, 26, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Rozners, E. Chemical Modifications of CRISPR RNAs to Improve Gene-Editing Activity and Specificity. J. Am. Chem. Soc. 2022, 144, 12584–12594. [Google Scholar] [CrossRef] [PubMed]
- Lemos, B.R.; Kaplan, A.C.; Bae, J.E.; Ferrazzoli, A.E.; Kuo, J.; Anand, R.P.; Waterman, D.P.; Haber, J.E. CRISPR/Cas9 Cleavages in Budding Yeast Reveal Templated Insertions and Strand-Specific Insertion/Deletion Profiles. Proc. Natl. Acad. Sci. USA 2018, 115, E2040–E2047. [Google Scholar] [CrossRef]
- Chen, X.; Li, M.; Feng, X.; Guang, S. Targeted Chromosomal Translocations and Essential Gene Knockout Using CRISPR/Cas9 Technology in Caenorhabditis elegans. Genetics 2015, 201, 1295–1306. [Google Scholar] [CrossRef]
- Zhang, N.; Roberts, H.M.; Van Eck, J.; Martin, G.B. Generation and Molecular Characterization of CRISPR/Cas9-Induced Mutations in 63 Immunity-Associated Genes in Tomato Reveals Specificity and a Range of Gene Modifications. Front. Plant Sci. 2020, 11, 10. [Google Scholar] [CrossRef]
- Tang, X.; Liu, G.; Zhou, J.; Ren, Q.; You, Q.; Tian, L.; Xin, X.; Zhong, Z.; Liu, B.; Zheng, X.; et al. A Large-Scale Whole-Genome Sequencing Analysis Reveals Highly Specific Genome Editing by both Cas9 and Cpf1 (Cas12a) Nucleases in Rice. Genome Biol. 2018, 19, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, H.; Li, T.; Chen, K.; Qiu, J.L.; Gao, C. Perfectly matched 20-nucleotide guide RNA Sequences Enable Robust Genome Editing Using High-Fidelity SpCas9 Nucleases. Genome Biol. 2017, 18, 191. [Google Scholar] [CrossRef]
- Polikarpova, A.V.; Egorova, T.V.; Lunev, E.A.; Tsitrina, A.A.; Vassilieva, S.G.; Savchenko, I.M.; Silaeva, Y.Y.; Deykin, A.V.; Bardina, M.V. CRISPR/Cas9-Generated Mouse Model with Humanizing Single-Base Substitution in the Gnao1 for Safety Studies of RNA Therapeutics. Front. Genome Ed. 2023, 5, 1034720. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Khalouei, S.; Hanafi, N.; Wood, J.A.; Lanza, D.G.; Lintott, L.G.; Willis, B.J.; Seavitt, J.R.; Braun, R.E.; Dickinson, M.E.; et al. Whole Genome Analysis for 163 gRNAs in Cas9-Edited Mice Reveals Minimal Off-Target Activity. Commun. Biol. 2023, 6, 626. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Grishin, D.; Wang, G.; Aach, J.; Zhang, C.Z.; Chari, R.; Homsy, J.; Cai, X.; Zhao, Y.; Fan, J.B.; et al. Targeted and Genome-Wide Sequencing Reveal Single Nucleotide Variations Impacting Specificity of Cas9 in Human Stem Cells. Nat. Commun. 2014, 5, 5507. [Google Scholar] [CrossRef]
- Tsuchida, C.A.; Brandes, N.; Bueno, R.; Trinidad, M.; Mazumder, T.; Yu, B.; Hwang, B.; Chang, C.; Liu, J.; Sun, Y.; et al. Mitigation of Chromosome Loss in Clinical CRISPR-Cas9-Engineered T Cells. Cell 2023, 186, 4567–4582.e20. [Google Scholar] [CrossRef]
- Tran, N.T.; Danner, E.; Li, X.; Graf, R.; Lebedin, M.; de la Rosa, K.; Kuhn, R.; Rajewsky, K.; Chu, V.T. Precise CRISPR-Cas-Mediated Gene Repair with Minimal off-Target and Unintended on-Target Mutations in Human Hematopoietic STEM cells. Sci. Adv. 2022, 8, eabm9106. [Google Scholar] [CrossRef]
- Atkins, A.; Chung, C.H.; Allen, A.G.; Dampier, W.; Gurrola, T.E.; Sariyer, I.K.; Nonnemacher, M.R.; Wigdahl, B. Off-Target Analysis in Gene Editing and Applications for Clinical Translation of CRISPR/Cas9 in HIV-1 Therapy. Front. Genome Ed. 2021, 3, 673022. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A Highly Sensitive In Vitro Screen for Genome-Wide CRISPR-Cas9 Nuclease off-Targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Giannoukos, G.; Ciulla, D.M.; Marco, E.; Abdulkerim, H.S.; Barrera, L.A.; Bothmer, A.; Dhanapal, V.; Gloskowski, S.W.; Jayaram, H.; Maeder, M.L.; et al. UDiTaS, A Genome Editing Detection Method for Indels and Genome Rearrangements. BMC Genom. 2018, 19, 212. [Google Scholar] [CrossRef]
- Valentine III, C.C.; Young, R.R.; Fielden, M.R.; Kulkarni, R.; Williams, L.N.; Li, T.; Minocherhomji, S.; Salk, J.J. Direct Quantification of In Vivo Mutagenesis and Carcinogenesis Using Duplex Sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 33414–33425. [Google Scholar] [CrossRef] [PubMed]
- Fijalkowska, I.J.; Dunn, R.L.; Schaaper, R.M. Mutants of Escherichia coli with Increased Fidelity of DNA Replication. Genetics 1993, 134, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Manghwar, H.; Sun, L.; Wang, P.; Wang, G.; Sheng, H.; Zhang, J.; Liu, H.; Qin, L.; Rui, H.; et al. Whole Genome Sequencing Reveals Rare Off-Target Mutations and Considerable Inherent Genetic or/and Somaclonal Variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2019, 17, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Zastrow-Hayes, G.; Deschamps, S.; Svitashev, S.; Zaremba, M.; Acharya, A.; Paulraj, S.; Peterson-Burch, B.; Schwartz, C.; Djukanovic, V.; et al. CRISPR-Cas9 Editing in Maize: Systematic Evaluation of Off-target Activity and Its Relevance in Crop Improvement. Sci. Rep. 2019, 9, 6729. [Google Scholar] [CrossRef]
- Malinin, N.L.; Lee, G.; Lazzarotto, C.R.; Li, Y.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Iafrate, A.J.; Le, L.P.; et al. Defining genome-wide CRISPR-Cas Genome-Editing Nuclease Activity with GUIDE-Seq. Nat. Protoc. 2021, 16, 5592–5615. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, C.R.; Malinin, N.L.; Li, Y.; Zhang, R.; Yang, Y.; Lee, G.; Cowley, E.; He, Y.; Lan, X.; Jividen, K.; et al. CHANGE-seq Reveals Genetic and Epigenetic Effects on CRISPR-Cas9 Genome-Wide Activity. Nat. Biotechnol. 2020, 38, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Raitskin, O.; Schudoma, C.; West, A.; Patron, N.J. Comparison of Efficiency and Specificity of CRISPR-Associated (Cas) Nucleases in plants: An Expanded Toolkit for Precision Genome Engineering. PLoS ONE 2019, 14, e0211598. [Google Scholar] [CrossRef]
- Vouillot, L.; Thelie, A.; Pollet, N. Comparison of T7E1 and Surveyor Mismatch Cleavage Assays to detect Mutations Triggered by Engineered Nucleases. G3 Genes Genomes Genet. 2015, 5, 407–415. [Google Scholar] [CrossRef]
- Modrzejewski, D.; Hartung, F.; Lehnert, H.; Sprink, T.; Kohl, C.; Keilwagen, J.; Wilhelm, R. Which Factors Affect the Occurrence of Off-Target Effects Caused by the Use of CRISPR/Cas: A Systematic Review in Plants. Front. Plant Sci. 2020, 11, 574959. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.L.; Bacinskaja, G.; Che, J.; Cheblal, A.; Elango, R.; Epshtein, A.; Fitzgerald, D.M.; Gomez-Gonzalez, B.; Khan, S.R.; Kumar, S.; et al. Guidelines for DNA Recombination and Repair Studies: Cellular Assays of DNA Repair Pathways. Microb. Cell 2019, 6, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration, H.H.S. International Conference on Harmonisation; guidance on S2(R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals intended for Human Use; Availability. Notice. Fed. Regist. 2012, 77, 33748–33749. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Nuclease | Species | PAM Sequences (5′ to 3′) |
|---|---|---|
| SpCas9 | Streptococcus pyogenes | NGG [14] |
| SaCas9 | Staphylococcus aureus | NNGRRT or NNGRR [27,28] |
| NmeCas9 | Neisseria meningitidis | NNNNGATT [29] |
| CjCas9 | Campylobacter jejuni | NNNNRYAC [30] |
| StCas9 | Streptococcus thermophilus | NNAGAAW [31] |
| Organism | Location | Mutation Type | Frequency |
|---|---|---|---|
| Saccharomyces cerevisiae [108] | On-target | +1 bp insertions | 0–74% |
| +2 bp insertions | 0–22% | ||
| +3 bp | 0–9% | ||
| Off-target | No data | ||
| Caenorhabditis elegans [109] | On-target | Chromosomal translocation | 12.6% |
| Off-target | No data | ||
| Tomato [110] | On-target | A- and T-inserts | 79.5% |
| Off-target | 18 predicted off-target sites | 0% | |
| Rice [111] | On-target | 7 gene indels | 15–100% |
| Off-target | Indels, SNVs | 0% | |
| Rice protoplasts [112] | On-target | Indel mutations | 25% |
| Off-target | Indel mutations | 1–8% | |
| Mice [113] | On-target | Single-base substitution G>A in the Gnao1 gene | 25% |
| Off-target | 10 predicted off-target sites | 0% | |
| Mice [114] | On-target | Exon deletion | 98% |
| Off-target | Indels, single-nucleotide variants (SNVs) | 0.2% | |
| Human embryonic stem cells [115] | On-target | 1–4 base pair deletions Tafazzin (TAZ) gene | 54% |
| Off-target | Indel mutations | 0.15% | |
| Human T-cells [116] | On-target | Loss of chromosomes 14 segment | 20% |
| Off-target | No data | ||
| Human hematopoietic stem cell lines [117] | On-target | Indel mutation of HBB locus | 59.07% |
| Indel mutation of ELANE locus | 21.21% | ||
| Indel mutation of PRF1 locus | 58.64% | ||
| Off-target | No data | ||
| HEK293T, HeLa, and U2OS [18] | On-target | Insertions of LINE-1 (L1) retrotransposons | 4–6% |
| Off-target | No data |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shumega, A.R.; Pavlov, Y.I.; Chirinskaite, A.V.; Rubel, A.A.; Inge-Vechtomov, S.G.; Stepchenkova, E.I. CRISPR/Cas9 as a Mutagenic Factor. Int. J. Mol. Sci. 2024, 25, 823. https://doi.org/10.3390/ijms25020823
Shumega AR, Pavlov YI, Chirinskaite AV, Rubel AA, Inge-Vechtomov SG, Stepchenkova EI. CRISPR/Cas9 as a Mutagenic Factor. International Journal of Molecular Sciences. 2024; 25(2):823. https://doi.org/10.3390/ijms25020823
Chicago/Turabian StyleShumega, Andrey R., Youri I. Pavlov, Angelina V. Chirinskaite, Aleksandr A. Rubel, Sergey G. Inge-Vechtomov, and Elena I. Stepchenkova. 2024. "CRISPR/Cas9 as a Mutagenic Factor" International Journal of Molecular Sciences 25, no. 2: 823. https://doi.org/10.3390/ijms25020823
APA StyleShumega, A. R., Pavlov, Y. I., Chirinskaite, A. V., Rubel, A. A., Inge-Vechtomov, S. G., & Stepchenkova, E. I. (2024). CRISPR/Cas9 as a Mutagenic Factor. International Journal of Molecular Sciences, 25(2), 823. https://doi.org/10.3390/ijms25020823