
Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review


Abstract
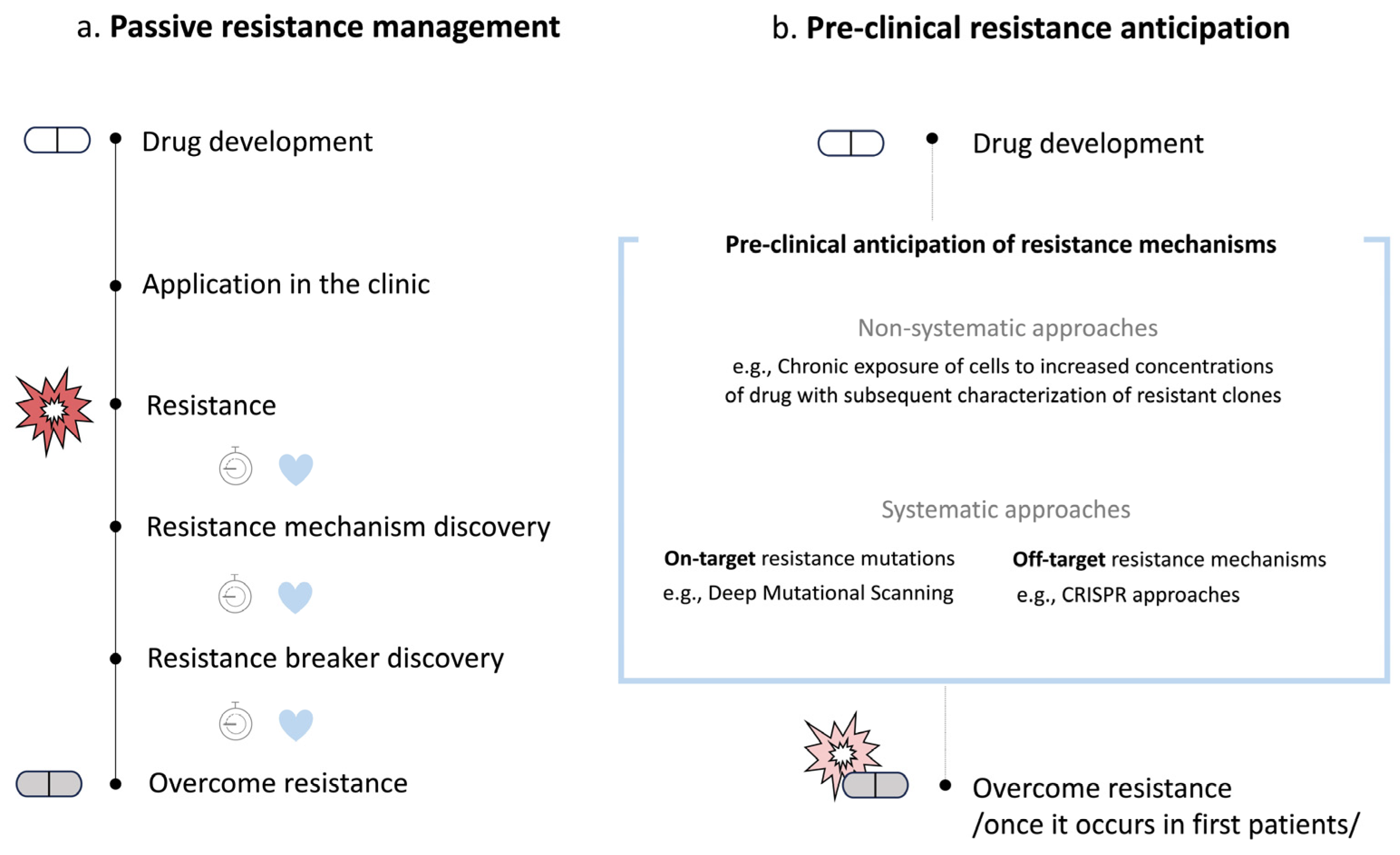
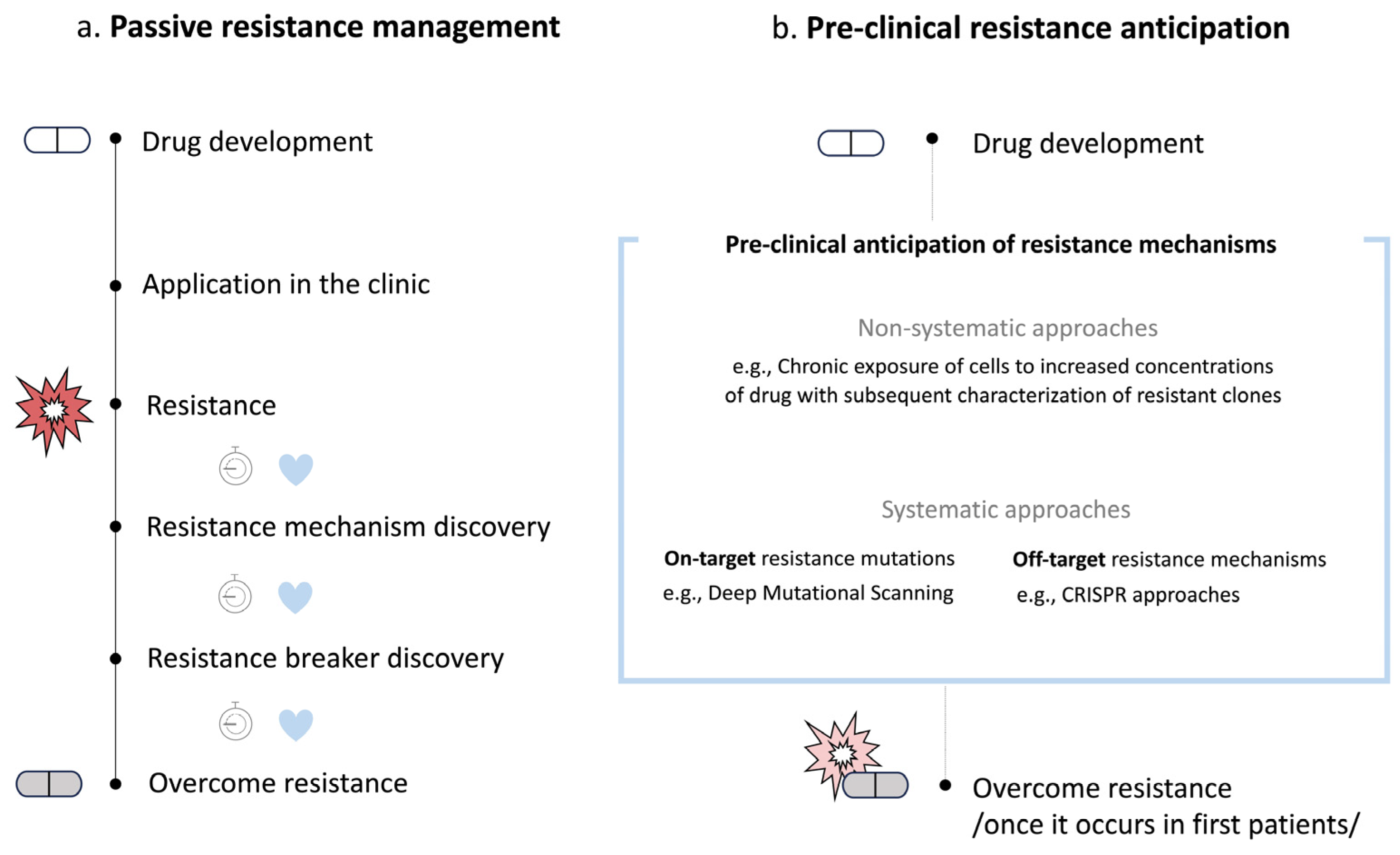
1. Introduction
2. Non-Systematic Preclinical Anticipation of Drug Resistance Mechanisms
2.1. Random Mutagenesis
2.2. Chronic Exposure of Tumor Models to Drug Regimens
3. Systematic Preclinical Anticipation of On-Target Drug Resistance Mechanisms
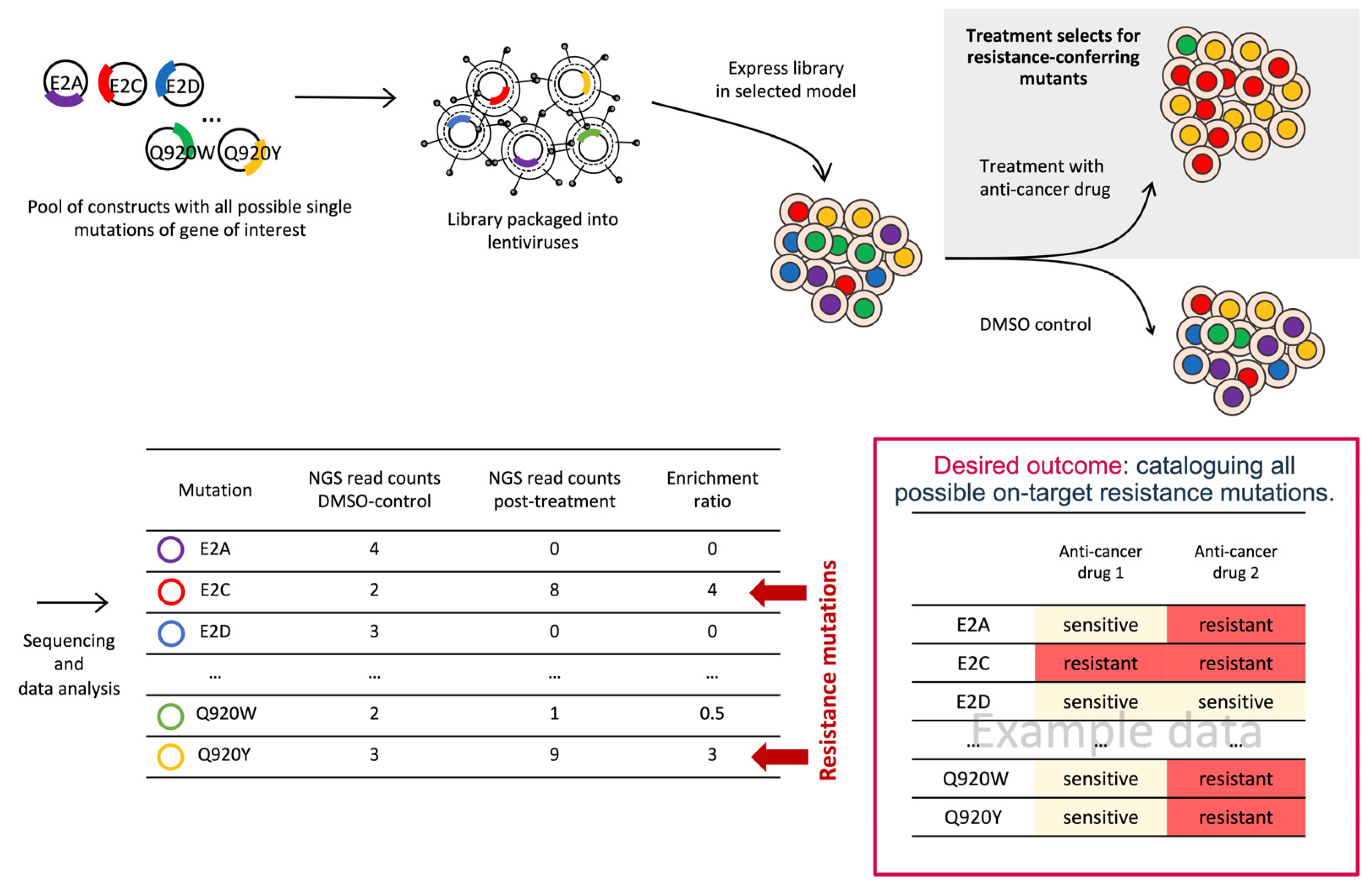
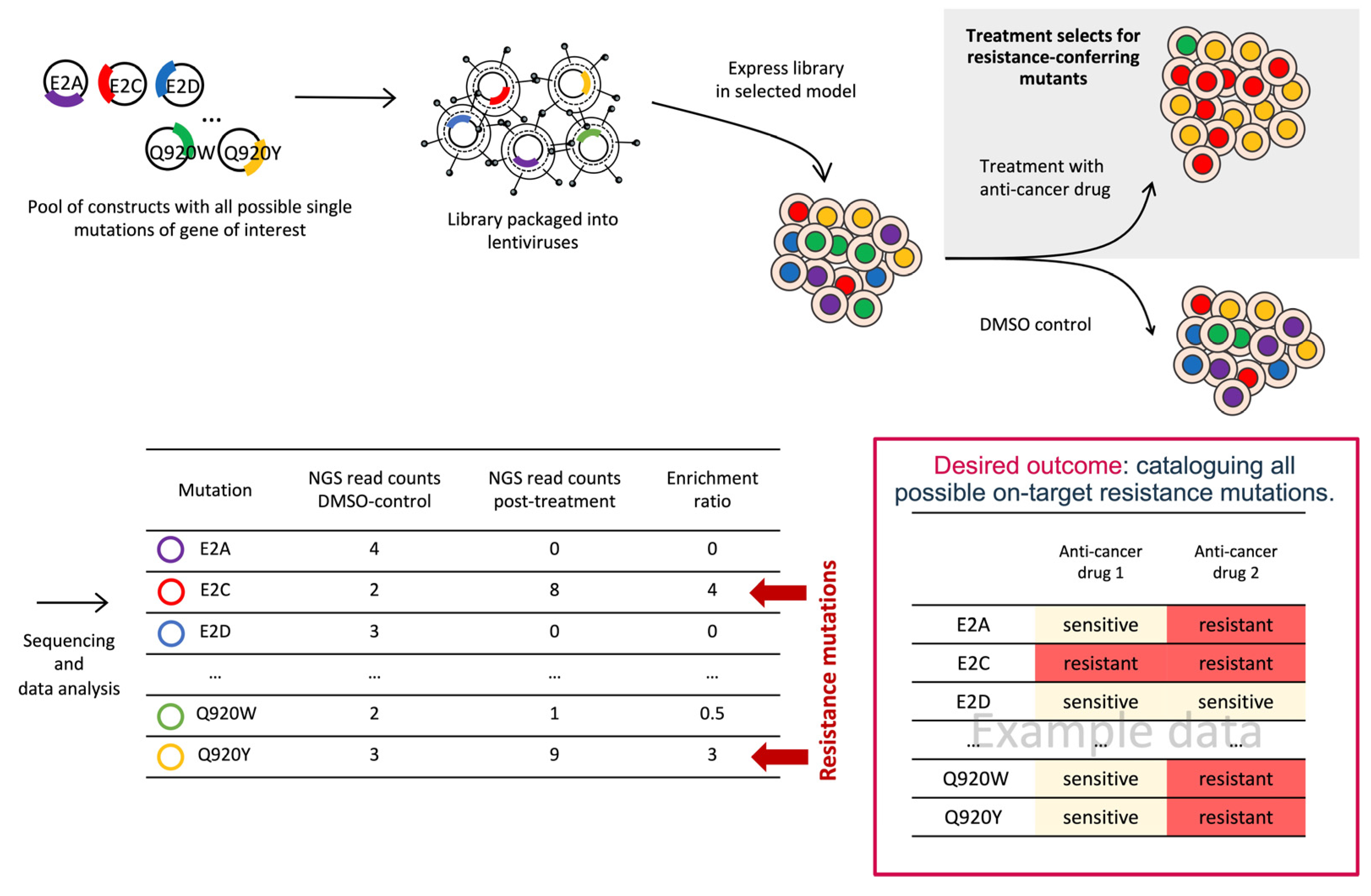
3.1. Deep Mutational Scanning (DMS)
3.2. CRISPR-Based Base-Editing Screens
3.3. Computational Methods
4. Systematic Preclinical Anticipation of Off-Target Drug Resistance Mechanisms
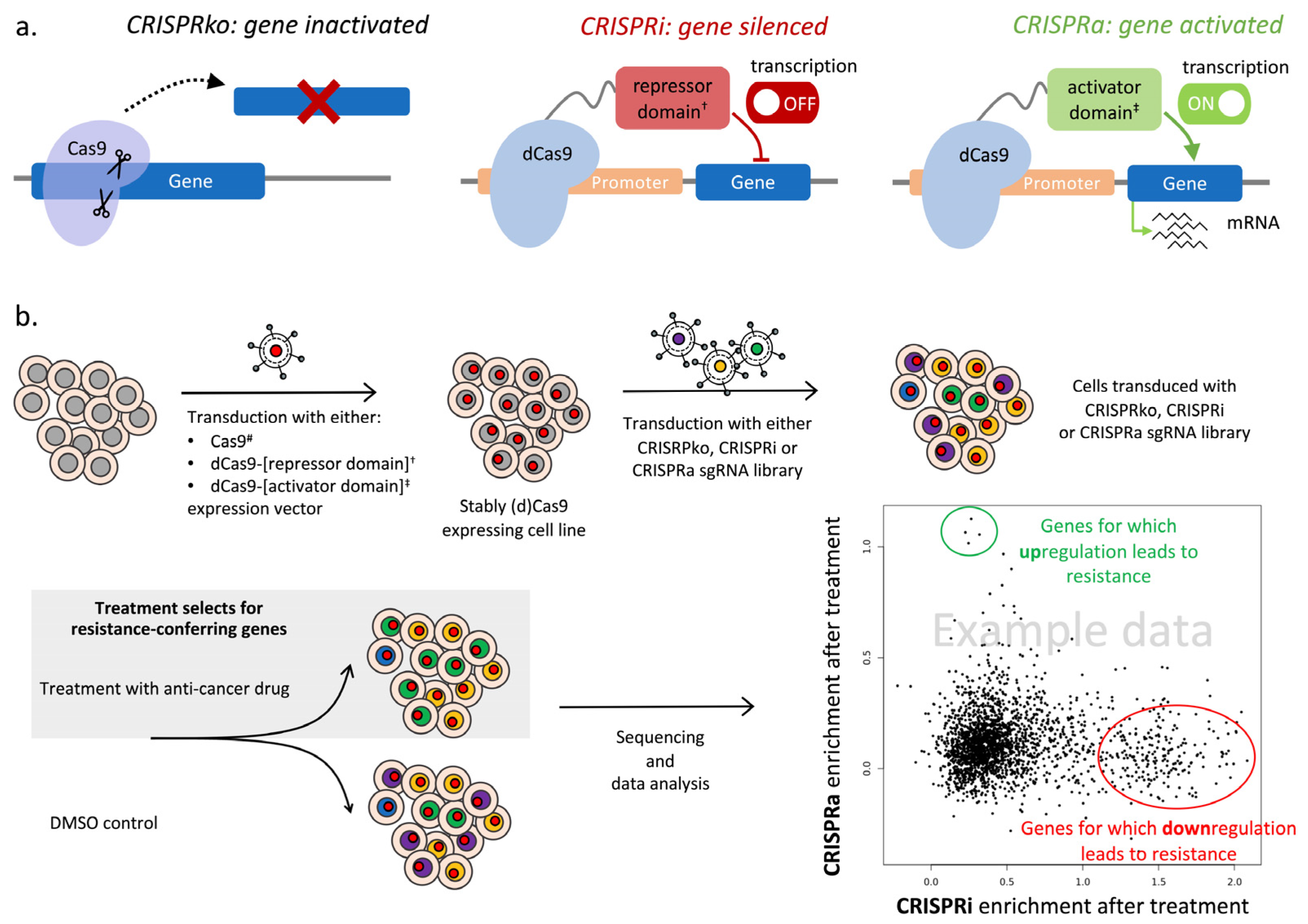
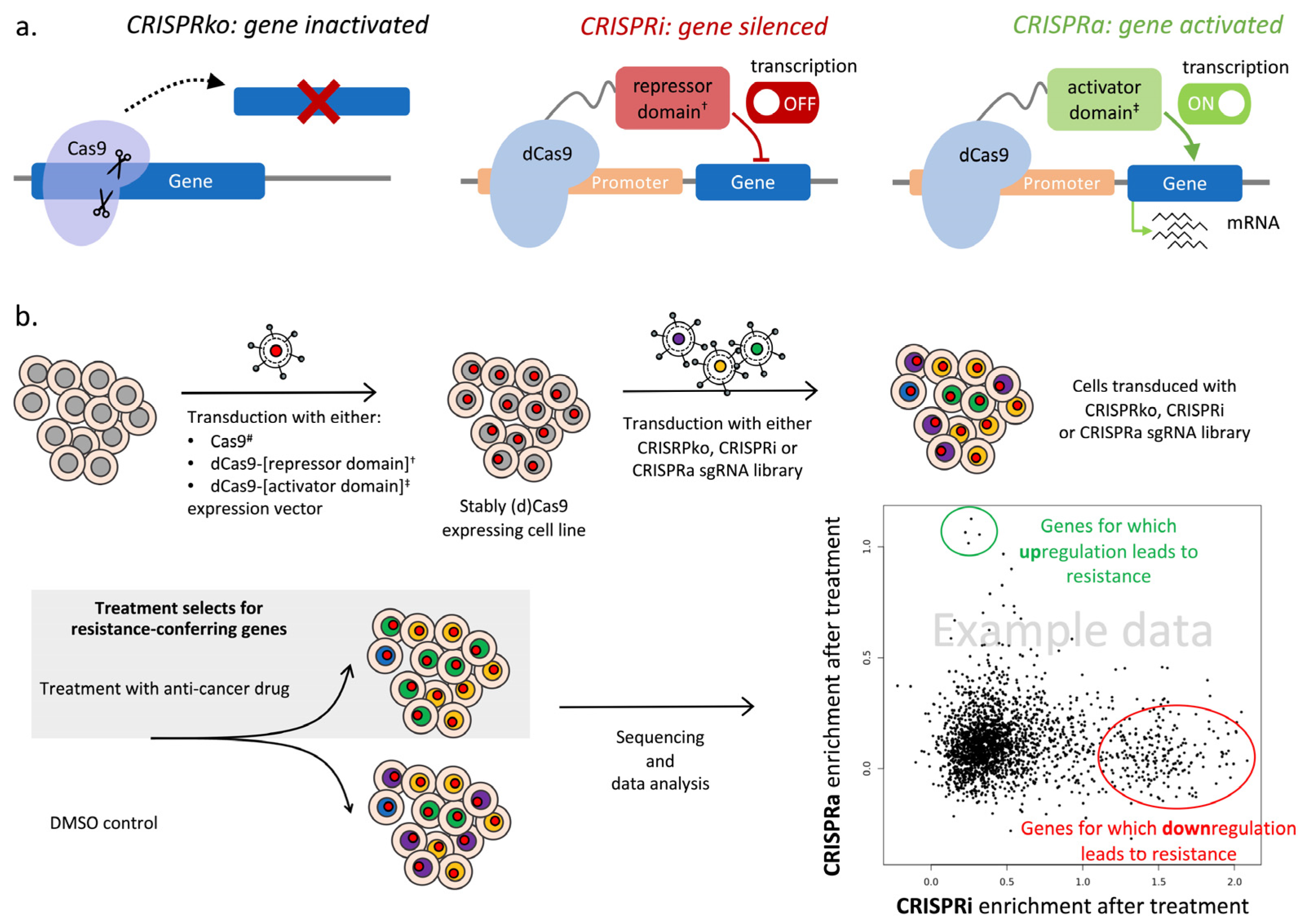
4.1. CRISPR/Cas9 Knockout Screens to Identify Genes and Pathways That Confer Drug Resistance
4.2. CRISPR Interference (CRISPRi) and CRISPR Activation (CRISPRa) Screens to Reveal Gene Expression Modulations That Confer Drug Resistance
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Muller, P.; Ji, Y. Innovative trial design in precision oncology. Semin. Cancer Biol. 2022, 84, 284–292. [Google Scholar] [CrossRef]
- Ye, J.C.; Horne, S.; Zhang, J.Z.; Jackson, L.; Heng, H.H. Therapy induced genome chaos: A novel mechanism of rapid cancer drug resistance. Front. Cell Dev. Biol. 2021, 9, 676344. [Google Scholar] [CrossRef]
- Heng, H.H.; Stevens, J.B.; Lawrenson, L.; Liu, G.; Ye, K.J.; Bremer, S.W.; Ye, C.J. Patterns of genome dynamics and cancer evolution. Cell Oncol. 2008, 30, 513–514. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef]
- Hendel, S.J.; Shoulders, M.D. Directed evolution in mammalian cells. Nat. Methods 2021, 18, 346–357. [Google Scholar] [CrossRef]
- Antony, R.; Emery, C.M.; Sawyer, A.M.; Garraway, L.A. C-RAF mutations confer resistance to RAF inhibitors. Cancer Res. 2013, 73, 4840–4851. [Google Scholar] [CrossRef]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef]
- Rosa, R.; Monteleone, F.; Zambrano, N.; Bianco, R. In vitro and in vivo models for analysis of resistance to anticancer molecular therapies. Curr. Med. Chem. 2014, 21, 1595–1606. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, K.; Zhao, Y.; Li, Y.; Su, W.; Li, S. Construction and applications of mammalian cell-based DNA-encoded peptide/protein libraries. ACS Synth. Biol. 2023, 12, 1874–1888. [Google Scholar] [CrossRef]
- Dobner, J.; Ramachandran, H.; Rossi, A. Genome editing in translational medicine: An inventory. Front. Biosci. (Landmark Ed.) 2022, 27, 241. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Kampmann, M. Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem. Commun. 2017, 53, 7162–7167. [Google Scholar] [CrossRef]
- Tsujino, T.; Komura, K.; Inamoto, T.; Azuma, H. CRISPR screen contributes to novel target discovery in prostate cancer. Int. J. Mol. Sci. 2021, 22, 12777. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, B. A review on CRISPR/Cas: A versatile tool for cancer screening, diagnosis, and clinic treatment. Funct. Integr. Genom. 2023, 23, 182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear receptors: Recent drug discovery for cancer therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schioth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent advances in prostate cancer treatment and drug discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Akamatsu, S.; Tsukahara, S.; Nagakawa, S.; Matsumoto, T.; Eto, M. Androgen receptor mutations for precision medicine in prostate cancer. Endocr. Relat. Cancer 2022, 29, R143–R155. [Google Scholar] [CrossRef]
- Branford, S.; Hughes, T.P. Mutational analysis in chronic myeloid leukemia: When and what to do? Curr. Opin. Hematol. 2011, 18, 111–116. [Google Scholar] [CrossRef]
- Forde, P.M.; Ettinger, D.S. Managing acquired resistance in EGFR-mutated non-small cell lung cancer. Clin. Adv. Hematol. Oncol. 2015, 13, 528–532. [Google Scholar]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.; Goh, B.C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef]
- Wu, L.; Ke, L.; Zhang, Z.; Yu, J.; Meng, X. Development of EGFR TKIs and options to manage resistance of third-generation EGFR TKI osimertinib: Conventional ways and immune checkpoint inhibitors. Front. Oncol. 2020, 10, 602762. [Google Scholar] [CrossRef]
- Sharma, B.; Singh, V.J.; Chawla, P.A. Epidermal growth factor receptor inhibitors as potential anticancer agents: An update of recent progress. Bioorganic Chem. 2021, 116, 105393. [Google Scholar] [CrossRef]
- Yu, Z.; Boggon, T.J.; Kobayashi, S.; Jin, C.; Ma, P.C.; Dowlati, A.; Kern, J.A.; Tenen, D.G.; Halmos, B. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res. 2007, 67, 10417–10427. [Google Scholar] [CrossRef]
- Yenerall, P.; Kollipara, R.K.; Avila, K.; Peyton, M.; Eide, C.A.; Bottomly, D.; McWeeney, S.K.; Liu, Y.; Westover, K.D.; Druker, B.J.; et al. Lentiviral-driven discovery of cancer drug resistance mutations. Cancer Res. 2021, 81, 4685–4695. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef]
- Barata, P.C.; Mendiratta, P.; Heald, B.; Klek, S.; Grivas, P.; Sohal, D.P.S.; Garcia, J.A. Targeted next-generation sequencing in men with metastatic prostate cancer: A pilot study. Target. Oncol. 2018, 13, 495–500. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Haendler, B. From omics to multi-omics approaches for in-depth analysis of the molecular mechanisms of prostate cancer. Int. J. Mol. Sci. 2022, 23, 6281. [Google Scholar] [CrossRef]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Bloom, J.D.; Meyer, M.M.; Meinhold, P.; Otey, C.R.; MacMillan, D.; Arnold, F.H. Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 2005, 15, 447–452. [Google Scholar] [CrossRef]
- Emond, S.; Mondon, P.; Pizzut-Serin, S.; Douchy, L.; Crozet, F.; Bouayadi, K.; Kharrat, H.; Potocki-Veronese, G.; Monsan, P.; Remaud-Simeon, M. A novel random mutagenesis approach using human mutagenic DNA polymerases to generate enzyme variant libraries. Protein Eng. Des. Sel. 2008, 21, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tee, K.L.; Wong, T.S. Polishing the craft of genetic diversity creation in directed evolution. Biotechnol. Adv. 2013, 31, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Vanhercke, T.; Ampe, C.; Tirry, L.; Denolf, P. Reducing mutational bias in random protein libraries. Anal. Biochem. 2005, 339, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Shivange, A.V.; Marienhagen, J.; Mundhada, H.; Schenk, A.; Schwaneberg, U. Advances in generating functional diversity for directed protein evolution. Curr. Opin. Chem. Biol. 2009, 13, 19–25. [Google Scholar] [CrossRef]
- Rasila, T.S.; Pajunen, M.I.; Savilahti, H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal. Biochem. 2009, 388, 71–80. [Google Scholar] [CrossRef] [PubMed]
- McCullum, E.O.; Williams, B.A.; Zhang, J.; Chaput, J.C. Random mutagenesis by error-prone PCR. Methods Mol. Biol. 2010, 634, 103–109. [Google Scholar] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Lee, S.S.; Kim, C.H.; Yoo, Y.D.; Lee, J.C. The role of MET activation in determining the sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors. Mol. Cancer Res. 2009, 7, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, Z.; Wang, J.; Zhang, M.; Li, Z.; Wang, S.; Dong, B.; Zhang, C.; Gao, J.; Shen, L. Mouse avatar models of esophageal squamous cell carcinoma proved the potential for EGFR-TKI afatinib and uncovered Src family kinases involved in acquired resistance. J. Hematol. Oncol. 2018, 11, 109. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of acquired BRAF inhibitor resistance in melanoma: A systematic review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Samarkina, A.; Youssef, M.K.; Ostano, P.; Ghosh, S.; Ma, M.; Tassone, B.; Proust, T.; Chiorino, G.; Levesque, M.P.; Goruppi, S.; et al. Androgen receptor is a determinant of melanoma targeted drug resistance. Nat. Commun. 2023, 14, 6498. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Qin, T.; You, L.; Lu, F.; Hu, D.; Xiao, R.; Qin, X.; Guo, E.; Yang, B.; et al. AZD5153 reverses palbociclib resistance in ovarian cancer by inhibiting cell cycle-related proteins and the MAPK/PI3K-AKT pathway. Cancer Lett. 2022, 528, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Wenwen, W.; Ohta, T.; Hayashi, S.I. Molecular targeted drugs resistance impairs double-strand break repair and sensitizes ER-positive breast cancer to PARP inhibitors. Breast Cancer 2022, 29, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.R.; Smith, A.R.; Zheng, G. Overcoming cancer drug resistance utilizing PROTAC technology. Front. Cell Dev. Biol. 2022, 10, 872729. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, J.; Kim, J.M. Targeted protein degradation to overcome resistance in cancer therapies: PROTAC and N-degron pathway. Biomedicines 2022, 10, 2100. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Palladino, C.; Thienger, P.; Britschgi, A.; Heichinger, C.; Berrera, M.; Julien-Laferriere, A.; Roudnicky, F.; Kam-Thong, T.; Bischoff, J.R.; et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chem. Biol. 2019, 14, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Kurimchak, A.M.; Herrera-Montavez, C.; Montserrat-Sangra, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal 2022, 15, eabn2707. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef]
- Karkampouna, S.; La Manna, F.; Benjak, A.; Kiener, M.; De Menna, M.; Zoni, E.; Grosjean, J.; Klima, I.; Garofoli, A.; Bolis, M.; et al. Patient-derived xenografts and organoids model therapy response in prostate cancer. Nat. Commun. 2021, 12, 1117. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Wang, P.; Wongvipat, J.; Karthaus, W.R.; Abida, W.; Armenia, J.; Rockowitz, S.; Drier, Y.; Bernstein, B.E.; Long, H.W.; et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. eLife 2017, 6, e27861. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef] [PubMed]
- Dharanipragada, P.; Zhang, X.; Liu, S.; Lomeli, S.H.; Hong, A.; Wang, Y.; Yang, Z.; Lo, K.Z.; Vega-Crespo, A.; Ribas, A.; et al. Blocking genomic instability prevents acquired resistance to MAPK inhibitor therapy in melanoma. Cancer Discov. 2023, 13, 880–909. [Google Scholar] [CrossRef] [PubMed]
- Tovari, J.; Vari-Mezo, D.; Surguta, S.E.; Ladanyi, A.; Kigyos, A.; Cserepes, M. Evolving acquired vemurafenib resistance in a BRAF V600E mutant melanoma PDTX model to reveal new potential targets. Cells 2023, 12, 1919. [Google Scholar] [CrossRef] [PubMed]
- Schueler, J.; Tschuch, C.; Klingner, K.; Bug, D.; Peille, A.L.; de Koning, L.; Oswald, E.; Klett, H.; Sommergruber, W. Induction of acquired resistance towards EGFR inhibitor gefitinib in a patient-derived xenograft model of non-small cell lung cancer and subsequent molecular characterization. Cells 2019, 8, 740. [Google Scholar] [CrossRef] [PubMed]
- Manas, A.; Aaltonen, K.; Andersson, N.; Hansson, K.; Adamska, A.; Seger, A.; Yasui, H.; van den Bos, H.; Radke, K.; Esfandyari, J.; et al. Clinically relevant treatment of PDX models reveals patterns of neuroblastoma chemoresistance. Sci. Adv. 2022, 8, eabq4617. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, J.; Li, X.; Niu, N.; Liu, Y.; Hajek, R.A.; Peng, G.; Westin, S.; Sood, A.K.; Liu, J. Targeting polyploid giant cancer cells potentiates a therapeutic response and overcomes resistance to PARP inhibitors in ovarian cancer. Sci. Adv. 2023, 9, eadf7195. [Google Scholar] [CrossRef]
- West, J.; Newton, P.K. Cellular interactions constrain tumor growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1918–1923. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.L.; Harle, A. Deciphering tumour heterogeneity: From tissue to liquid biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Schenck, R.O.; Gatenbee, C.; Robertson-Tessi, M.; Anderson, A.R.A. Normal tissue architecture determines the evolutionary course of cancer. Nat. Commun. 2021, 12, 2060. [Google Scholar] [CrossRef]
- Melnikov, A.; Rogov, P.; Wang, L.; Gnirke, A.; Mikkelsen, T.S. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014, 42, e112. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Front. Genet. 2023, 14, 1087267. [Google Scholar] [CrossRef]
- Persky, N.S.; Hernandez, D.; Do Carmo, M.; Brenan, L.; Cohen, O.; Kitajima, S.; Nayar, U.; Walker, A.; Pantel, S.; Lee, Y.; et al. Defining the landscape of ATP-competitive inhibitor resistance residues in protein kinases. Nat. Struct. Mol. Biol. 2020, 27, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wang, Y.; Wu, G.; Wang, Z.; Shi, Z.; Liu, C.; Wang, C.; Yi, M.; Niu, C.; Duan, S.; et al. Defining the sensitivity landscape of EGFR variants to tyrosine kinase inhibitors. Transl. Res. 2023, 255, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Li, Y.; Zhang, K.; Fan, Y.; Li, B.; Su, W.; Li, S. piggyBac-mediated genomic integration of linear dsDNA-based library for deep mutational scanning in mammalian cells. Cell Mol. Life Sci. 2023, 80, 321. [Google Scholar] [CrossRef]
- Christensen, S.; Wernersson, C.; Andre, I. Facile method for high-throughput identification of stabilizing mutations. J. Mol. Biol. 2023, 435, 168209. [Google Scholar] [CrossRef]
- Kaplanis, J.; Akawi, N.; Gallone, G.; McRae, J.F.; Prigmore, E.; Wright, C.F.; Fitzpatrick, D.R.; Firth, H.V.; Barrett, J.C.; Hurles, M.E.; et al. Exome-wide assessment of the functional impact and pathogenicity of multinucleotide mutations. Genome Res. 2019, 29, 1047–1056. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Yip, B.H. Recent advances in CRISPR/Cas9 delivery strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Balon, K.; Sheriff, A.; Jackow, J.; Laczmanski, L. Targeting cancer with CRISPR/Cas9-based therapy. Int. J. Mol. Sci. 2022, 23, 573. [Google Scholar] [CrossRef]
- Lue, N.Z.; Liau, B.B. Base editor screens for in situ mutational scanning at scale. Mol. Cell 2023, 83, 2167–2187. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Kim, D.; Lim, K.; Kim, S.T.; Yoon, S.H.; Kim, K.; Ryu, S.M.; Kim, J.S. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 2017, 35, 475–480. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Sousa, A.A.; Walton, R.T.; Tak, Y.E.; Hsu, J.Y.; Clement, K.; Welch, M.M.; Horng, J.E.; Malagon-Lopez, J.; Scarfo, I.; et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 2019, 37, 276–282. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Sola-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, B.; Chen, L.; Xie, L.; Yu, W.; Wang, Y.; Li, L.; Yin, S.; Yang, L.; Hu, H.; et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020, 38, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Xue, N.; Liu, X.; Zhang, D.; Wu, Y.; Zhong, Y.; Wang, J.; Fan, W.; Jiang, H.; Zhu, B.; Ge, X.; et al. Improving adenine and dual base editors through introduction of TadA-8e and Rad51DBD. Nat. Commun. 2023, 14, 1224. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Xue, N.; Hong, M.; Zhang, X.; Zhang, D.; Yang, J.; Bai, S.; Huang, Y.; Meng, H.; et al. Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. 2023, 19, 101–110. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021, 39, 35–40. [Google Scholar] [CrossRef]
- Sahu, S.; Poplawska, M.; Lim, S.H.; Dutta, D. CRISPR-based precision medicine for hematologic disorders: Advancements, challenges, and prospects. Life Sci. 2023, 333, 122165. [Google Scholar]
- Kweon, J.; Jang, A.H.; Shin, H.R.; See, J.E.; Lee, W.; Lee, J.W.; Chang, S.; Kim, K.; Kim, Y. A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene 2020, 39, 30–35. [Google Scholar] [CrossRef]
- Hanna, R.E.; Hegde, M.; Fagre, C.R.; DeWeirdt, P.C.; Sangree, A.K.; Szegletes, Z.; Griffith, A.; Feeley, M.N.; Sanson, K.R.; Baidi, Y.; et al. Massively parallel assessment of human variants with base editor screens. Cell 2021, 184, 1064–1080.e20. [Google Scholar] [CrossRef]
- Sangree, A.K.; Griffith, A.L.; Szegletes, Z.M.; Roy, P.; DeWeirdt, P.C.; Hegde, M.; McGee, A.V.; Hanna, R.E.; Doench, J.G. Benchmarking of SpCas9 variants enables deeper base editor screens of BRCA1 and BCL2. Nat. Commun. 2022, 13, 1318. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.; Cooper, S.; Strauss, M.E.; Karakoc, E.; Bhosle, S.; Goncalves, E.; Picco, G.; Burgold, T.; Cattaneo, C.M.; Veninga, V.; et al. Base editing screens map mutations affecting interferon-gamma signaling in cancer. Cancer Cell 2023, 41, 288–303.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, J.; Huang, S.; Li, M.; Yu, W.; Zhao, Y.; Guo, J.; Zhang, P.; Huang, X.; Qiao, Y. Functional phosphoproteomics in cancer chemoresistance using CRISPR-mediated base editors. Adv. Sci. 2022, 9, e2200717. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, B. Mathematical modeling and computational prediction of cancer drug resistance. Brief. Bioinform. 2018, 19, 1382–1399. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Molecular dynamics simulations of biomolecules. Acc. Chem. Res. 2002, 35, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Potts, S.J.; Kantarjian, H.; Cortes, J.; Giles, F.; Albitar, M. Molecular basis explanation for imatinib resistance of BCR-ABL due to T315I and P-loop mutations from molecular dynamics simulations. Cancer 2008, 112, 1744–1753. [Google Scholar] [CrossRef]
- Ikemura, S.; Yasuda, H.; Matsumoto, S.; Kamada, M.; Hamamoto, J.; Masuzawa, K.; Kobayashi, K.; Manabe, T.; Arai, D.; Nakachi, I.; et al. Molecular dynamics simulation-guided drug sensitivity prediction for lung cancer with rare EGFR mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 10025–10030. [Google Scholar] [CrossRef]
- Ogunleye, A.Z.; Piyawajanusorn, C.; Goncalves, A.; Ghislat, G.; Ballester, P.J. Interpretable machine learning models to predict the resistance of breast cancer patients to doxorubicin from their microRNA profiles. Adv. Sci. 2022, 9, e2201501. [Google Scholar] [CrossRef]
- Sammut, S.J.; Crispin-Ortuzar, M.; Chin, S.F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629. [Google Scholar] [CrossRef]
- Trebeschi, S.; Drago, S.G.; Birkbak, N.J.; Kurilova, I.; Calin, A.M.; Delli Pizzi, A.; Lalezari, F.; Lambregts, D.M.J.; Rohaan, M.W.; Parmar, C.; et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann. Oncol. 2019, 30, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Sakellaropoulos, T.; Vougas, K.; Narang, S.; Koinis, F.; Kotsinas, A.; Polyzos, A.; Moss, T.J.; Piha-Paul, S.; Zhou, H.; Kardala, E.; et al. A deep learning framework for predicting response to therapy in cancer. Cell Rep. 2019, 29, 3367–3373.e4. [Google Scholar] [CrossRef] [PubMed]
- Rafique, R.; Islam, S.M.R.; Kazi, J.U. Machine learning in the prediction of cancer therapy. Comput. Struct. Biotechnol. J. 2021, 19, 4003–4017. [Google Scholar] [CrossRef]
- Baker, R.E.; Pena, J.M.; Jayamohan, J.; Jerusalem, A. Mechanistic models versus machine learning, a fight worth fighting for the biological community? Biol. Lett. 2018, 14, 20170660. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Basit, S.A.; Shamsi, J.A.; Fan, X.; Nawaz, M.; Yan, H.; Alam, T. Machine learning based personalized drug response prediction for lung cancer patients. Sci. Rep. 2022, 12, 18935. [Google Scholar] [CrossRef] [PubMed]
- Alyateem, G.; Wade, H.M.; Bickert, A.A.; Lipsey, C.C.; Mondal, P.; Smith, M.D.; Labib, R.M.; Mock, B.A.; Robey, R.W.; Gottesman, M.M. Use of CRISPR-based screens to identify mechanisms of chemotherapy resistance. Cancer Gene Ther. 2023, 30, 1043–1050. [Google Scholar] [CrossRef]
- Chakravarti, R.; Lenka, S.K.; Gautam, A.; Singh, R.; Ravichandiran, V.; Roy, S.; Ghosh, D. A review on CRISPR-mediated epigenome editing: A future directive for therapeutic management of cancer. Curr. Drug Targets 2022, 23, 836–853. [Google Scholar]
- La Russa, M.F.; Qi, L.S. The new state of the art: Cas9 for gene activation and repression. Mol. Cell. Biol. 2015, 35, 3800–3809. [Google Scholar] [CrossRef]
- Li, M.; Sun, J.; Shi, G. Application of CRISPR screen in mechanistic studies of tumor development, tumor drug resistance, and tumor immunotherapy. Front. Cell Dev. Biol. 2023, 11, 1220376. [Google Scholar] [CrossRef]
- McLean, B.; Istadi, A.; Clack, T.; Vankan, M.; Schramek, D.; Neely, G.G.; Pajic, M. A CRISPR path to finding vulnerabilities and solving drug resistance: Targeting the diverse cancer landscape and its ecosystem. Adv. Genet. 2022, 3, 2200014. [Google Scholar] [CrossRef]
- Shirani-Bidabadi, S.; Tabatabaee, A.; Tavazohi, N.; Hariri, A.; Aref, A.R.; Zarrabi, A.; Casarcia, N.; Bishayee, A.; Mirian, M. CRISPR technology: A versatile tool to model, screen, and reverse drug resistance in cancer. Eur. J. Cell Biol. 2023, 102, 151299. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Sanchez, A.; Pogacar, Z.; Vecchione, L. Importance of genetic screens in precision oncology. ESMO Open 2019, 4, e000505. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, M.J.; Bloom, J.S.; Day, L.; Siegel, J.J.; Kosuri, S.; Kruglyak, L. Highly parallel genome variant engineering with CRISPR-Cas9. Nat. Genet. 2018, 50, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Wright, J.; Zheng, K.; Shalem, O.; Fontanillas, P.; Joung, J.; Cheng, C.; Regev, A.; Zhang, F. High-resolution interrogation of functional elements in the noncoding genome. Science 2016, 353, 1545–1549. [Google Scholar] [CrossRef]
- Hany, D.; Vafeiadou, V.; Picard, D. CRISPR-Cas9 screen reveals a role of purine synthesis for estrogen receptor alpha activity and tamoxifen resistance of breast cancer cells. Sci. Adv. 2023, 9, eadd3685. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Cai, Y.; Xu, G.; Wu, F.; Michelini, F.; Chan, C.; Qu, X.; Selenica, P.; Ladewig, E.; Castel, P.; Cheng, Y.; et al. Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit the sensitivity to PI3Kα inhibitors. Cancer Res. 2021, 81, 2470–2480. [Google Scholar] [CrossRef]
- Palit, S.A.; Vis, D.; Stelloo, S.; Lieftink, C.; Prekovic, S.; Bekers, E.; Hofland, I.; Sustic, T.; Wolters, L.; Beijersbergen, R.; et al. TLE3 loss confers AR inhibitor resistance by facilitating GR-mediated human prostate cancer cell growth. eLife 2019, 8, e47430. [Google Scholar] [CrossRef]
- Haldrup, J.; Weiss, S.; Schmidt, L.; Sorensen, K.D. Investigation of enzalutamide, docetaxel, and cabazitaxel resistance in the castration resistant prostate cancer cell line C4 using genome-wide CRISPR/Cas9 screening. Sci. Rep. 2023, 13, 9043. [Google Scholar] [CrossRef] [PubMed]
- Palit, S.A.L.; van Dorp, J.; Vis, D.; Lieftink, C.; Linder, S.; Beijersbergen, R.; Bergman, A.M.; Zwart, W.; van der Heijden, M.S. A kinome-centered CRISPR-Cas9 screen identifies activated BRAF to modulate enzalutamide resistance with potential therapeutic implications in BRAF-mutated prostate cancer. Sci. Rep. 2021, 11, 13683. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Y.; He, D.; Jones, K.M.; Tang, S.; Allison, D.B.; Zhang, Y.; Chen, J.; Zhang, Q.; Wang, X.; et al. A kinome-wide CRISPR screen identifies CK1α as a target to overcome enzalutamide resistance of prostate cancer. Cell Rep. Med. 2023, 4, 101015. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat. Commun. 2023, 14, 252. [Google Scholar] [CrossRef]
- Ipsen, M.B.; Sorensen, E.M.G.; Thomsen, E.A.; Weiss, S.; Haldrup, J.; Dalby, A.; Palmfeldt, J.; Bross, P.; Rasmussen, M.; Fredsoe, J.; et al. A genome-wide CRISPR-Cas9 knockout screen identifies novel PARP inhibitor resistance genes in prostate cancer. Oncogene 2022, 41, 4271–4281. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.W.; Serrano-Benitez, A.; Thomas, J.C.; Gupta, V.; Jackson, S.P. Revolutionizing DNA repair research and cancer therapy with CRISPR-Cas screens. Nat. Rev. Mol. Cell Biol. 2023, 24, 477–494. [Google Scholar] [CrossRef]
- Zeng, H.; Castillo-Cabrera, J.; Manser, M.; Lu, B.; Yang, Z.; Strande, V.; Begue, D.; Zamponi, R.; Qiu, S.; Sigoillot, F.; et al. Genome-wide CRISPR screening reveals genetic modifiers of mutant EGFR dependence in human NSCLC. eLife 2019, 8, e50223. [Google Scholar] [CrossRef]
- Lee, J.; Choi, A.; Cho, S.Y.; Jun, Y.; Na, D.; Lee, A.; Jang, G.; Kwon, J.Y.; Kim, J.; Lee, S.; et al. Genome-scale CRISPR screening identifies cell cycle and protein ubiquitination processes as druggable targets for erlotinib-resistant lung cancer. Mol. Oncol. 2021, 15, 487–502. [Google Scholar] [CrossRef]
- Yu, C.; Luo, D.; Yu, J.; Zhang, M.; Zheng, X.; Xu, G.; Wang, J.; Wang, H.; Xu, Y.; Jiang, K.; et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 2022, 41, 191–203. [Google Scholar] [CrossRef]
- Jin, H.; Shi, Y.; Lv, Y.; Yuan, S.; Ramirez, C.F.A.; Lieftink, C.; Wang, L.; Wang, S.; Wang, C.; Dias, M.H.; et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021, 595, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liang, S.Q.; Yang, H.; Xu, D.; Bruggmann, R.; Gao, Y.; Deng, H.; Berezowska, S.; Hall, S.R.R.; Marti, T.M.; et al. CRISPR-mediated kinome editing prioritizes a synergistic combination therapy for FGFR1-amplified lung cancer. Cancer Res. 2021, 81, 3121–3133. [Google Scholar] [CrossRef]
- Drosos, Y.; Myers, J.A.; Xu, B.; Mathias, K.M.; Beane, E.C.; Radko-Juettner, S.; Mobley, R.J.; Larsen, M.E.; Piccioni, F.; Ma, X.; et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol. Cell 2022, 82, 2472–2489 e8. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Geer, M.J.; Guo, X.; Dolgalev, I.; Sanjana, N.E.; Neel, B.G. Genome-wide CRISPR/Cas9 screens reveal shared and cell-specific mechanisms of resistance to SHP2 inhibition. J. Exp. Med. 2023, 220, e20221563. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, R.; Matthews, G.M.; Gandolfi, S.; de Matos Simoes, R.; Buckley, D.L.; Raja Vora, J.; Sievers, Q.L.; Bruggenthies, J.B.; Dashevsky, O.; Poarch, H.; et al. Functional genomics identify distinct and overlapping genes mediating resistance to different classes of heterobifunctional degraders of oncoproteins. Cell Rep. 2021, 34, 108532. [Google Scholar] [CrossRef] [PubMed]
- Waldeck, K.; Van Zuylekom, J.; Cullinane, C.; Gulati, T.; Simpson, K.J.; Tothill, R.W.; Blyth, B.; Hicks, R.J. A genome-wide CRISPR/Cas9 screen identifies DNA-PK as a sensitiser to 177Lutetium-DOTA-octreotate radionuclide therapy. Theranostics 2023, 13, 4745–4761. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.; Roudnicky, F. Functional genomics for cancer drug target discovery. Cancer Cell 2020, 38, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Liu, X.; Li, Z.; Dai, X. The application of CRISPR/Cas9 technology for cancer Immunotherapy: Current status and problems. Front. Oncol. 2021, 11, 704999. [Google Scholar] [CrossRef]
- Li, Y.R.; Lyu, Z.; Tian, Y.; Fang, Y.; Zhu, Y.; Chen, Y.; Yang, L. Advancements in CRISPR screens for the development of cancer immunotherapy strategies. Mol. Ther. Oncolytics 2023, 31, 100733. [Google Scholar] [CrossRef]
- Scheidmann, M.C.; Castro-Giner, F.; Strittmatter, K.; Krol, I.; Paasinen-Sohns, A.; Scherrer, R.; Donato, C.; Gkountela, S.; Szczerba, B.M.; Diamantopoulou, Z.; et al. An in vivo CRISPR screen identifies stepwise genetic dependencies of metastatic progression. Cancer Res. 2022, 82, 681–694. [Google Scholar] [CrossRef]
- Dai, M.; Yan, G.; Wang, N.; Daliah, G.; Edick, A.M.; Poulet, S.; Boudreault, J.; Ali, S.; Burgos, S.A.; Lebrun, J.J. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021, 12, 3055. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Amendola, M.; Brusson, M.; Miccio, A. CRISPRthripsis: The risk of CRISPR/Cas9-induced chromothripsis in gene therapy. Stem Cells Transl. Med. 2022, 11, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Chen, G.Q.; Li, X.F.; Wang, M.; Liu, K.M.; Yang, X.Y.; Liu, S.C.; Feng, Y.L.; Liu, P.Y.; Lin, H.; et al. Small extrachromosomal circular DNA harboring targeted tumor suppressor gene mutations supports intratumor heterogeneity in mouse liver cancer induced by multiplexed CRISPR/Cas9. Genome Med. 2023, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; Durin, M.A.; Thomas, R.; Moralli, D.; O’Cathail, S.M.; Tomlinson, I.; Green, C.M.; Lewis, A. CRISPR-Cas9 causes chromosomal instability and rearrangements in cancer cell lines, detectable by cytogenetic methods. CRISPR J. 2019, 2, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Palenikova, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Castanon, O.; Said, K.; Volf, V.; Khoshakhlagh, P.; Hornick, A.; Ferreira, R.; Wu, C.T.; Guell, M.; Garg, S.; et al. Enabling large-scale genome editing at repetitive elements by reducing DNA nicking. Nucleic Acids Res. 2020, 48, 5183–5195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Cai, R.; Lv, R.; Shi, X.; Yang, G.; Jin, J. CRISPR/dCas9 tools: Epigenetic mechanism and application in gene transcriptional regulation. Int. J. Mol. Sci. 2023, 24, 14865. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]
- Alerasool, N.; Segal, D.; Lee, H.; Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods 2020, 17, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; PR Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef] [PubMed]
- le Sage, C.; Lawo, S.; Panicker, P.; Scales, T.M.E.; Rahman, S.A.; Little, A.S.; McCarthy, N.J.; Moore, J.D.; Cross, B.C.S. Dual direction CRISPR transcriptional regulation screening uncovers gene networks driving drug resistance. Sci. Rep. 2017, 7, 17693. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.J.H.; Wong, J.H.; El Farran, C.; Tan, B.X.; Coffill, C.R.; Loh, Y.H.; Lane, D.; Arumugam, P. Identification of pathways modulating vemurafenib resistance in melanoma cells via a genome-wide CRISPR/Cas9 screen. G3 2021, 11, jkaa069. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Luo, F.; Cohen, O.; Goodale, A.B.; Lee, Y.; Pantel, S.; Bagul, M.; Piccioni, F.; Root, D.E.; Garraway, L.A.; et al. A functional landscape of resistance to MEK1/2 and CDK4/6 inhibition in NRAS-mutant melanoma. Cancer Res. 2019, 79, 2352–2366. [Google Scholar] [CrossRef] [PubMed]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Quiceno, L.G.; Chen, T.; Kim, T.J.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER stress signaling promotes the survival of cancer “persister cells” Tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 2018, 78, 1044–1057. [Google Scholar] [CrossRef]
- Kabir, S.; Cidado, J.; Andersen, C.; Dick, C.; Lin, P.C.; Mitros, T.; Ma, H.; Baik, S.H.; Belmonte, M.A.; Drew, L.; et al. The CUL5 ubiquitin ligase complex mediates resistance to CDK9 and MCL1 inhibitors in lung cancer cells. eLife 2019, 8, e44288. [Google Scholar] [CrossRef]
- Ding, Y.; Gong, C.; Huang, D.; Chen, R.; Sui, P.; Lin, K.H.; Liang, G.; Yuan, L.; Xiang, H.; Chen, J.; et al. Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat. Commun. 2018, 9, 4274. [Google Scholar] [CrossRef]
- Zheng, C.; Wei, Y.; Zhang, Q.; Sun, M.; Wang, Y.; Hou, J.; Zhang, P.; Lv, X.; Su, D.; Jiang, Y.; et al. Multiomics analyses reveal DARS1-AS1/YBX1-controlled posttranscriptional circuits promoting glioblastoma tumorigenesis/radioresistance. Sci. Adv. 2023, 9, eadf3984. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Unno, K.; Truica, M.I.; Chalmers, Z.R.; Yoo, Y.A.; Vatapalli, R.; Sagar, V.; Yu, J.; Lysy, B.; Hussain, M.; et al. A genome-wide CRISPR activation screen identifies PRRX2 as a regulator of enzalutamide resistance in prostate cancer. Cancer Res. 2022, 82, 2110–2123. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Sathe, A.; Ebner, B.; Qi, P.; Veltkamp, C.; Gschwend, J.E.; Holm, P.S.; Nawroth, R. Functional genomics identifies predictive markers and clinically actionable resistance mechanisms to CDK4/6 inhibition in bladder cancer. J. Exp. Clin. Cancer Res. 2019, 38, 322. [Google Scholar] [CrossRef]
- Freeman-Cook, K.; Hoffman, R.L.; Miller, N.; Almaden, J.; Chionis, J.; Zhang, Q.; Eisele, K.; Liu, C.; Zhang, C.; Huser, N.; et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 2021, 39, 1404–1421.e11. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Diepstraten, S.T.; Potts, M.A.; Giner, G.; Trezise, S.; Ng, A.P.; Healey, G.; Kane, S.R.; Cooray, A.; Behrens, K.; et al. Generation of a CRISPR activation mouse that enables modelling of aggressive lymphoma and interrogation of venetoclax resistance. Nat. Commun. 2022, 13, 4739. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, S.; Kloetgen, A.; Petroulia, S.; Hockemeyer, K.; Sievers, S.; Tsirigos, A.; Aifantis, I.; Imig, J. Integrative CRISPR activation and small molecule inhibitor screening for lncRNA mediating BRAF inhibitor resistance in melanoma. Biomedicines 2023, 11, 2054. [Google Scholar] [CrossRef]
- Naghizadeh, S.; Mansoori, B.; Mohammadi, A.; Sakhinia, E.; Baradaran, B. Gene silencing strategies in cancer therapy: An update for drug resistance. Curr. Med. Chem. 2019, 26, 6282–6303. [Google Scholar] [CrossRef]
- Weber, J.; Braun, C.J.; Saur, D.; Rad, R. In vivo functional screening for systems-level integrative cancer genomics. Nat. Rev. Cancer 2020, 20, 573–593. [Google Scholar] [CrossRef]
- Drobna-Sledzinska, M.; Mackowska-Maslak, N.; Jaksik, R.; Dabek, P.; Witt, M.; Dawidowska, M. CRISPRi for specific inhibition of miRNA clusters and miRNAs with high sequence homology. Sci. Rep. 2022, 12, 6297. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, J.; Karim, K.; Chen, X.; Wang, T.; Iwama, S.; Carobbio, S.; Keen, P.; Vidal-Puig, A.; Kotter, M.R.; et al. Massively parallel characterization of CRISPR activator efficacy in human induced pluripotent stem cells and neurons. Mol. Cell 2023, 83, 1125–1139 e8. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.S.; Liu, Y.; Gaido, O.E.R.; Konig, M.F.; Mog, B.J.; Shen, L.L.; Aviles-Vazquez, F.; Marin-Gonzalez, A.; Ha, T. Improving the sensitivity of in vivo CRISPR off-target detection with DISCOVER-Seq. Nat. Methods 2023, 20, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Nisar, S.; Mukherjee, S.; Saha, N.; Yarravarapu, N.; Lone, S.N.; Masoodi, T.; Chauhan, R.; Maacha, S.; Bagga, P.; et al. Integration of CRISPR/Cas9 with artificial intelligence for improved cancer therapeutics. J. Transl. Med. 2022, 20, 534. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 2016, 167, 1853–1866 e17. [Google Scholar] [CrossRef]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef]
- Hou, J.; Liang, S.; Xu, C.; Wei, Y.; Wang, Y.; Tan, Y.; Sahni, N.; McGrail, D.J.; Bernatchez, C.; Davies, M.; et al. Single-cell CRISPR immune screens reveal immunological roles of tumor intrinsic factors. NAR Cancer 2022, 4, zcac038. [Google Scholar] [CrossRef]
- Zhou, Y.; Luo, K.; Liang, L.; Chen, M.; He, X. A new Bayesian factor analysis method improves detection of genes and biological processes affected by perturbations in single-cell CRISPR screening. Nat. Methods 2023, 20, 1693–1703. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, X.; Ke, R. Spatial transcriptomics for tumor heterogeneity analysis. Front. Genet. 2022, 13, 906158. [Google Scholar] [CrossRef]
- Ancos-Pintado, R.; Bragado-Garcia, I.; Morales, M.L.; Garcia-Vicente, R.; Arroyo-Barea, A.; Rodriguez-Garcia, A.; Martinez-Lopez, J.; Linares, M.; Hernandez-Sanchez, M. High-throughput CRISPR screening in hematological neoplasms. Cancers 2022, 14, 3612. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bano, S.; Kapse, P.; Kundu, G.C. CRISPR based therapeutics: A new paradigm in cancer precision medicine. Mol. Cancer 2022, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.N.; Wang, K. CRISPR/Cas9: A powerful strategy to improve CAR-T cell persistence. Int. J. Mol. Sci. 2023, 24, 12317. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Xi, J.; Zi, Y.; Chen, M.; Zou, Q.; Zhou, X.; Tang, C. Prospects and challenges of CRISPR/Cas9 gene-editing technology in cancer research. Clin. Genet. 2023, 104, 613–624. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Pleguezuelos-Manzano, C.; Oka, R.; Carrillo, L.; Andersson-Rolf, A.; Boretto, M.; Brunsveld, J.E.; van Boxtel, R.; Beekman, J.M.; et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance 2021, 4, e202000940. [Google Scholar] [CrossRef]
- Iyer, D.N.; Schimmer, A.D.; Chang, H. Applying CRISPR-Cas9 screens to dissect hematological malignancies. Blood Adv. 2023, 7, 2252–2270. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Type of Resistance | Approach | Method |
|---|---|---|
| On-target and/or off-target | Non-systematic | Random mutagenesis |
| Chronic exposure of cells to increased concentrations and characterization of resistant clones | ||
| On-target | Systematic | Deep Mutational Scanning (DMS) |
| CRISPR base editing (BE) | ||
| Computational methods | ||
| Off-target | Systematic | CRISPR knockout (CRISPRko) |
| CRISPR interference (CRISPRi) | ||
| CRISPR activation (CRISPRa) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziubańska-Kusibab, P.J.; Nevedomskaya, E.; Haendler, B. Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 705. https://doi.org/10.3390/ijms25020705
Dziubańska-Kusibab PJ, Nevedomskaya E, Haendler B. Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review. International Journal of Molecular Sciences. 2024; 25(2):705. https://doi.org/10.3390/ijms25020705
Chicago/Turabian StyleDziubańska-Kusibab, Paulina J., Ekaterina Nevedomskaya, and Bernard Haendler. 2024. "Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review" International Journal of Molecular Sciences 25, no. 2: 705. https://doi.org/10.3390/ijms25020705
APA StyleDziubańska-Kusibab, P. J., Nevedomskaya, E., & Haendler, B. (2024). Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review. International Journal of Molecular Sciences, 25(2), 705. https://doi.org/10.3390/ijms25020705