

8-Oxoadenine: A «New» Player of the Oxidative Stress in Mammals?

 , , and
, , and
Abstract
1. Introduction
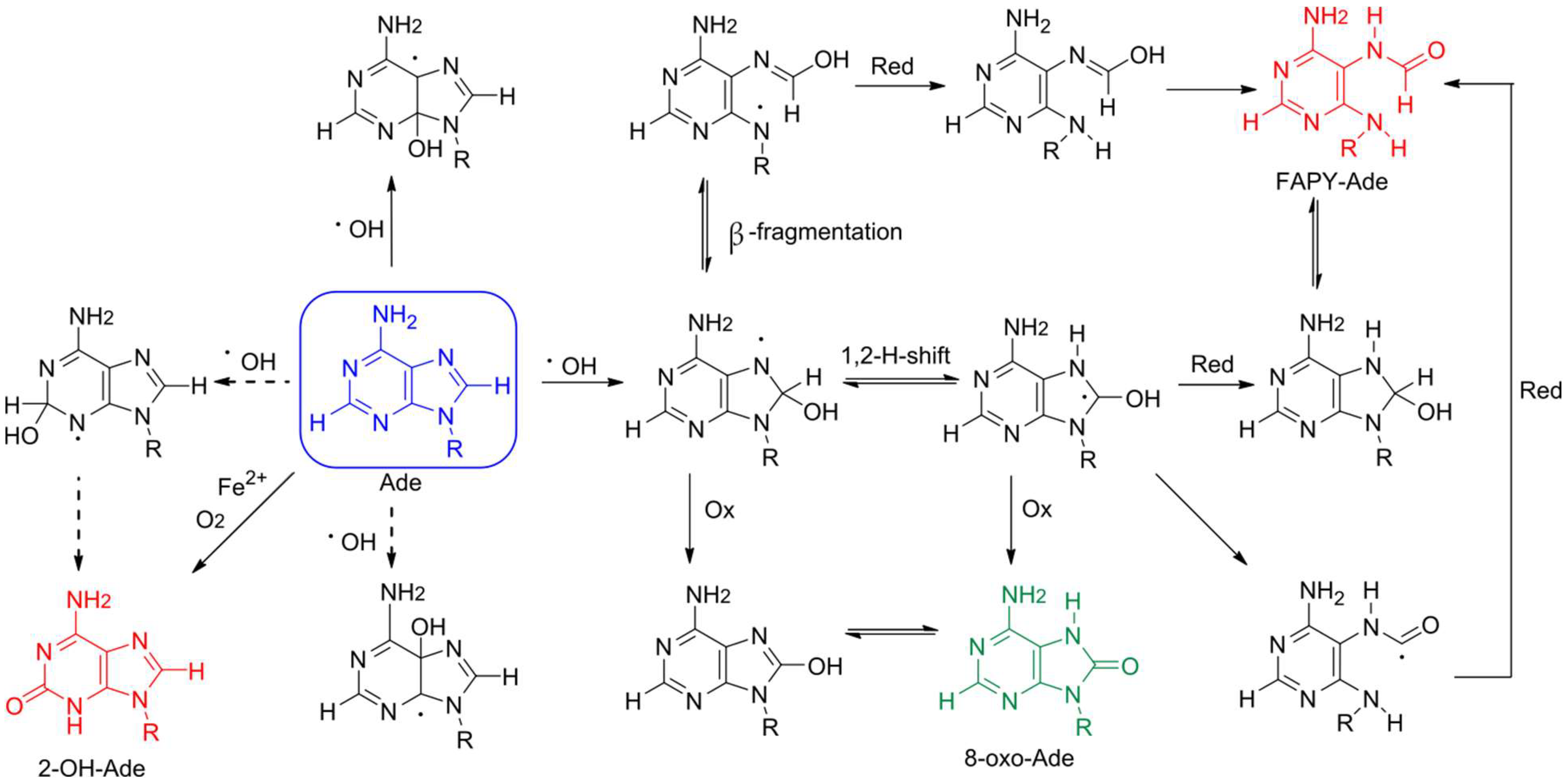
2. Formation of 8-oxoA and Its Derivatives
3. Mutagenic Potential of 8-oxoA
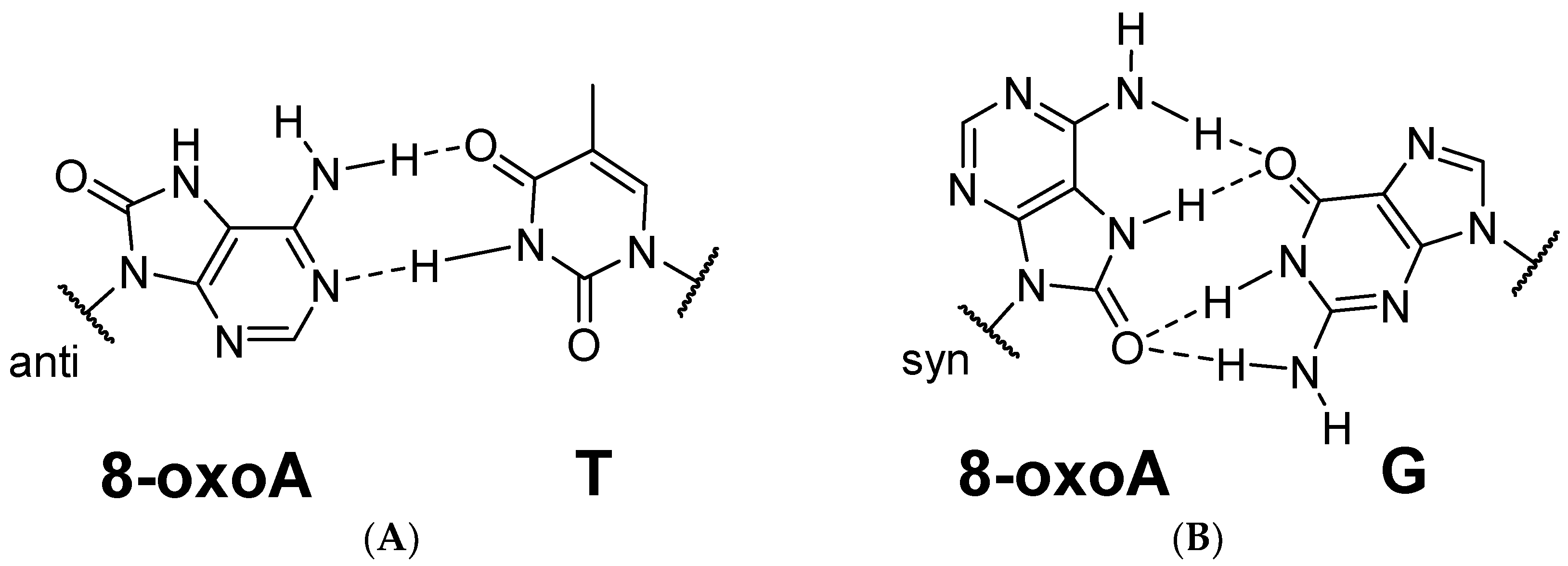
3.1. Pro-Mutagenic Nature of 8-oxoA
3.2. 8-oxoA-Induced Mutagenesis in Prokaryotes
3.3. 8-oxoA-Induced Mutagenesis in Mammalian Cells
3.4. Incorporation of 8-oxodATP
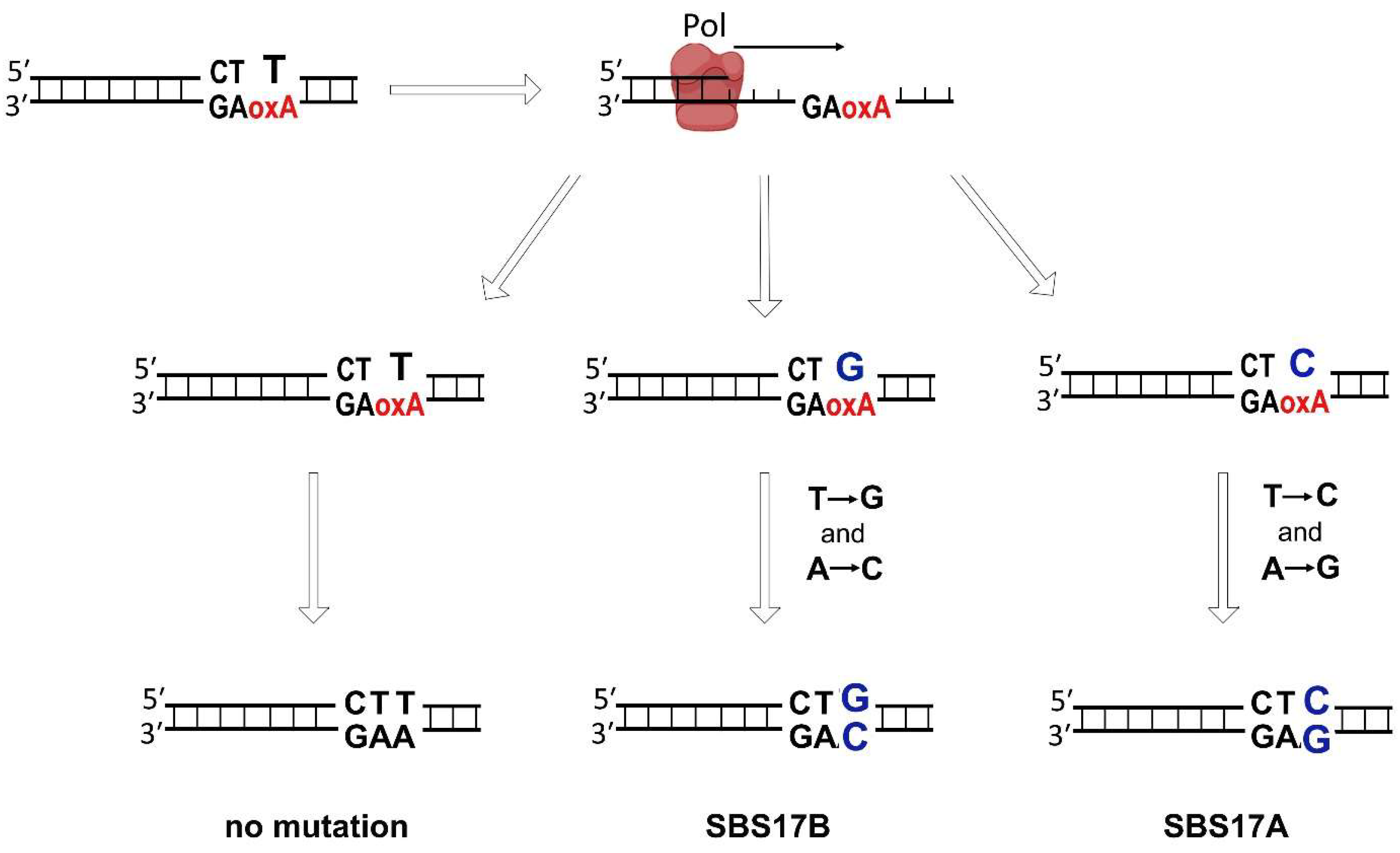
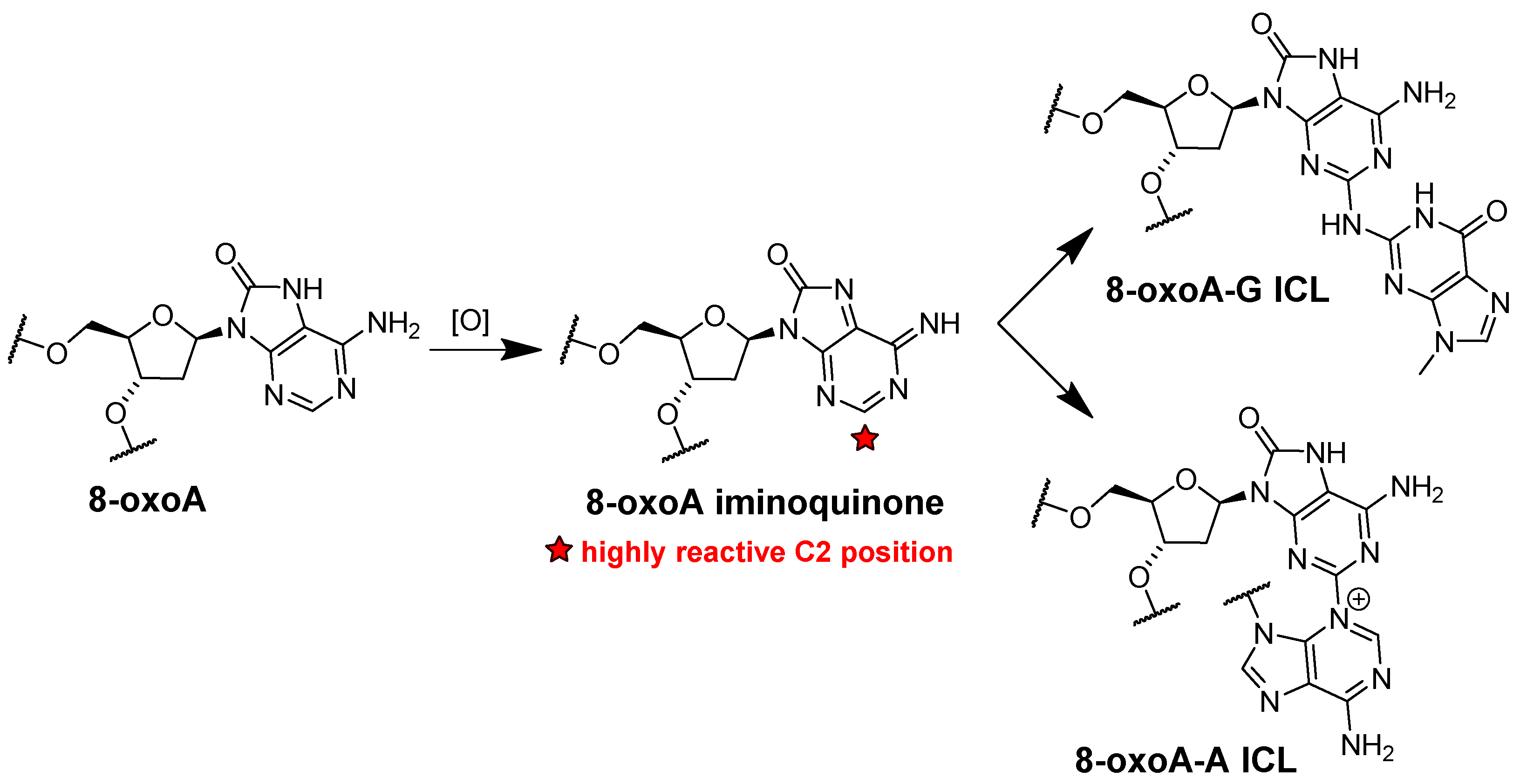
4. The Effects of 8-oxoA Persistence in Genome
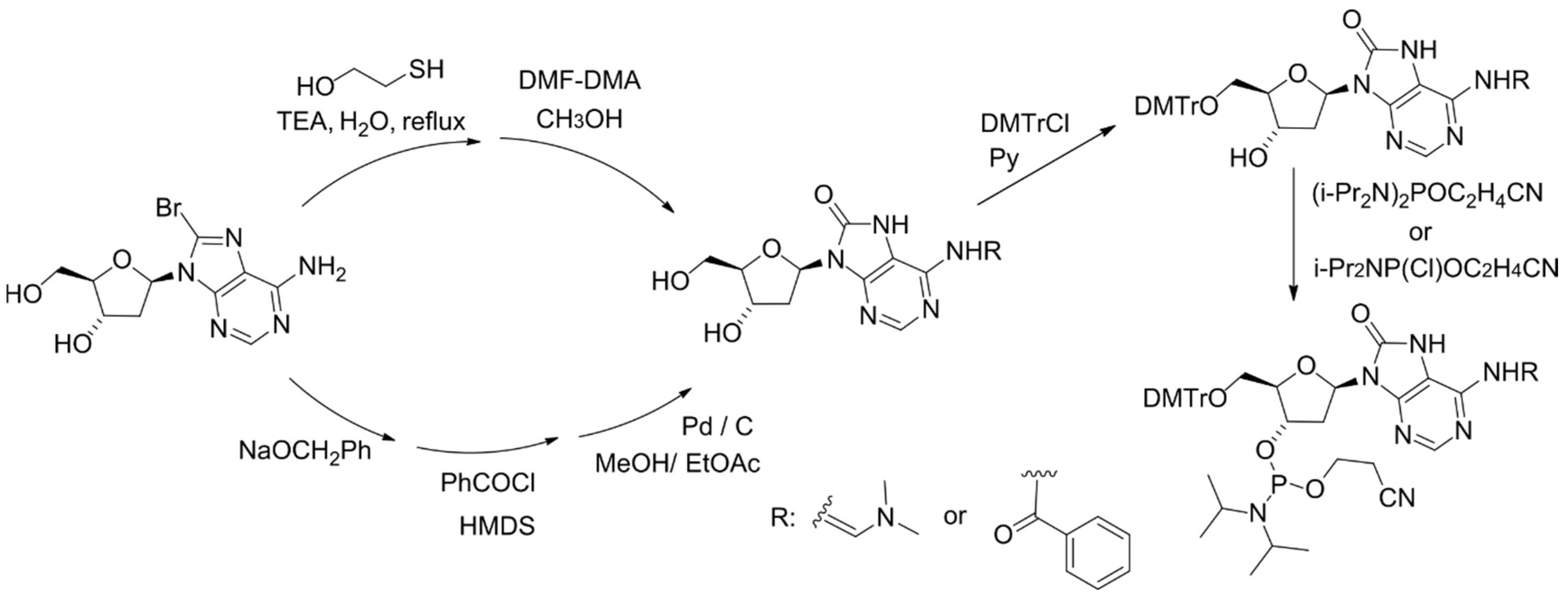
5. Repair of 8-oxoA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | DNA Glycosylase | Base Pair | Reference |
|---|---|---|---|
| Prokaryotes | MUG | 8A:T | [129] |
| 8A:G | |||
| 8A:C | |||
| 8A:A (low activity) | [130] | ||
| 8A:C (low activity) | |||
| Fpg | 8A:C (low activity) | [106,107,108,109] | |
| γ-irradiated DNA (low activity) | |||
| Eukaryotes | OGG1 | 8A:C | [106,117,122,127] |
| 8A:5-mC | |||
| TDG | 8A:T | [90,129] | |
| 8A:G | |||
| 8A:C | |||
| T:8A | |||
| 8A:G (+++) | [130] | ||
| 8A:C | |||
| 8A:A | |||
| 8A:T | |||
| NEIL1 | 8A:C | [128] | |
| Unidentified mitochondrial enzyme, distinct from OGG1 | 8A:G | [122] |
6. Translesion DNA Synthesis Opposite 8-oxoA: The Second Chance to Avoid Harmful Effects
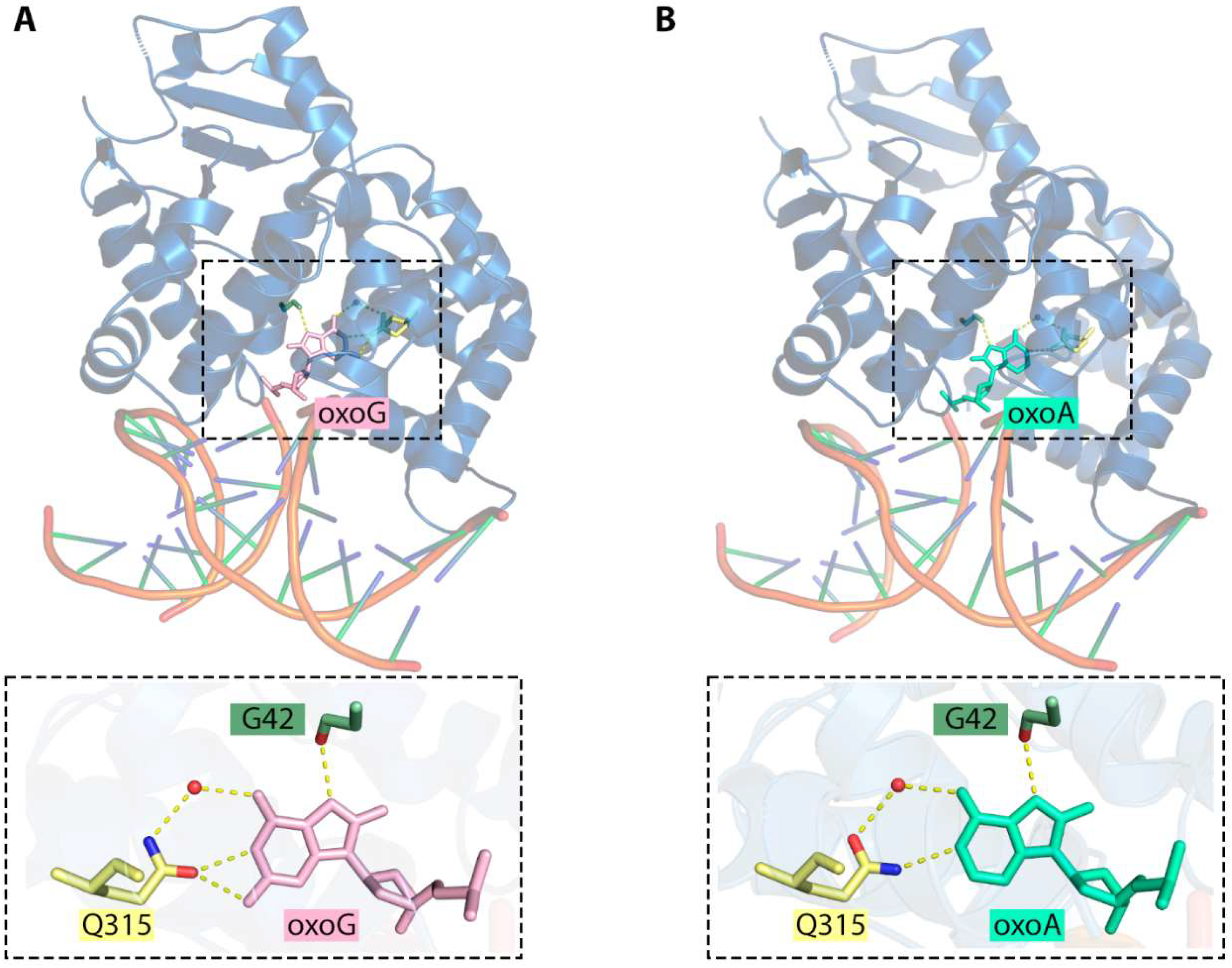
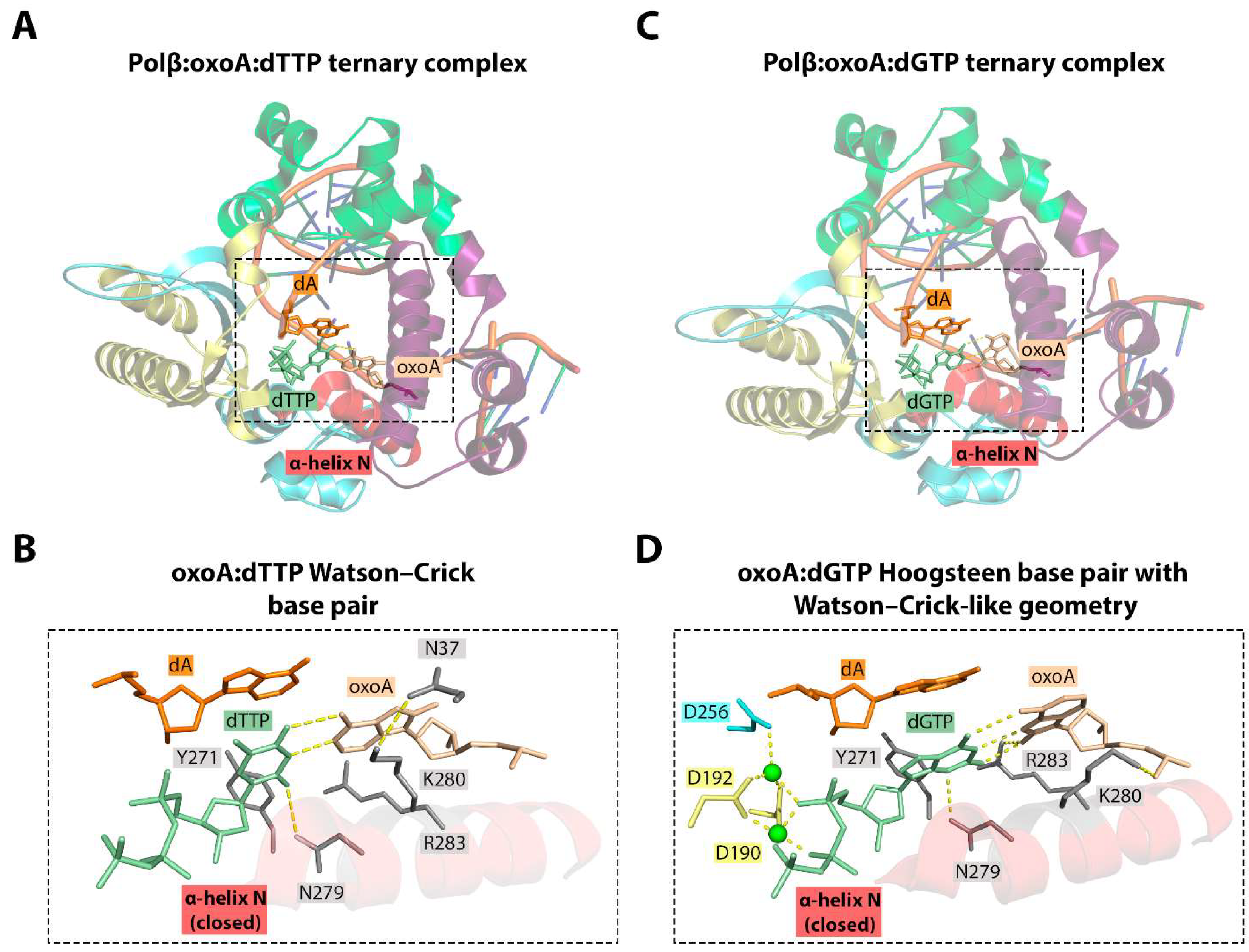
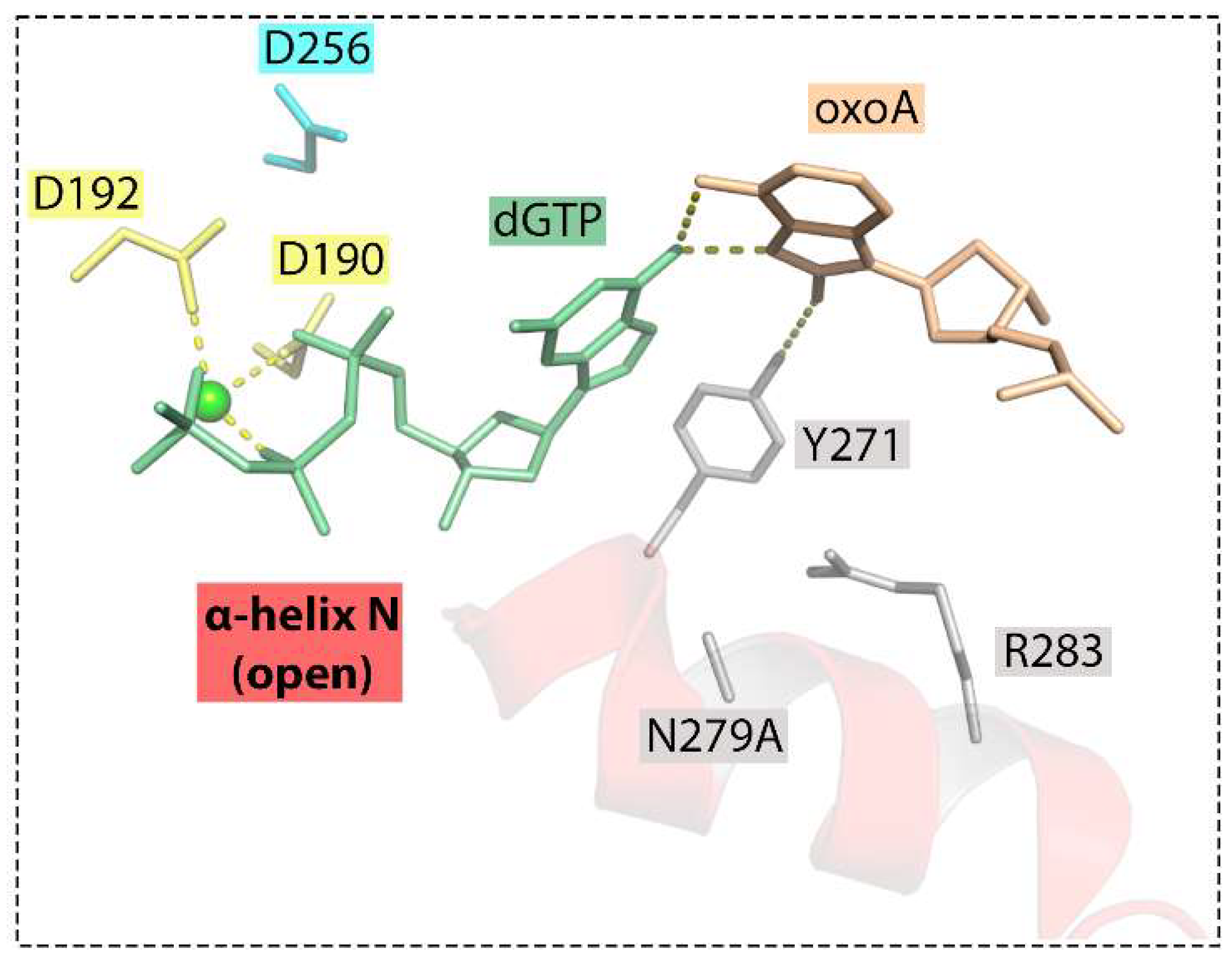
6.1. 8-oxoA Bypass by DNA Polymerase β
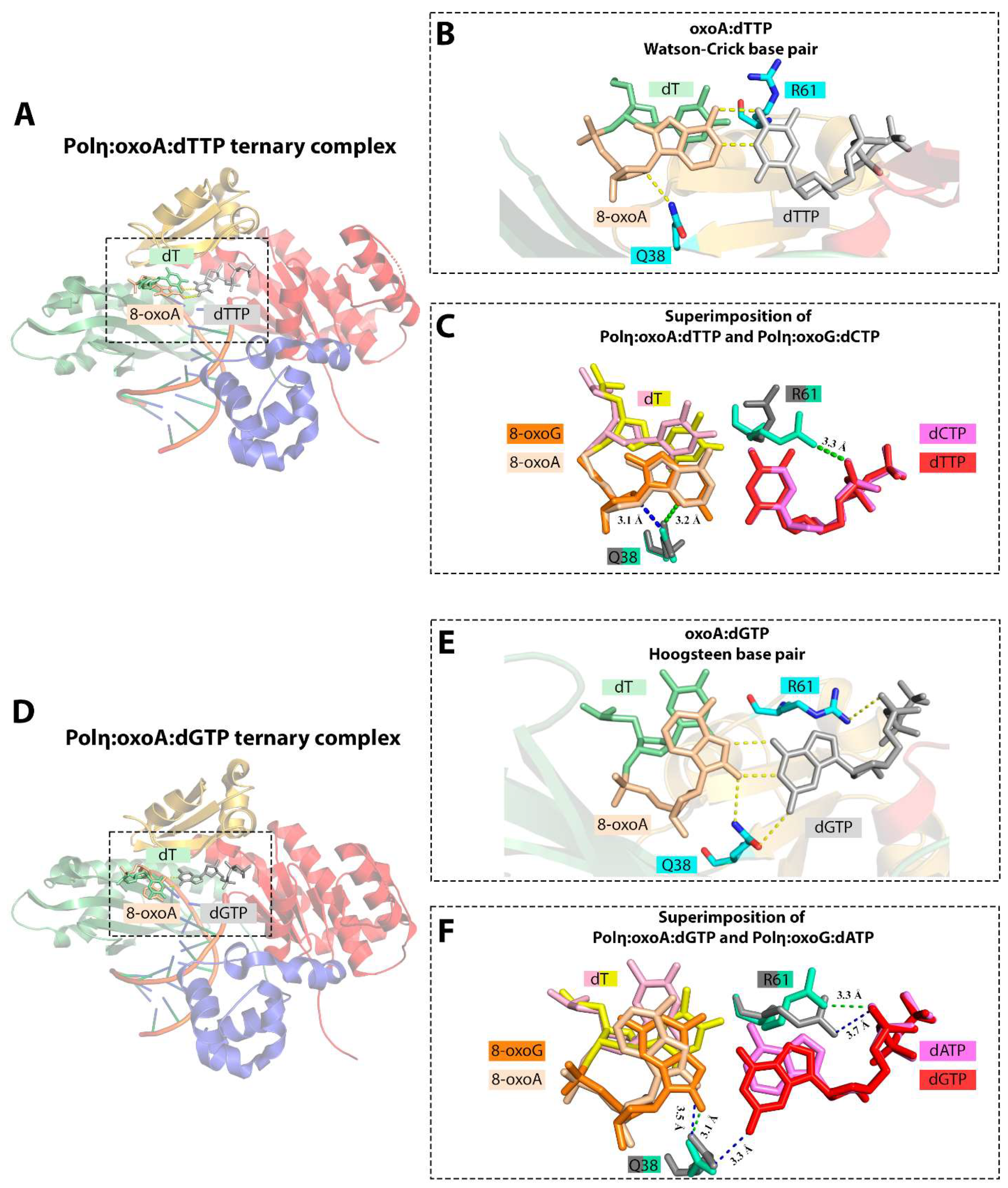
6.2. 8-oxoA Bypass by DNA Polymerase η
6.3. 8-oxoA Bypass by Dpo4
7. Unresolved Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cadet, J.; Davies, K.J.A.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and Repair of Oxidatively Generated Damage in Cellular DNA. Free Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Koag, M.-C.; Jung, H.; Lee, S. Mutagenesis Mechanism of the Major Oxidative Adenine Lesion 7,8-Dihydro-8-Oxoadenine. Nucleic Acids Res. 2020, 48, 5119–5134. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidative Damage to DNA in Mammalian Chromatin. Mutat. Res. 1992, 275, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hori, Y.; Dizdaroglu, M. DNA Base Damage Generated in Vivo in Hepatic Chromatin of Mice Upon Whole Body γ-Irradiation. Int. J. Radiat. Biol. 1993, 64, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Close, D.M.; Gorb, L.; Leszczynski, J. Determination of Redox Potentials for the Watson−Crick Base Pairs, DNA Nucleosides, and Relevant Nucleoside Analogues. J. Phys. Chem. B 2007, 111, 5386–5395. [Google Scholar] [CrossRef]
- Cadet, J. Oxidative Damage to DNA: Formation, Measurement and Biochemical Features. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 531, 5–23. [Google Scholar] [CrossRef]
- Jałoszyński, P.; Jaruga, P.; Oliński, R.; Biczysko, W.; Szyfter, W.; Nagy, E.; Möller, L.; Szyfter, K. Oxidative DNA Base Modifications and Polycyclic Aromatic Hydrocarbon DNA Adducts in Squamous Cell Carcinoma of Larynx. Free Radic. Res. 2003, 37, 231–240. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Conlay, J.J. Effect of Ionizing Radiation on Adenine in Aerated and De-Aerated Aqueous Solutions. Nature 1963, 197, 555–557. [Google Scholar] [CrossRef]
- Van Hemmen, J.J.; Bleichrodt, J.F. The Decomposition of Adenine by Ionizing Radiation. Radiat. Res. 1971, 46, 444. [Google Scholar] [CrossRef] [PubMed]
- Fuciarelli, A.F.; Wegher, B.J.; Gajewski, E.; Dizdaroglu, M.; Blakely, W.F. Quantitative Measurement of Radiation-Induced Base Products in DNA Using Gas Chromatography-Mass Spectrometry. Radiat. Res. 1989, 119, 219. [Google Scholar] [CrossRef] [PubMed]
- Bonicel, A.; Mariaggi, N.; Hughes, E.; Teoule, R. In Vitro γ Irradiation of DNA: Identification of Radioinduced Chemical Modifications of the Adenine Moiety. Radiat. Res. 1980, 83, 19. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Zastawny, T.; Budzbon, J.; Skokowski, J.; Zegarski, W.; Dizdaroglu, M. DNA Base Modifications in Chromatin of Human Cancerous Tissues. FEBS Lett. 1992, 309, 193–198. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. Formation of 2-Hydroxydeoxyadenosine Triphosphate, an Oxidatively Damaged Nucleotide, and Its Incorporation by DNA Polymerases. J. Biol. Chem. 1995, 270, 19446–19450. [Google Scholar] [CrossRef] [PubMed]
- Frelon, S.; Douki, T.; Cadet, J. Radical Oxidation of the Adenine Moiety of Nucleoside and DNA: 2-Hydroxy-2′-Deoxyadenosine Is a Minor Decomposition Product. Free Radic. Res. 2002, 36, 499–508. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. Effects of Sequence Contexts on Misincorporation of Nucleotides Opposite 2-hydroxyadenine. FEBS Lett. 1996, 391, 113–116. [Google Scholar] [CrossRef]
- Kamiya, H. 2-Hydroxy-dATP Is Incorporated Opposite G by Escherichia coli DNA Polymerase III Resulting in High Mutagenicity. Nucleic Acids Res. 2000, 28, 1640–1646. [Google Scholar] [CrossRef]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of Human MutY Homolog (hMYH) as a Repair Enzyme for 2-Hydroxyadenine in DNA and Detection of Multiple Forms of hMYH Located in Nuclei and Mitochondria. Nucleic Acids Res. 2015, 43, 3870–3871. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Kamiya, H.; Yakushiji, H.; Fujii, Y.; Nakabeppu, Y.; Kasai, H. The Oxidized Forms of dATP Are Substrates for the Human MutT Homologue, the hMTH1 Protein. J. Biol. Chem. 1999, 274, 18201–18205. [Google Scholar] [CrossRef] [PubMed]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-26120-9. [Google Scholar]
- Vieira, A.J.S.C.; Steenken, S. Pattern of Hydroxy Radical Reaction with Adenine and Its Nucleosides and Nucleotides. Characterization of Two Types of Isomeric Hydroxy Adduct and Their Unimolecular Transformation Reactions. J. Am. Chem. Soc. 1990, 112, 6986–6994. [Google Scholar] [CrossRef]
- Bodepudi, V.; Shibutani, S.; Johnson, F. Synthesis of 2’-Deoxy-7,8-Dihydro-8-Oxoguanosine and 2’-Deoxy-7,8-Dihydro-8-Oxoadenosinaend Their Incorporation into Oligomeric DNA. Chem. Res. Toxicol. 1992, 5, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Navacchia, M.L.; Postigo, A. A Facile One-Pot Synthesis of 8-Oxo-7,8-Dihydro-(2′-Deoxy)Adenosine in Water. Tetrahedron Lett. 2006, 47, 711–714. [Google Scholar] [CrossRef]
- Bande, O.; Braddick, D.; Agnello, S.; Jang, M.; Pezo, V.; Schepers, G.; Rozenski, J.; Lescrinier, E.; Marlière, P.; Herdewijn, P. Base Pairing Involving Artificial Bases in Vitro and in Vivo. Chem. Sci. 2016, 7, 995–1010. [Google Scholar] [CrossRef]
- Dolinnaya, N.G.; Kubareva, E.A.; Romanova, E.A.; Trikin, R.M.; Oretskaya, T.S. Thymidine Glycol: The Effect on DNA Molecular Structure and Enzymatic Processing. Biochimie 2013, 95, 134–147. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis: DNA Damage and Repair. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Guschlbauer, W.; Duplaa, A.-M.; Guy, A.; Téoule, R.; Fazakerley, G.V. Structure and in Vitro Replication of DNA Templates Containing 7,8-Dihydro-8-Oxoadenine. Nucleic Acids Res. 1991, 19, 1753–1758. [Google Scholar] [CrossRef]
- Barone, F.; Cellai, L.; Giordano, C.; Sala, G.L.; Mazzei, F. Influence of an 8-Oxoadenine Lesion on the Structural and Dynamic Features of a 30-Mer DNA Fragment with and without a Mismatch. Int. J. Radiat. Biol. 2002, 78, 9–16. [Google Scholar] [CrossRef]
- Malins, D.C.; Polissar, N.L.; Ostrander, G.K.; Vinson, M.A. Single 8-Oxo-Guanine and 8-Oxo-Adenine Lesions Induce Marked Changes in the Backbone Structure of a 25-Base DNA Strand. Proc. Natl. Acad. Sci. USA 2000, 97, 12442–12445. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.A.; Guy, A.; Brown, T.; Thule, R.; Hunter, W.N. Conformation of Guanine.8-Oxoadenine Base Pairs in the Crystal Structure of (dCGCGAATT(08A)GCG). Biochemistry 1992, 31, 8415–8420. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.P.; Evans, F.E. Structure of Oxidatively Damaged Nucleic Acid Adducts. 3. Tautomerism, Ionization and Protonation of 8-Hydroxyadenosine Studied by 15 N NMR Spectroscopy. Nucleic Acids Res. 1991, 19, 1041–1046. [Google Scholar] [CrossRef]
- Chen, H.; Johnson, F.; Grollman, A.P.; Patel, D.J. Structural Studies of the Ionizing Radiation Adduct 7,8-Dihydro-8-Oxoadenine (Aoxo) Positioned Opposite Thymine in a DNA Duplex. Magn. Reson. Chem. 1996, 34, S23–S32. [Google Scholar] [CrossRef]
- Koag, M.-C.; Jung, H.; Lee, S. Mutagenic Replication of the Major Oxidative Adenine Lesion 7,8-Dihydro-8-Oxoadenine by Human DNA Polymerases. J. Am. Chem. Soc. 2019, 141, 4584–4596. [Google Scholar] [CrossRef] [PubMed]
- Burak, M.J.; Guja, K.E.; Hambardjieva, E.; Derkunt, B.; Garcia-Diaz, M. A Fidelity Mechanism in DNA Polymerase Lambda Promotes Error-free Bypass of 8-oxo-dG. EMBO J. 2016, 35, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Rechkoblit, O.; Johnson, R.E.; Gupta, Y.K.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural Basis of DNA Synthesis Opposite 8-Oxoguanine by Human PrimPol Primase-Polymerase. Nat. Commun. 2021, 12, 4020. [Google Scholar] [CrossRef]
- Patra, A.; Nagy, L.D.; Zhang, Q.; Su, Y.; Müller, L.; Guengerich, F.P.; Egli, M. Kinetics, Structure, and Mechanism of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine Bypass by Human DNA Polymerase η. J. Biol. Chem. 2014, 289, 16867–16882. [Google Scholar] [CrossRef]
- Batra, V.K.; Shock, D.D.; Beard, W.A.; McKenna, C.E.; Wilson, S.H. Binary Complex Crystal Structure of DNA Polymerase β Reveals Multiple Conformations of the Templating 8-Oxoguanine Lesion. Proc. Natl. Acad. Sci. USA 2012, 109, 113–118. [Google Scholar] [CrossRef]
- Wood, M.L.; Esteve, A.; Morningstar, M.L.; Kuziemko, G.M.; Essigmann, J.M. Genetic Effects of Oxidative DNA Damage: Comparative Mutagenesis of 7,8-Dihydro-8-Oxoguanine and 7,8-Dihydro-8-Oxoadenine in Escherichia Coli. Nucleic Acids Res. 1992, 20, 6023–6032. [Google Scholar] [CrossRef]
- Kamiya, H.; Miura, H.; Murata-Kamiya, N.; Ishikawa, H.; Sakaguchi, T.; Inoue, H.; Sasaki, T.; Masutanl, C.; Hanaoka, F.; Nishimura, S.; et al. 8-Hydroxyadenine (7, 8-Dihydro-8-Oxoadenine) Induces Misincorporation in in Vitro DNA Synthesis and Mutations in NIH 3T3 Cells. Nucleic Acids Res. 1995, 23, 2893–2899. [Google Scholar] [CrossRef]
- Shibutani, S.; Bodepudi, V.; Johnson, F.; Grollman, A.P. Translesional Synthesis on DNA Templates Containing 8-Oxo-7,8-Dihydrodeoxyadenosine. Biochemistry 1993, 32, 4615–4621. [Google Scholar] [CrossRef]
- Duarte, V.; Muller, J.G.; Burrows, C.J. Insertion of dGMP and dAMP during in Vitro DNA Synthesis Opposite an Oxidized Form of 7,8-Dihydro-8-Oxoguanine. Nucleic Acids Res. 1999, 27, 496–502. [Google Scholar] [CrossRef]
- Shibutani, S. Quantitation of Base Substitutions and Deletions Induced by Chemical Mutagens during DNA Synthesis in Vitro. Chem. Res. Toxicol. 1993, 6, 625–629. [Google Scholar] [CrossRef]
- Jung, H.; Lee, S. Promutagenic Bypass of 7,8-Dihydro-8-Oxoadenine by Translesion Synthesis DNA Polymerase Dpo4. Biochem. J. 2020, 477, 2859–2871. [Google Scholar] [CrossRef]
- Jung, H.; Lee, S. Insights into the Mismatch Discrimination Mechanism of Y-Family DNA Polymerase Dpo4. Biochem. J. 2021, 478, 1769–1781. [Google Scholar] [CrossRef]
- Shibutani, S.; Grollman, A.P. Miscoding during DNA Synthesis on Damaged DNA Templates Catalysed by Mammalian Cell Extracts. Cancer Lett. 1994, 83, 315–322. [Google Scholar] [CrossRef]
- Kamiya, H.; Murata-Kamiya, N.; Koizume, S.; Inoue, H.; Nishimura, S.; Ohtsuka, E. 8-Hydroxyguanine (7,8-Dihydro-8-Oxoguanine) in Hot Spots of the c-Ha- Ras Gene: Effects of Sequence Contexts on Mutation Spectra. Carcinogenesis 1995, 16, 883–889. [Google Scholar] [CrossRef]
- Tan, X.; Grollman, A.P.; Shibutani, S. Comparison of the Mutagenic Properties of 8-Oxo-7,8-Dihydro-2′-Deoxyadenosine and 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine DNA Lesions in Mammalian Cells. Carcinogenesis 1999, 20, 2287–2292. [Google Scholar] [CrossRef]
- Yudkina, A.V.; Shilkin, E.S.; Endutkin, A.V.; Makarova, A.V.; Zharkov, D.O. Reading and Misreading 8-Oxoguanine, a Paradigmatic Ambiguous Nucleobase. Crystals 2019, 9, 269. [Google Scholar] [CrossRef]
- Pavlov, Y.I.; Minnick, D.T.; Izuta, S.; Kunkel, T.A. DNA Replication Fidelity with 8-Oxodeoxyguanosine Triphosphate. Biochemistry 1994, 33, 4695–4701. [Google Scholar] [CrossRef]
- Hayakawa, H.; Taketomi, A. Generation and Elimination of 8-Oxo-7,8-Dihydro-2’-Deoxyguanosine 5’-Triphosphate, a Mutagenic Substrate for DNA Synthesis, in Human Cells. Biochemistry 1995, 34, 89–95. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Ohta, E.; Abolhassani, N. MTH1 as a Nucleotide Pool Sanitizing Enzyme: Friend or Foe? Free Radic. Biol. Med. 2017, 107, 151–158. [Google Scholar] [CrossRef]
- Grin, I.R.; Vasilyeva, S.V.; Dovgerd, A.P.; Silnikov, V.N.; Zharkov, D.O. Human and Bacterial DNA Polymerases Discriminate against 8-Oxo-2’-Deoxyadenosine- 5’-Triphosphate. Biopolym. Cell 2012, 28, 306–309. [Google Scholar] [CrossRef][Green Version]
- Purmal, A.A.; Kow, Y.W.; Wallace, S.S. 5-Hydroxypyrimidine Deoxynucleoside Triphosphates Are More Efficiently Incorporated into DNA by Exonuclease-Free Klenow Fragment than 8-Oxopurine Deoxynucleoside Triphosphates. Nucleic Acids Res. 1994, 22, 3930–3935. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.-M. 8-Oxoguanine and 8-Oxodeoxyguanosine Biomarkers of Oxidative DNA Damage: A Review on HPLC–ECD Determination. Molecules 2022, 27, 1620. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, M. Determination of Steady-State Levels of Oxidative DNA Base Modifications in Mammalian Cells by Means of Repair Endonucleases. Carcinogenesis 1997, 18, 2225–2231. [Google Scholar] [CrossRef]
- Helbock, H.J.; Beckman, K.B.; Shigenaga, M.K.; Walter, P.B.; Woodall, A.A.; Yeo, H.C.; Ames, B.N. DNA Oxidation Matters: The HPLC–Electrochemical Detection Assay of 8-Oxo-Deoxyguanosine and 8-Oxo-Guanine. Proc. Natl. Acad. Sci. USA 1998, 95, 288–293. [Google Scholar] [CrossRef]
- Collins, A.R. Oxidative DNA Damage, Antioxidants, and Cancer. Bioessays 1999, 21, 238–246. [Google Scholar] [CrossRef]
- Møller, P.; Cooke, M.S.; Collins, A.; Olinski, R.; Rozalski, R.; Loft, S. Harmonising Measurements of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine in Cellular DNA and Urine. Free Radic. Res. 2012, 46, 541–553. [Google Scholar] [CrossRef]
- Tuo, J.; Jaruga, P.; Rodriguez, H.; Dizdaroglu, M.; Bohr, V.A. The Cockayne Syndrome Group B Gene Product Is Involved in Cellular Repair of 8-Hydroxyadenine in DNA. J. Biol. Chem. 2002, 277, 30832–30837. [Google Scholar] [CrossRef] [PubMed]
- Jaruga, P. Repair of Products of Oxidative DNA Base Damage in Human Cells. Nucleic Acids Res. 1996, 24, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Ho, Y.-S.; Lo, M.-J.; Lin, J. Oxidative Modification of DNA Bases in Rat Liver and Lung during Chemical Carcinogenesis and Aging. Chem. Biol. Interact. 1995, 94, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Miura, K.; Ishikawa, H.; Inoue, H.; Nishimura, S.; Ohtsuka, E. C-Ha-Ras Containing 8-Hydroxyguanine at Codon 12 Induces Point Mutations at the Modified and Adjacent Positions1. Cancer Res. 1992, 52, 3483–3485. [Google Scholar] [PubMed]
- Poetsch, A.R. The Genomics of Oxidative DNA Damage, Repair, and Resulting Mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-Defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Pich, O.; Devonshire, G.; Zamani, S.A.; Katz-Summercorn, A.; Killcoyne, S.; Cheah, C.; Nutzinger, B.; Grehan, N.; Lopez-Bigas, N.; et al. Mutational Signature Dynamics Shaping the Evolution of Oesophageal Adenocarcinoma. Nat. Commun. 2023, 14, 4239. [Google Scholar] [CrossRef]
- Tomkova, M.; Tomek, J.; Kriaucionis, S.; Schuster-Böckler, B. Mutational Signature Distribution Varies with DNA Replication Timing and Strand Asymmetry. Genome Biol. 2018, 19, 129. [Google Scholar] [CrossRef]
- Christensen, S.; Van Der Roest, B.; Besselink, N.; Janssen, R.; Boymans, S.; Martens, J.W.M.; Yaspo, M.-L.; Priestley, P.; Kuijk, E.; Cuppen, E.; et al. 5-Fluorouracil Treatment Induces Characteristic T>G Mutations in Human Cancer. Nat. Commun. 2019, 10, 4571. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. APE1 Deficiency Promotes Cellular Senescence and Premature Aging Features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in Mouse Myh and Ogg1 Result in Tumor Predisposition and G to T Mutations in Codon 12 of the K-Ras Oncogene in Lung Tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Pin, E.; Russo, M.T.; Barone, F.; Degan, P.; Sanchez, M.; Quaia, M.; Minoprio, A.; Turco, E.; Mazzei, F.; et al. Loss of MUTYH Function in Human Cells Leads to Accumulation of Oxidative Damage and Genetic Instability. Oncogene 2013, 32, 4500–4508. [Google Scholar] [CrossRef]
- Sakamoto, K.; Tominaga, Y.; Yamauchi, K.; Nakatsu, Y.; Sakumi, K.; Yoshiyama, K.; Egashira, A.; Kura, S.; Yao, T.; Tsuneyoshi, M.; et al. MUTYH-Null Mice Are Susceptible to Spontaneous and Oxidative Stress–Induced Intestinal Tumorigenesis. Cancer Res. 2007, 67, 6599–6604. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Schomacher, L.; Schüle, K.M.; Mallick, M.; Musheev, M.U.; Karaulanov, E.; Krebs, L.; Von Seggern, A.; Niehrs, C. NEIL1 and NEIL2 DNA Glycosylases Protect Neural Crest Development against Mitochondrial Oxidative Stress. eLife 2019, 8, e49044. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous Tumorigenesis in Mice Defective in the MTH1 Gene Encoding 8-Oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Leon, J.; Sakumi, K.; Abolhassani, N.; Sheng, Z.; Tsuchimoto, D.; LaFerla, F.M.; Nakabeppu, Y. MTH1 and OGG1 Maintain a Low Level of 8-Oxoguanine in Alzheimer’s Brain, and Prevent the Progression of Alzheimer’s Pathogenesis. Sci. Rep. 2021, 11, 5819. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Je, G.; Kim, Y.-S. Transcriptional Mutagenesis by 8-oxodG in α-Synuclein Aggregation and the Pathogenesis of Parkinson’s Disease. Exp. Mol. Med. 2015, 47, e179. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Jaruga, P.; Rodriguez, H.; Bohr, V.A.; Dizdaroglu, M. Primary Fibroblasts of Cockayne Syndrome Patients Are Defective in Cellular Repair of 8-hydroxyguanine and 8-hydroxyadenine Resulting from Oxidative Stress. FASEB J. 2003, 17, 668–674. [Google Scholar] [CrossRef]
- Rozelle, A.L.; Cheun, Y.; Vilas, C.K.; Koag, M.-C.; Lee, S. DNA Interstrand Cross-Links Induced by the Major Oxidative Adenine Lesion 7,8-Dihydro-8-Oxoadenine. Nat. Commun. 2021, 12, 1897. [Google Scholar] [CrossRef]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively Generated Complex DNA Damage: Tandem and Clustered Lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef]
- Hada, M.; Georgakilas, A.G. Formation of Clustered DNA Damage after High-LET Irradiation: A Review. JRR 2008, 49, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Jaruga, P.; Dizdaroglu, M.; Neumann, R.D.; Winters, T.A. Molecular Analysis of Base Damage Clustering Associated with a Site-Specific Radiation- Induced DNA Double-Strand Break. Radiat. Res. 2006, 166, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication Fork Collapse Is a Major Cause of the High Mutation Frequency at Three-Base Lesion Clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef]
- Rodriguez, G.P.; Song, J.B.; Crouse, G.F. In Vivo Bypass of 8-oxodG. PLoS Genet. 2013, 9, e1003682. [Google Scholar] [CrossRef] [PubMed]
- Shikazono, N.; Akamatsu, K. Mutagenic Potential of 8-Oxo-7,8-Dihydroguanine (8-oxoG) Is Influenced by Nearby Clustered Lesions. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2018, 810, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Salje, H.; Cunniffe, S.; O’Neill, P. 8-OxoA Inhibits the Incision of an AP Site by the DNA Glycosylases Fpg, Nth and the AP Endonuclease HAP1. Radiat. Res. 2005, 163, 79–84. [Google Scholar] [CrossRef]
- David-Cordonnier, M.-H.; Laval, J.; O’Neill, P. Clustered DNA Damage, Influence on Damage Excision by XRS5 Nuclear Extracts and Escherichia Coli Nth and Fpg Proteins. J. Biol. Chem. 2000, 275, 11865–11873. [Google Scholar] [CrossRef]
- David-Cordonnier, M.-H.; Laval, J.; O’Neill, P. Recognition and Kinetics for Excision of a Base Lesion within Clustered DNA Damage by the Escherichia Coli Proteins Fpg and Nth. Biochemistry 2001, 40, 5738–5746. [Google Scholar] [CrossRef] [PubMed]
- Talhaoui, I.; Couve, S.; Gros, L.; Ishchenko, A.A.; Matkarimov, B.; Saparbaev, M.K. Aberrant Repair Initiated by Mismatch-Specific Thymine-DNA Glycosylases Provides a Mechanism for the Mutational Bias Observed in CpG Islands. Nucleic Acids Res. 2014, 42, 6300–6313. [Google Scholar] [CrossRef]
- Machwe, A.; Ganunis, R.; Bohr, V.A.; Orren, D.K. Selective Blockage of the 3’->5’ Exonuclease Activity of WRN Protein by Certain Oxidative Modifications and Bulky Lesions in DNA. Nucleic Acids Res. 2000, 28, 2762–2770. [Google Scholar] [CrossRef]
- Orren, D.K.; Machwe, A.; Karmakar, P.; Piotrowski, J.; Cooper, M.P.; Bohr, V.A. A Functional Interaction of Ku with Werner Exonuclease Facilitates Digestion of Damaged DNA. Nucleic Acids Res. 2001, 29, 1926–1934. [Google Scholar] [CrossRef]
- Kamiya, H.; Makino, T.; Suzuki, T.; Kobayashi, M.; Matsuoka, I. Mutations Induced by 8-Oxo-7,8-Dihydroguanine in WRN- and DNA Polymerase λ-Double Knockdown Cells. Mutagenesis 2018, 33, 301–310. [Google Scholar] [CrossRef]
- Sabourin, M.; Osheroff, N. Sensitivity of Human Type II Topoisomerases to DNA Damage: Stimulation of Enzyme-Mediated DNA Cleavage by Abasic, Oxidized and Alkylated Lesions. Nucleic Acids Res. 2000, 28, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, L.; Tian, L.; Sayer, J.M.; Kalena, G.P.; Kroth, H.; Jerina, D.M.; Shuman, S. Site-Specific DNA Transesterification by Vaccinia Topoisomerase. J. Biol. Chem. 2003, 278, 42170–42177. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.P.G.; Toomire, K.J.; Strauss, P.R. DNA Modifications Repaired by Base Excision Repair Are Epigenetic. DNA Repair 2013, 12, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread Transcriptional Gene Inactivation Initiated by a Repair Intermediate of 8-Oxoguanine. Nucleic Acids Res. 2016, 44, gkw473. [Google Scholar] [CrossRef] [PubMed]
- Hailer-Morrison, M.K.; Kotler, J.M.; Martin, B.D.; Sugden, K.D. Oxidized Guanine Lesions as Modulators of Gene Transcription. Altered P50 Binding Affinity and Repair Shielding by 7,8-Dihydro-8-Oxo-2‘-Deoxyguanosine Lesions in the NF-κB Promoter Element. Biochemistry 2003, 42, 9761–9770. [Google Scholar] [CrossRef]
- Ramon, O.; Sauvaigo, S.; Gasparutto, D.; Faure, P.; Favier, A.; Cadet, J. Effects of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine on the Binding of the Transcription Factor Sp1 to Its Cognate Target DNA Sequence (GC Box). Free Radic. Res. 1999, 31, 217–229. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, W.; Hu, Y.; Zhao, Z.; Wu, J.; Zhang, X.; Hao, F.; Han, J.; Xu, J.; Hao, W.; et al. DNA Repair Byproduct 8-Oxoguanine Base Promotes Myoblast Differentiation. Redox Biol. 2023, 61, 102634. [Google Scholar] [CrossRef]
- Konovalov, K.A.; Pardo-Avila, F.; Tse, C.K.M.; Oh, J.; Wang, D.; Huang, X. 8-Oxo-Guanine DNA Damage Induces Transcription Errors by Escaping Two Distinct Fidelity Control Checkpoints of RNA Polymerase II. J. Biol. Chem. 2019, 294, 4924–4933. [Google Scholar] [CrossRef]
- Dai, D.-P.; Gan, W.; Hayakawa, H.; Zhu, J.-L.; Zhang, X.-Q.; Hu, G.-X.; Xu, T.; Jiang, Z.-L.; Zhang, L.-Q.; Hu, X.-D.; et al. Transcriptional Mutagenesis Mediated by 8-oxoG Induces Translational Errors in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2018, 115, 4218–4222. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.A.; Ezerskyte, M.; Bottai, M.; Dreij, K. Transcriptional Mutagenesis Reduces Splicing Fidelity in Mammalian Cells. Nucleic Acids Res. 2017, 45, 6520–6529. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Suzuki, K.; Ito, S.; Hayashida, M.; Kwei, J.S.M.; Ikegami, T.; Handa, H.; Nakabeppu, Y.; Tanaka, K. RNA Polymerase II Bypasses 8-Oxoguanine in the Presence of Transcription Elongation Factor TFIIS. DNA Repair 2007, 6, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Bodepudi, V.; Shibutani, S.; Antoshechkin, I.; Miller, J.; Grollman, A.P.; Johnson, F. Substrate Specificity of Fpg Protein. Recognition and Cleavage of Oxidatively Damaged DNA. J. Biol. Chem. 1994, 269, 15318–15324. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.M.; D’Ham, C.; Cadet, J.; Boiteux, S. Opposite Base-Dependent Excision of 7,8-Dihydro-8-Oxoadenine by the Ogg1 Protein of Saccharomyces Cerevisiae. Carcinogenesis 1998, 19, 1299–1305. [Google Scholar] [CrossRef]
- Kathe, S.D.; Barrantes-Reynolds, R.; Jaruga, P.; Newton, M.R.; Burrows, C.J.; Bandaru, V.; Dizdaroglu, M.; Bond, J.P.; Wallace, S.S. Plant and Fungal Fpg Homologs Are Formamidopyrimidine DNA Glycosylases but Not 8-Oxoguanine DNA Glycosylases. DNA Repair 2009, 8, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Gajewski, E.; Laval, J.; Dizdaroglu, M. Substrate Specificity of the Escherichia Coli Fpg Protein Formamidopyrimidine-DNA Glycosylase: Excision of Purine Lesions in DNA Produced by Ionizing Radiation or Photosensitization. Biochemistry 1992, 31, 106–110. [Google Scholar] [CrossRef]
- Guo, Y.; Bandaru, V.; Jaruga, P.; Zhao, X.; Burrows, C.J.; Iwai, S.; Dizdaroglu, M.; Bond, J.P.; Wallace, S.S. The Oxidative DNA Glycosylases of Mycobacterium Tuberculosis Exhibit Different Substrate Preferences from Their Escherichia coli Counterparts. DNA Repair 2010, 9, 177–190. [Google Scholar] [CrossRef]
- Fromme, J.C.; Verdine, G.L. DNA Lesion Recognition by the Bacterial Repair Enzyme MutM. J. Biol. Chem. 2003, 278, 51543–51548. [Google Scholar] [CrossRef]
- Coste, F.; Ober, M.; Carell, T.; Boiteux, S.; Zelwer, C.; Castaing, B. Structural Basis for the Recognition of the FapydG Lesion (2,6-Diamino-4-Hydroxy-5-Formamidopyrimidine) by Formamidopyrimidine-DNA Glycosylase. J. Biol. Chem. 2004, 279, 44074–44083. [Google Scholar] [CrossRef]
- Perlow-Poehnelt, R.A.; Zharkov, D.O.; Grollman, A.P.; Broyde, S. Substrate Discrimination by Formamidopyrimidine-DNA Glycosylase: Distinguishing Interactions within the Active Site. Biochemistry 2004, 43, 16092–16105. [Google Scholar] [CrossRef] [PubMed]
- Bulychev, N.V.; Varaprasad, C.V.; Dormán, G.; Miller, J.H.; Eisenberg, M.; Grollman, A.P.; Johnson, F. Substrate Specificity of Escherichia coli MutY Protein. Biochemistry 1996, 35, 13147–13156. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Formation and Processing of DNA Damage Substrates for the hNEIL Enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Endutkin, A.V.; Zharkov, D.O. Substrate Specificities of DNA Glycosylases In Vitro and In Vivo. In Chemical Biology; Dizdaroglu, M., Lloyd, R.S., Eds.; Royal Society of Chemistry: Cambridge, UK, 2020; Volume 1, pp. 175–203. ISBN 978-1-78801-889-0. [Google Scholar]
- Lu, R.; Nash, H.M.; Verdine, G.L. A Mammalian DNA Repair Enzyme That Excises Oxidatively Damaged Guanines Maps to a Locus Frequently Lost in Lung Cancer. Curr. Biol. 1997, 7, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate Specificity and Reaction Mechanism of Murine 8-Oxoguanine-DNA Glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef]
- Karahalil, B. Substrate Specificity of the Ogg1 Protein of Saccharomyces Cerevisiae: Excision of Guanine Lesions Produced in DNA by Ionizing Radiation- or Hydrogen Peroxide/Metal Ion-Generated Free Radicals. Nucleic Acids Res. 1998, 26, 1228–1233. [Google Scholar] [CrossRef]
- Dherin, C.; Radicella, J.P.; Dizdaroglu, M.; Boiteux, S. Excision of Oxidatively Damaged DNA Bases by the Human α-hOgg1 Protein and the Polymorphic α-hOgg1(Ser326Cys) Protein Which Is Frequently Found in Human Populations. Nucleic Acids Res. 1999, 27, 4001–4007. [Google Scholar] [CrossRef]
- Dherin, C. Repair of Oxidative DNA Damage in Drosophila Melanogaster: Identification and Characterization of dOgg1, a Second DNA Glycosylase Activity for 8-Hydroxyguanine and Formamidopyrimidines. Nucleic Acids Res. 2000, 28, 4583–4592. [Google Scholar] [CrossRef]
- Morales-Ruiz, T.; Birincioglu, M.; Jaruga, P.; Rodriguez, H.; Roldan-Arjona, T.; Dizdaroglu, M. Arabidopsis Thaliana Ogg1 Protein Excises 8-Hydroxyguanine and 2,6-Diamino-4-Hydroxy-5-Formamidopyrimidine from Oxidatively Damaged DNA Containing Multiple Lesions. Biochemistry 2003, 42, 3089–3095. [Google Scholar] [CrossRef]
- Jensen, A.; Calvayrac, G.; Karahalil, B.; Bohr, V.A.; Stevnsner, T. Mammalian 8-Oxoguanine DNA Glycosylase 1 Incises 8-Oxoadenine Opposite Cytosine in Nuclei and Mitochondria, While a Different Glycosylase Incises 8-Oxoadenine Opposite Guanine in Nuclei. J. Biol. Chem. 2003, 278, 19541–19548. [Google Scholar] [CrossRef]
- Bruner, S.D.; Norman, D.P.G.; Verdine, G.L. Structural Basis for Recognition and Repair of the Endogenous Mutagen 8-Oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W. Stimulation of Human 8-Oxoguanine-DNA Glycosylase by AP-Endonuclease: Potential Coordination of the Initial Steps in Base Excision Repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E. Mechanism of Stimulation of the DNA Glycosylase Activity of hOGG1 by the Major Human AP Endonuclease: Bypass of the AP Lyase Activity Step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Shinmura, K.; Yamaguchi, S.; Tani, M.; Seki, S.; Murakami, H.; Nojima, Y.; Yokota, J. Enhancement of OGG1 Protein AP Lyase Activity by Increase of APEX Protein. Mutat. Res. DNA Repair 2001, 486, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of Stimulation of Human 8-Oxoguanine–DNA Glycosylase by AP Endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Grin, I.R.; Dianov, G.L.; Zharkov, D.O. The Role of Mammalian NEIL1 Protein in the Repair of 8-Oxo-7,8-Dihydroadenine in DNA. FEBS Lett. 2010, 584, 1553–1557. [Google Scholar] [CrossRef]
- Talhaoui, I.; Couvé, S.; Ishchenko, A.A.; Kunz, C.; Schär, P.; Saparbaev, M. 7,8-Dihydro-8-Oxoadenine, a Highly Mutagenic Adduct, Is Repaired by Escherichia Coli and Human Mismatch-Specific Uracil/Thymine-DNA Glycosylases. Nucleic Acids Res. 2013, 41, 912–923. [Google Scholar] [CrossRef]
- Servius, H.W.; Pidugu, L.S.; Sherman, M.E.; Drohat, A.C. Rapid Excision of Oxidized Adenine by Human Thymine DNA Glycosylase. J. Biol. Chem. 2023, 299, 102756. [Google Scholar] [CrossRef]
- Tuo, J.; Chen, C.; Zeng, X.; Christiansen, M.; Bohr, V.A. Functional Crosstalk between hOgg1 and the Helicase Domain of Cockayne Syndrome Group B Protein. DNA Repair 2002, 1, 913–927. [Google Scholar] [CrossRef]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; Von Thaler, A.-K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dollé, M.E.T.; Kuiper, R.; Majora, M.; et al. Proteins of Nucleotide and Base Excision Repair Pathways Interact in Mitochondria to Protect from Loss of Subcutaneous Fat, a Hallmark of Aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Teson, M.; De Jesus, B.M.B.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjørås, M.; Crescenzi, M.; Pedrini, A.M.; et al. New Functions of XPC in the Protection of Human Skin Cells from Oxidative Damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Batra, V.K.; Beard, W.A.; Hou, E.W.; Pedersen, L.C.; Prasad, R.; Wilson, S.H. Mutagenic Conformation of 8-Oxo-7,8-Dihydro-2′-dGTP in the Confines of a DNA Polymerase Active Site. Nat. Struct. Mol. Biol. 2010, 17, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C. Mechanisms of Accurate Translesion Synthesis by Human DNA Polymerase Eta. EMBO J. 2000, 19, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Masutani, C.; Iwai, S.; Hanaoka, F. Complementation of Defective Translesion Synthesis and UV Light Sensitivity in Xeroderma Pigmentosum Variant Cells by Human and Mouse DNA Polymerase. Nucleic Acids Res. 2000, 28, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Ziv, O.; Gueranger, Q.; Geacintov, N.; Livneh, Z. Reduced Efficiency and Increased Mutagenicity of Translesion DNA Synthesis across a TT Cyclobutane Pyrimidine Dimer, but Not a TT 6-4 Photoproduct, in Human Cells Lacking DNA Polymerase η. DNA Repair 2008, 7, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Clark, A.B.; McCulloch, S.D.; Yuan, T.; Bronson, R.T.; Kunkel, T.A.; Kucherlapati, R. Increased Susceptibility to UV-Induced Skin Carcinogenesis in Polymerase η–Deficient Mice. Cancer Res. 2006, 66, 87–94. [Google Scholar] [CrossRef]
- Mudrak, S.V.; Welz-Voegele, C.; Jinks-Robertson, S. The Polymerase η Translesion Synthesis DNA Polymerase Acts Independently of the Mismatch Repair System To Limit Mutagenesis Caused by 7,8-Dihydro-8-Oxoguanine in Yeast. Mol. Cell. Biol. 2009, 29, 5316–5326. [Google Scholar] [CrossRef]
- De Padula, M. The Post-Replication Repair RAD18 and RAD6 Genes Are Involved in the Prevention of Spontaneous Mutations Caused by 7,8-Dihydro-8-Oxoguanine in Saccharomyces Cerevisiae. Nucleic Acids Res. 2004, 32, 5003–5010. [Google Scholar] [CrossRef]
- Kamiya, H.; Yamaguchi, A.; Suzuki, T.; Harashima, H. Roles of Specialized DNA Polymerases in Mutagenesis by 8-Hydroxyguanine in Human Cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 686, 90–95. [Google Scholar] [CrossRef]
- Avkin, S.; Livneh, Z. Efficiency, Specificity and DNA Polymerase-Dependence of Translesion Replication across the Oxidative DNA Lesion 8-Oxoguanine in Human Cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 510, 81–90. [Google Scholar] [CrossRef]
- Tolentino, J.H.; Burke, T.J.; Mukhopadhyay, S.; McGregor, W.G.; Basu, A.K. Inhibition of DNA Replication Fork Progression and Mutagenic Potential of 1, N6-Ethenoadenine and 8-Oxoguanine in Human Cell Extracts. Nucleic Acids Res. 2007, 36, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Zhang, Y.; Rajpal, D.K.; Wu, X.; Guo, D.; Wang, M.; Taylor, J.-S.; Wang, Z. Specificity of DNA Lesion Bypass by the Yeast DNA Polymerase η. J. Biol. Chem. 2000, 275, 8233–8239. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K.D.; Washington, M.T. Mechanism of Efficient and Accurate Nucleotide Incorporation Opposite 7,8-Dihydro-8-Oxoguanine by Saccharomyces Cerevisiae DNA Polymerase η. Mol. Cell. Biol. 2005, 25, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Yu, S.-L.; Johnson, R.E.; Prakash, L.; Prakash, S. Efficient and Accurate Replication in the Presence of 7,8-Dihydro-8-Oxoguanine by DNA Polymerase η. Nat. Genet. 2000, 25, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, T.D.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural Basis for Error-Free Replication of Oxidatively Damaged DNA by Yeast DNA Polymerase η. Structure 2010, 18, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Rechkoblit, O.; Taylor, J.-S.; Geacintov, N.E.; Wang, Z. Error-Prone Lesion Bypass by Human DNA Polymerase η. Nucleic Acids Res. 2000, 28, 4717–4724. [Google Scholar] [CrossRef] [PubMed]
- Gokce, G.; Ozsarlak-Sozer, G.; Oktay, G.; Kirkali, G.; Jaruga, P.; Dizdaroglu, M.; Kerry, Z. Glutathione Depletion by Buthionine Sulfoximine Induces Oxidative Damage to DNA in Organs of Rabbits in Vivo. Biochemistry 2009, 48, 4980–4987. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Villani, G.; Crespan, E.; Wimmer, U.; Ferrari, E.; Bertocci, B.; Hübscher, U. 8-Oxo-Guanine Bypass by Human DNA Polymerases in the Presence of Auxiliary Proteins. Nature 2007, 447, 606–608. [Google Scholar] [CrossRef]
- Van Der Kemp, P.A.; De Padula, M.; Burguiere-Slezak, G.; Ulrich, H.D.; Boiteux, S. PCNA Monoubiquitylation and DNA Polymerase Ubiquitin-Binding Domain Are Required to Prevent 8-Oxoguanine-Induced Mutagenesis in Saccharomyces Cerevisiae. Nucleic Acids Res. 2009, 37, 2549–2559. [Google Scholar] [CrossRef]
- Calvo, P.A.; Sastre-Moreno, G.; Perpiñá, C.; Guerra, S.; Martínez-Jiménez, M.I.; Blanco, L. The Invariant Glutamate of Human PrimPol DxE Motif Is Critical for Its Mn2+-Dependent Distinctive Activities. DNA Repair 2019, 77, 65–75. [Google Scholar] [CrossRef]
- Makarova, A.V.; Boldinova, E.O.; Belousova, E.A.; Lavrik, O.I. In Vitro Lesion Bypass by Human PrimPol. DNA Repair 2018, 70, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.G.; Woodgate, R. Increased Catalytic Activity and Altered Fidelity of Human DNA Polymerase ι in the Presence of Manganese. J. Biol. Chem. 2007, 282, 24689–24696. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Patra, A.; Yeom, M.; Lee, Y.-S.; Zhang, Q.; Egli, M.; Guengerich, F.P. Kinetic and Structural Impact of Metal Ions and Genetic Variations on Human DNA Polymerase ι. J. Biol. Chem. 2016, 291, 21063–21073. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Ignatov, A.; Miropolskaya, N.; Kulbachinskiy, A. Roles of the Active Site Residues and Metal Cofactors in Noncanonical Base-Pairing during Catalysis by Human DNA Polymerase Iota. DNA Repair 2014, 22, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Blanca, G.; Shevelev, I.; Ramadan, K.; Villani, G.; Spadari, S.; Hübscher, U.; Maga, G. Human DNA Polymerase λ Diverged in Evolution from DNA Polymerase β toward Specific Mn ++ Dependence: A Kinetic and Thermodynamic Study. Biochemistry 2003, 42, 7467–7476. [Google Scholar] [CrossRef] [PubMed]
- Koag, M.-C.; Nam, K.; Lee, S. The Spontaneous Replication Error and the Mismatch Discrimination Mechanisms of Human DNA Polymerase β. Nucleic Acids Res. 2014, 42, 11233–11245. [Google Scholar] [CrossRef] [PubMed]
- Yung, C.; Suzuki, T.; Okugawa, Y.; Kawakami, A.; Loakes, D.; Negishi, K.; Negishi, T. Nucleotide Incorporation against 7,8-Dihydro-8-Oxoguanine Is Influenced by Neighboring Base Sequences in TLS DNA Polymerase Reaction. Nucleic Acids Symp. Ser. 2007, 51, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.W.; Hirst, W.G.; Szewczyk, G.A.; Simmons, L.A. The Effect of Local Sequence Context on Mutational Bias of Genes Encoded on the Leading and Lagging Strands. Curr. Biol. 2016, 26, 692–697. [Google Scholar] [CrossRef]
- Sung, W.; Ackerman, M.S.; Gout, J.-F.; Miller, S.F.; Williams, E.; Foster, P.L.; Lynch, M. Asymmetric Context-Dependent Mutation Patterns Revealed through Mutation–Accumulation Experiments. Mol. Biol. Evol. 2015, 32, 1672–1683. [Google Scholar] [CrossRef]
- Vaziri, C.; Rogozin, I.B.; Gu, Q.; Wu, D.; Day, T.A. Unravelling Roles of Error-Prone DNA Polymerases in Shaping Cancer Genomes. Oncogene 2021, 40, 6549–6565. [Google Scholar] [CrossRef]
- Zhao, Y.; Gregory, M.T.; Biertümpfel, C.; Hua, Y.-J.; Hanaoka, F.; Yang, W. Mechanism of Somatic Hypermutation at the WA Motif by Human DNA Polymerase η. Proc. Natl. Acad. Sci. USA 2013, 110, 8146–8151. [Google Scholar] [CrossRef] [PubMed]
- Schaibley, V.M.; Zawistowski, M.; Wegmann, D.; Ehm, M.G.; Nelson, M.R.; St. Jean, P.L.; Abecasis, G.R.; Novembre, J.; Zöllner, S.; Li, J.Z. The Influence of Genomic Context on Mutation Patterns in the Human Genome Inferred from Rare Variants. Genome Res. 2013, 23, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Petruska, J.; Goodman, M.F. Influence of Neighboring Bases on DNA Polymerase Insertion and Proofreading Fidelity. J. Biol. Chem. 1985, 260, 7533–7539. [Google Scholar] [CrossRef] [PubMed]
- Arnedo-Pac, C.; Muiños, F.; Gonzalez-Perez, A.; Lopez-Bigas, N. Hotspot Propensity across Mutational Processes. bioRxiv 2023. [Google Scholar] [CrossRef]









| In Vitro/ In Vivo | Enzyme/ Organism | Sequence | Assay | Accuracy | Ref. |
|---|---|---|---|---|---|
| in vitro | KF | 3′-…TCGGXTGGT…-5′ | Primer extension | Effectively bypasses | [30] |
| 5′-CCTTCXCTAC…-3′ 5′-GTTGXGTAC…-3′ | Primer extension and steady-state kinetics | Preferentially inserts dTMP and small amounts of dAMP and dGMP; Fins dTTP~500–1000x > Fins dAMP/dGMP; Fext oxoA:T~630–3000x > Fext 8-oxoA:A and 8-oxoA:C | [43] | ||
| 5′-…GGCCXAG-3′ (HRAS template) | Primer extension | Inserts only dTMP | [42] | ||
| 5′-…GGTCXTCGG-3′ | Primer extension | Preferentially inserts dTMP and small amounts of dAMP | [44] | ||
| Taq pol | 3′-…TCGGXTGGT…-5′ | Primer extension | Inserts only dTMP | [30] | |
| 5′-…GGCCXAG-3′ (HRAS template) | Primer extension | Inserts only dTMP | [42] | ||
| Dpo4 | 5′-TCATXGAAT…-3′ 5′-TTCATXGAAT…-3′ | Steady-state kinetics | Fins dTMP~14x > Fins dGMP Fins 8-oxoA:dGMP~320x > Fins A:dGMP Fext 8-oxoA:T~5x > Fext 8-oxoA:G | [46,47] | |
| in vivo | E. coli | 5′-GCTXG-3′ | Mutagenesis assay | MF~0.2–0.3% | [41] |
| In Vitro/ In Vivo | Enzyme/ Cell Line | Sequence | Assay | Accuracy | Ref. |
|---|---|---|---|---|---|
| In vitro | Pol α | 5′-CCTTCXCTAC…-3′ 5′-GTTGXGTAC…-3′ | Primer extension and steady-state kinetics | Preferentially inserts dTMP and small amounts of dGMP; Fins dTTP~10x > dGTP; Fext 8-oxoA:T and 8-oxoA:G~280–3500x < than for KF | [43] |
| 5′-…GGCCXAG-3′ (HRAS template) | Primer extension | dTMP > dGMP * | [42] | ||
| Pol β | 5′-CCTTCXCTAC…-3′ 5′-GTTGXGTAC…-3′ | Primer extension and steady-state kinetics | Preferentially inserts dTMP and small amounts of dGMP; Fins dTTP~18x > dGTP | [43] | |
| 5′-…GGCCXAG-3′ (HRAS template) | Primer extension | dTMP > dGMP > dAMP * | [42] | ||
| 5′-…TACGXCGCA…-3′ | Steady-state kinetics | Fins 8-oxoA:dTMP ~2.5x < Fins A:dTMP kcat/Km 8-oxoA:T~4x > kcat/Km 8-oxoA:G | [36] | ||
| Pol η | 5′-…TACGXCGCA…-3′ | Steady-state kinetics | Fins dTTP~2x > dGTP; kcat/Km 8-oxoA:T~1.1x > kcat/Km 8-oxoA:G | [36] | |
| HeLa and COS-7 cell extracts | 5′-CCTTCXCTAC…-3′ | Primer extension | Inserts only dTMP | [48] | |
| In vivo | NIH 3T3 cells | 5′-…GGCCXAG-3′ (HRAS template) | Mutagenesis assay | A → C in 55% clones, A → G in 10% clones | [42] |
| COS-7 cells | 5′-…TCCTXGCCT…-3′ (non-coding strand of HRAS template) | Mutagenesis assay | MF~1.2% | [50] | |
| 5′-…CCTGXCCTC…-3′ | MF~0.24% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruchinin, A.A.; Kamzeeva, P.N.; Zharkov, D.O.; Aralov, A.V.; Makarova, A.V. 8-Oxoadenine: A «New» Player of the Oxidative Stress in Mammals? Int. J. Mol. Sci. 2024, 25, 1342. https://doi.org/10.3390/ijms25021342
Kruchinin AA, Kamzeeva PN, Zharkov DO, Aralov AV, Makarova AV. 8-Oxoadenine: A «New» Player of the Oxidative Stress in Mammals? International Journal of Molecular Sciences. 2024; 25(2):1342. https://doi.org/10.3390/ijms25021342
Chicago/Turabian StyleKruchinin, Alexander A., Polina N. Kamzeeva, Dmitry O. Zharkov, Andrey V. Aralov, and Alena V. Makarova. 2024. "8-Oxoadenine: A «New» Player of the Oxidative Stress in Mammals?" International Journal of Molecular Sciences 25, no. 2: 1342. https://doi.org/10.3390/ijms25021342
APA StyleKruchinin, A. A., Kamzeeva, P. N., Zharkov, D. O., Aralov, A. V., & Makarova, A. V. (2024). 8-Oxoadenine: A «New» Player of the Oxidative Stress in Mammals? International Journal of Molecular Sciences, 25(2), 1342. https://doi.org/10.3390/ijms25021342