
Distinct Alterations in Dendritic Spine Morphology in the Absence of β-Neurexins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
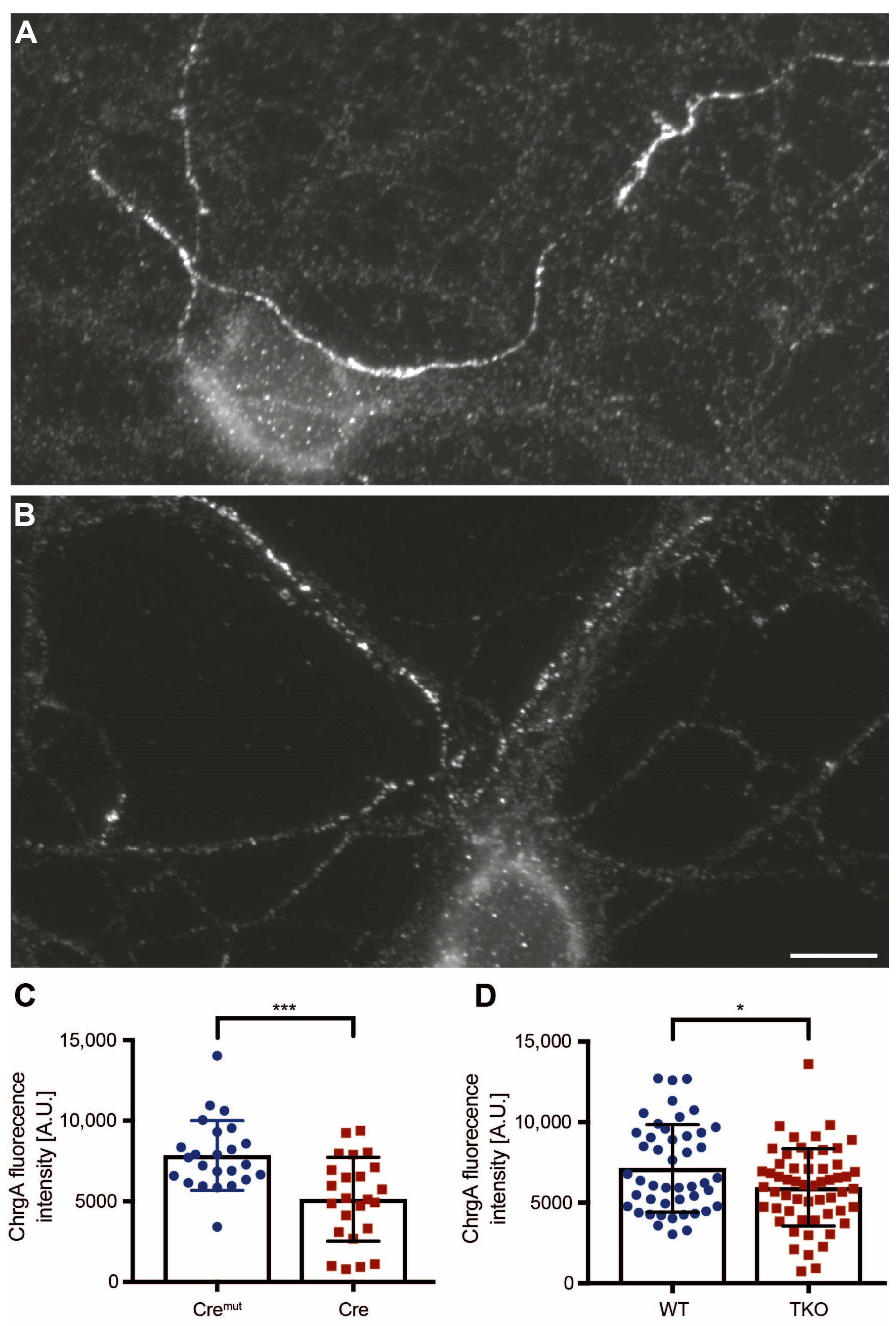
2.1. DCV Distribution Is Altered in Cultured Neurons of Two βNrxn Deletion Mouse Models
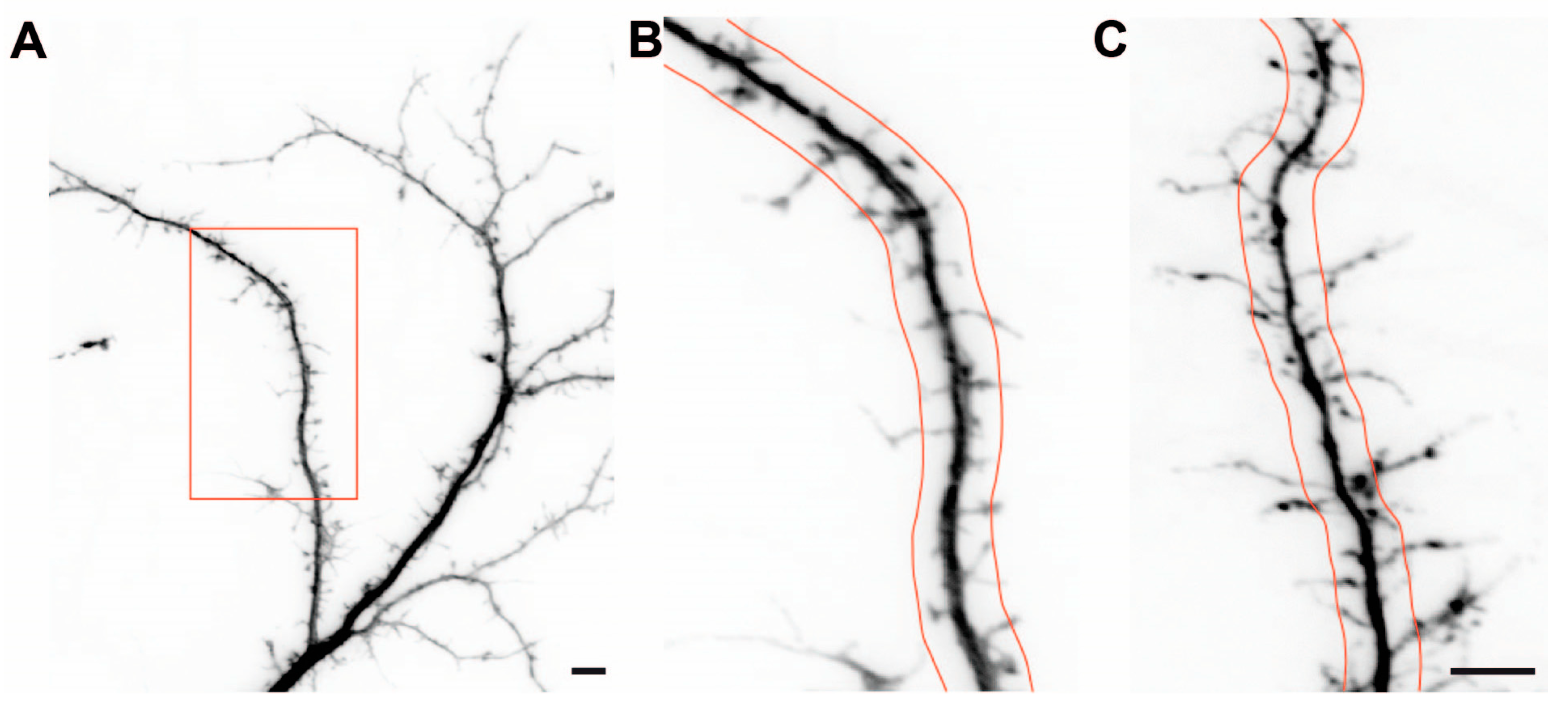
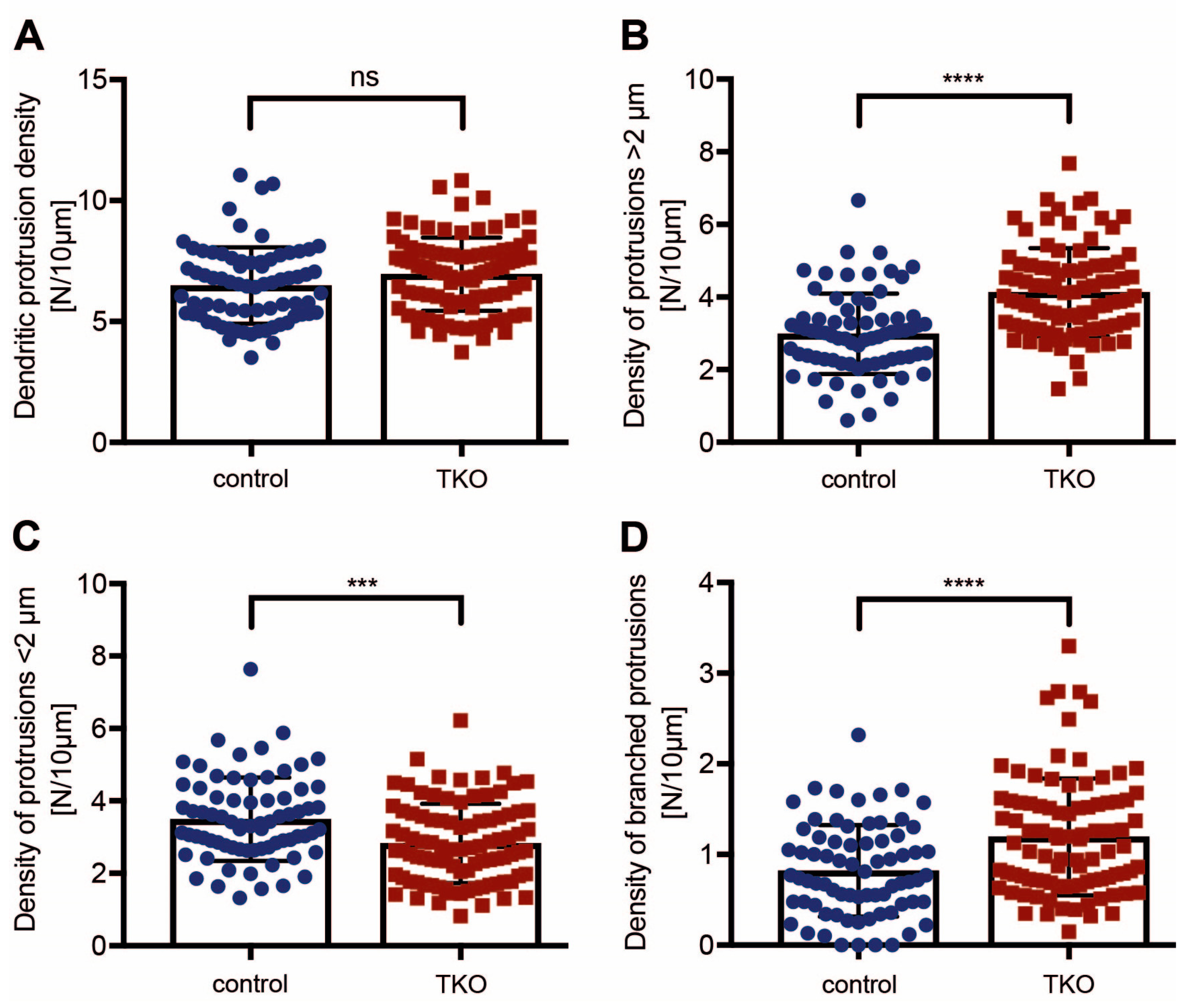
2.2. Dendritic Spine Alterations in Cultured βNrxn-Deficient Neurons
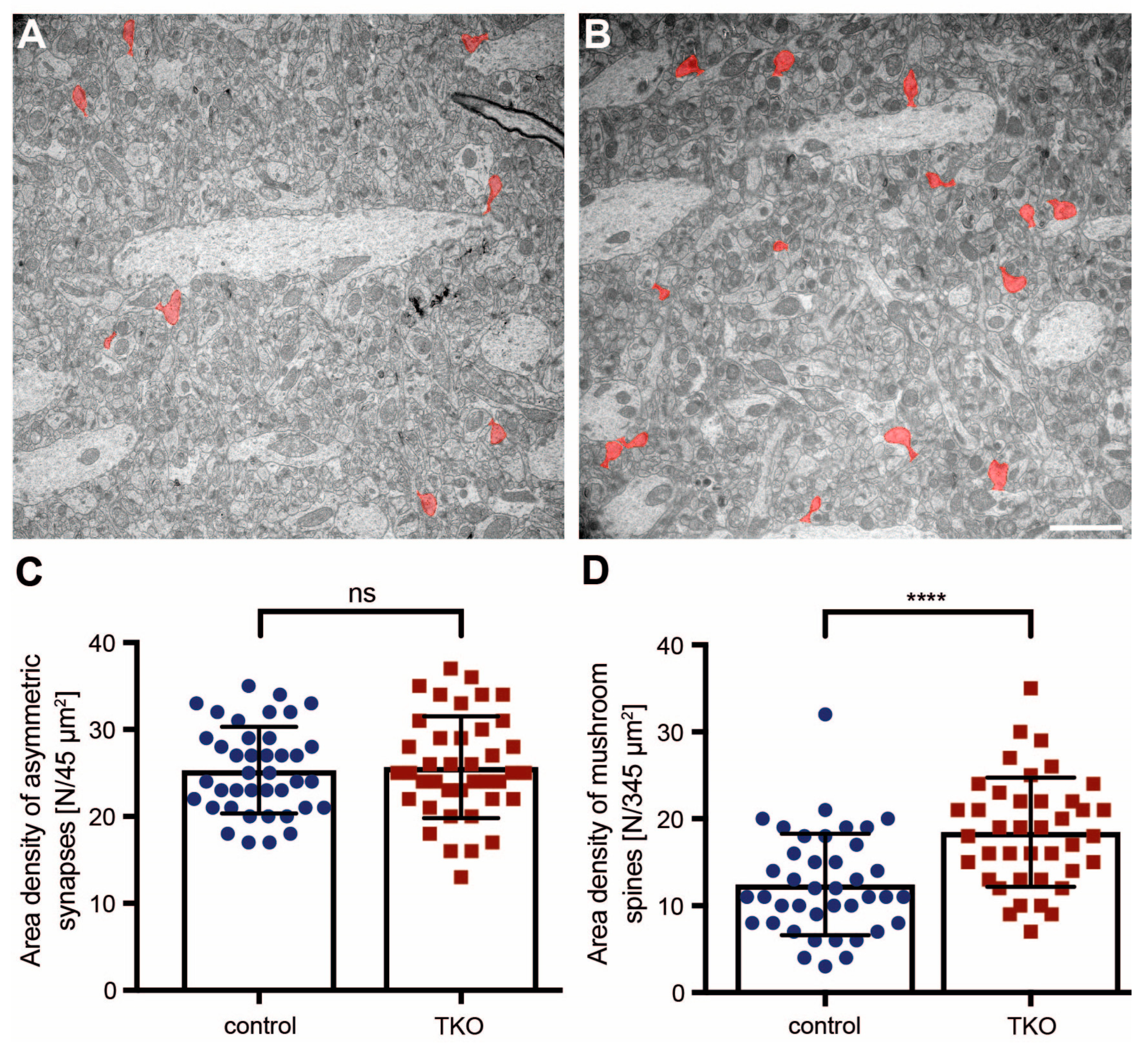
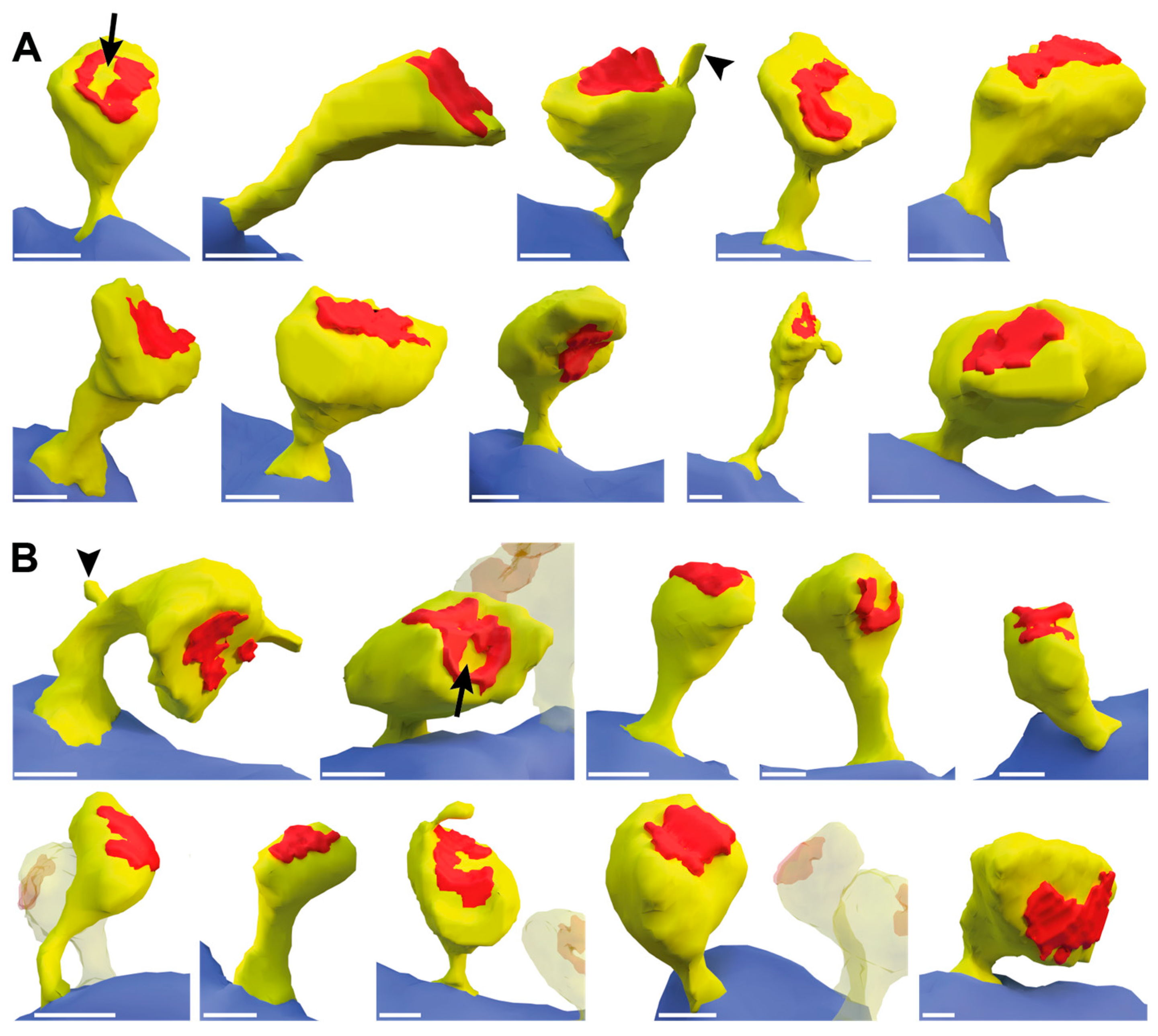
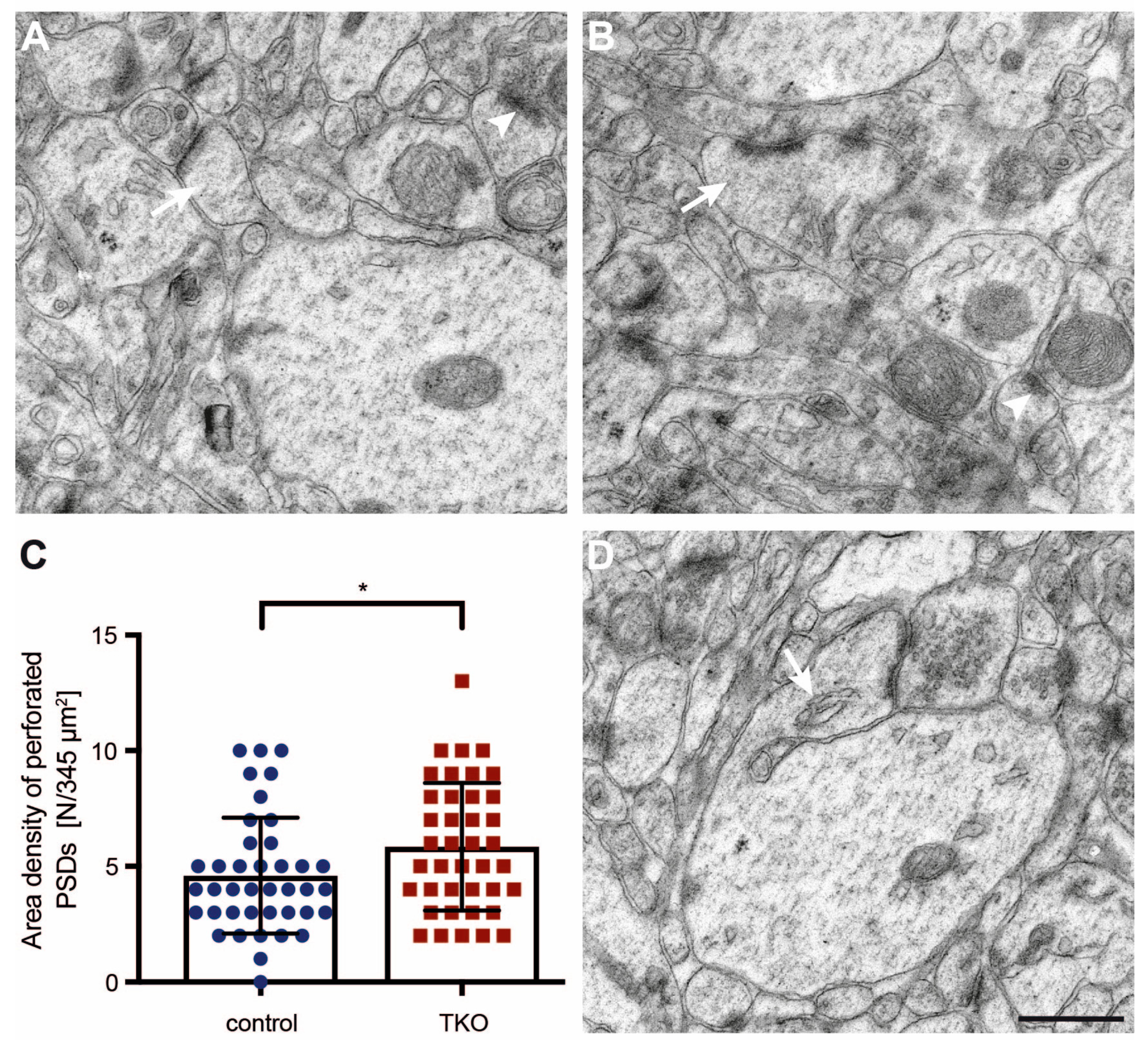
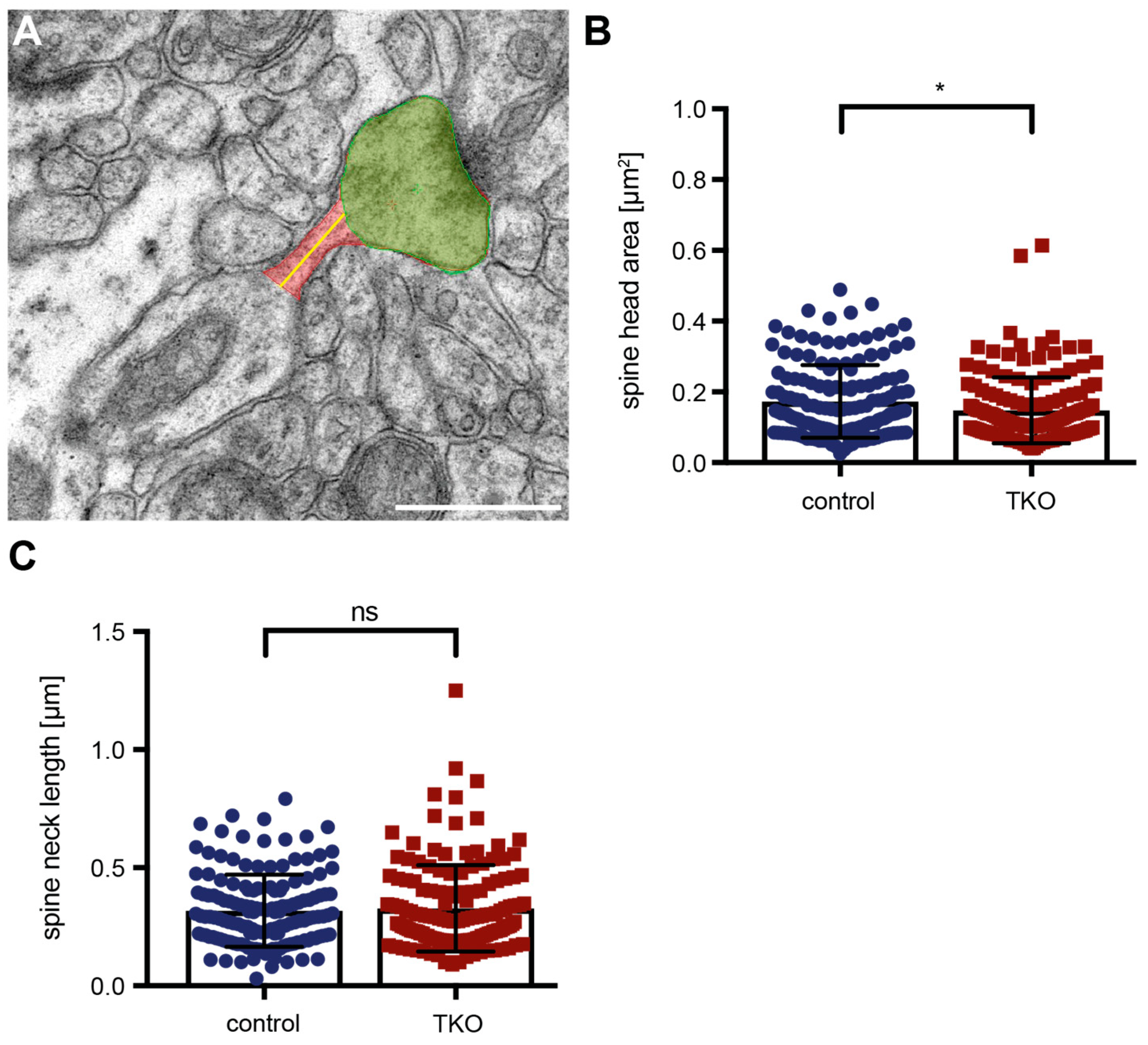
2.3. Dendritic Spine Alterations in the Hippocampal Tissue of βNrxn-Deficient Mice
3. Discussion
3.1. Our Strategy for Detecting the ßNrxn Spine Phenotype Is Sensitive and Reliable
3.2. Putative Mechanisms for Explaining the ßNrxn Spine Phenotype
3.3. Disease Associations of the ßNrxn Phenotype
4. Materials and Methods
4.1. Animals
4.2. Analysis of βNrxn-Deficient Neurons in Culture
4.2.1. Neuronal Cell Culture
4.2.2. Lentivirus Production and Transduction
4.2.3. Transfection of Cell Cultures
4.2.4. Immunocytochemistry of Cell Cultures
4.2.5. Fluorescent Microscopy of Cell Cultures
4.2.6. Analysis of ChrgA-Stained Axons
4.2.7. Analysis of RFP-Labeled Dendrites and Spines
4.3. Electron Microscopic Analysis of βNrxn-Deficient Brain Tissue
4.3.1. Preparation of Mouse Hippocampi
4.3.2. Three-Dimensional Reconstructions
4.3.3. Ultrastructural Image Analysis
4.4. Software and Statistics
4.4.1. Three-Dimensional Reconstruction
4.4.2. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Post-Synapses in the Brain: Role of Dendritic and Spine Structures. Biomedicines 2022, 10, 1859. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Ziv, N.E.; Okazaki, H.; Yagishita, S.; Toyoizumi, T. Spine dynamics in the brain, mental disorders and artificial neural networks. Nat. Rev. Neurosci. 2021, 22, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Bourne, J.N.; Harris, K.M. Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 2008, 31, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Yuste, R. Dendritic spines and distributed circuits. Neuron 2011, 71, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Jensen, F.E.; Tsao, B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: Implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 1992, 12, 2685–2705. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Duan, X.; Taylor, M.R.; Martin, E.A.; Muralidhar, S.; Wang, Y.; Gangi-Wellman, L.; Das, S.C.; Yamagata, M.; West, P.J.; et al. Heterophilic Type II Cadherins Are Required for High-Magnitude Synaptic Potentiation in the Hippocampus. Neuron 2017, 96, 160–176.e8. [Google Scholar] [CrossRef] [PubMed]
- Bourne, J.; Harris, K.M. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 2007, 17, 381–386. [Google Scholar] [CrossRef]
- Chirillo, M.A.; Waters, M.S.; Lindsey, L.F.; Bourne, J.N.; Harris, K.M. Local resources of polyribosomes and SER promote synapse enlargement and spine clustering after long-term potentiation in adult rat hippocampus. Sci. Rep. 2019, 9, 3861. [Google Scholar] [CrossRef]
- Baczynska, E.; Pels, K.K.; Basu, S.; Wlodarczyk, J.; Ruszczycki, B. Quantification of Dendritic Spines Remodeling under Physiological Stimuli and in Pathological Conditions. Int. J. Mol. Sci. 2021, 22, 4053. [Google Scholar] [CrossRef]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 2012, 4, a005587. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.A.; Sabatini, B.L. Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 2007, 30, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Fukuda, M.; Watanabe, S.; Hayashi-Takagi, A.; Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010, 33, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.E.; Zuo, Y. Cortical dendritic spine development and plasticity: Insights from in vivo imaging. Curr. Opin. Neurobiol. 2018, 53, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Higashi, T.; Obashi, K.; Sato, Y.; Komiyama, N.H.; Grant, S.G.N.; Okabe, S. Computational geometry analysis of dendritic spines by structured illumination microscopy. Nat. Commun. 2019, 10, 1285. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Koleske, A.J. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 2010, 33, 349–378. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, M.; Lanoue, V.; Hotulainen, P. Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef]
- Gomez, A.M.; Traunmuller, L.; Scheiffele, P. Neurexins: Molecular codes for shaping neuronal synapses. Nat. Rev. Neurosci. 2021, 22, 137–151. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 213. [Google Scholar] [CrossRef]
- Sudhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, D.; Nguyen, T.M.; Russo, G.; Heber, S.; Patrignani, A.; Ahrne, E.; Scheiffele, P. Targeted combinatorial alternative splicing generates brain region-specific repertoires of neurexins. Neuron 2014, 84, 386–398. [Google Scholar] [CrossRef]
- Treutlein, B.; Gokce, O.; Quake, S.R.; Sudhof, T.C. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E1291–E1299. [Google Scholar] [CrossRef] [PubMed]
- Biederer, T.; Kaeser, P.S.; Blanpied, T.A. Transcellular Nanoalignment of Synaptic Function. Neuron 2017, 96, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Sogabe, T.; Hayashi, A.; Motohashi, J.; Miura, E.; Arai, I.; Yuzaki, M. In vivo nanoscopic landscape of neurexin ligands underlying anterograde synapse specification. Neuron 2022, 110, 3168–3185 e3168. [Google Scholar] [CrossRef]
- Lloyd, B.A.; Han, Y.; Roth, R.; Zhang, B.; Aoto, J. Neurexin-3 subsynaptic densities are spatially distinct from Neurexin-1 and essential for excitatory synapse nanoscale organization in the hippocampus. Nat. Commun. 2023, 14, 4706. [Google Scholar] [CrossRef]
- Anderson, G.R.; Aoto, J.; Tabuchi, K.; Foldy, C.; Covy, J.; Yee, A.X.; Wu, D.; Lee, S.J.; Chen, L.; Malenka, R.C.; et al. beta-Neurexins Control Neural Circuits by Regulating Synaptic Endocannabinoid Signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef]
- Chen, L.Y.; Jiang, M.; Zhang, B.; Gokce, O.; Sudhof, T.C. Conditional Deletion of All Neurexins Defines Diversity of Essential Synaptic Organizer Functions for Neurexins. Neuron 2017, 94, 611–625.e14. [Google Scholar] [CrossRef]
- Missler, M.; Zhang, W.; Rohlmann, A.; Kattenstroth, G.; Hammer, R.E.; Gottmann, K.; Sudhof, T.C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 2003, 423, 939–948. [Google Scholar] [CrossRef]
- Dudanova, I.; Tabuchi, K.; Rohlmann, A.; Sudhof, T.C.; Missler, M. Deletion of alpha-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J. Comp. Neurol. 2007, 502, 261–274. [Google Scholar] [CrossRef]
- Klatt, O.; Repetto, D.; Brockhaus, J.; Reissner, C.; El Khallouqi, A.; Rohlmann, A.; Heine, M.; Missler, M. Endogenous beta-neurexins on axons and within synapses show regulated dynamic behavior. Cell Rep. 2021, 35, 109266. [Google Scholar] [CrossRef] [PubMed]
- Boucard, A.A.; Chubykin, A.A.; Comoletti, D.; Taylor, P.; Sudhof, T.C. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 2005, 48, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Ichtchenko, K.; Hata, Y.; Nguyen, T.; Ullrich, B.; Missler, M.; Moomaw, C.; Sudhof, T.C. Neuroligin 1: A splice site-specific ligand for beta-neurexins. Cell 1995, 81, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Reissner, C.; Klose, M.; Fairless, R.; Missler, M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc. Natl. Acad. Sci. USA 2008, 105, 15124–15129. [Google Scholar] [CrossRef] [PubMed]
- de Wit, J.; Sylwestrak, E.; O’Sullivan, M.L.; Otto, S.; Tiglio, K.; Savas, J.N.; Yates, J.R., 3rd; Comoletti, D.; Taylor, P.; Ghosh, A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 2009, 64, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Fuccillo, M.V.; Malenka, R.C.; Sudhof, T.C. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 2009, 64, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, T.J.; Pancaroglu, R.; Kang, Y.; Rooyakkers, A.; Craig, A.M. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J. Neurosci. 2010, 30, 7495–7506. [Google Scholar] [CrossRef]
- Reissner, C.; Stahn, J.; Breuer, D.; Klose, M.; Pohlentz, G.; Mormann, M.; Missler, M. Dystroglycan Binding to alpha-Neurexin Competes with Neurexophilin-1 and Neuroligin in the Brain. J. Biol. Chem. 2014, 289, 27585–27603. [Google Scholar] [CrossRef]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Sudhof, T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001, 154, 435–445. [Google Scholar] [CrossRef]
- Boucard, A.A.; Ko, J.; Sudhof, T.C. High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 produces an intercellular adhesion complex. J. Biol. Chem. 2012, 287, 9399–9413. [Google Scholar] [CrossRef]
- Matsuda, K.; Yuzaki, M. Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur. J. Neurosci. 2011, 33, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Lee, S.J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.Y.; Lee, S.J.; Uemura, T.; Yoshida, T.; Yasumura, M.; Watanabe, M.; Mishina, M. Differential interactions of cerebellin precursor protein (Cbln) subtypes and neurexin variants for synapse formation of cortical neurons. Biochem. Biophys. Res. Commun. 2011, 406, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Brockhaus, J.; Schreitmuller, M.; Repetto, D.; Klatt, O.; Reissner, C.; Elmslie, K.; Heine, M.; Missler, M. alpha-Neurexins Together with alpha2delta-1 Auxiliary Subunits Regulate Ca(2+) Influx through Cav2.1 Channels. J. Neurosci. 2018, 38, 8277–8294. [Google Scholar] [CrossRef] [PubMed]
- Kattenstroth, G.; Tantalaki, E.; Sudhof, T.C.; Gottmann, K.; Missler, M. Postsynaptic N-methyl-D-aspartate receptor function requires alpha-neurexins. Proc. Natl. Acad. Sci. USA 2004, 101, 2607–2612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Rohlmann, A.; Sargsyan, V.; Aramuni, G.; Hammer, R.E.; Sudhof, T.C.; Missler, M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J. Neurosci. 2005, 25, 4330–4342. [Google Scholar] [CrossRef]
- Ferdos, S.; Brockhaus, J.; Missler, M.; Rohlmann, A. Deletion of beta-Neurexins in Mice Alters the Distribution of Dense-Core Vesicles in Presynapses of Hippocampal and Cerebellar Neurons. Front. Neuroanat. 2022, 15, 757017. [Google Scholar] [CrossRef]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef]
- Puntman, D.C.; Arora, S.; Farina, M.; Toonen, R.F.; Verhage, M. Munc18-1 is essential for neuropeptide secretion in neurons. J. Neurosci. 2021, 41, 5980–5993. [Google Scholar] [CrossRef]
- Persoon, C.M.; Moro, A.; Nassal, J.P.; Farina, M.; Broeke, J.H.; Arora, S.; Dominguez, N.; van Weering, J.R.; Toonen, R.F.; Verhage, M. Pool size estimations for dense-core vesicles in mammalian CNS neurons. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Tacke, C.; Korte, M. BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res. 2020, 382, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Bharat, V.; Siebrecht, M.; Burk, K.; Ahmed, S.; Reissner, C.; Kohansal-Nodehi, M.; Steubler, V.; Zweckstetter, M.; Ting, J.T.; Dean, C. Capture of Dense Core Vesicles at Synapses by JNK-Dependent Phosphorylation of Synaptotagmin-4. Cell Rep. 2017, 21, 2118–2133. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, N.; van Weering, J.R.T.; Borges, R.; Toonen, R.F.G.; Verhage, M. Dense-core vesicle biogenesis and exocytosis in neurons lacking chromogranins A and B. J. Neurochem. 2018, 144, 241–254. [Google Scholar] [CrossRef]
- Sorra, K.E.; Mishra, A.; Kirov, S.A.; Harris, K.M. Dense core vesicles resemble active-zone transport vesicles and are diminished following synaptogenesis in mature hippocampal slices. Neuroscience 2006, 141, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Petrak, L.J.; Harris, K.M.; Kirov, S.A. Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J. Comp. Neurol. 2005, 484, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, J.; Katona, G.; Rozsa, B.; Nagerl, U.V. Spine neck plasticity regulates compartmentalization of synapses. Nat. Neurosci. 2014, 17, 678–685. [Google Scholar] [CrossRef]
- Cornejo, V.H.; Ofer, N.; Yuste, R. Voltage compartmentalization in dendritic spines in vivo. Science 2022, 375, 82–86. [Google Scholar] [CrossRef]
- Kinney, J.P.; Spacek, J.; Bartol, T.M.; Bajaj, C.L.; Harris, K.M.; Sejnowski, T.J. Extracellular sheets and tunnels modulate glutamate diffusion in hippocampal neuropil. J. Comp. Neurol. 2013, 521, 448–464. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Nishida, H.; Okabe, S. Direct astrocytic contacts regulate local maturation of dendritic spines. J. Neurosci. 2007, 27, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Muller, D.; Nikonenko, I. Astrocyte-synapse structural plasticity. Neural Plast. 2014, 2014, 232105. [Google Scholar] [CrossRef]
- Verbich, D.; Prenosil, G.A.; Chang, P.K.; Murai, K.K.; McKinney, R.A. Glial glutamate transport modulates dendritic spine head protrusions in the hippocampus. Glia 2012, 60, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.G. Axo-somatic and axo-dendritic synapses of the cerebral cortex: An electron microscope study. J. Anat. 1959, 93, 420–433. [Google Scholar]
- Arellano, J.I.; Benavides-Piccione, R.; Defelipe, J.; Yuste, R. Ultrastructure of dendritic spines: Correlation between synaptic and spine morphologies. Front. Neurosci. 2007, 1, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.; Bui, V.D.; Haas, C.A.; Beck, J.; Queisser, G.; Vlachos, A. Calcium modeling of spine apparatus-containing human dendritic spines demonstrates an “all-or-nothing” communication switch between the spine head and dendrite. PLoS Comput. Biol. 2022, 18, e1010069. [Google Scholar] [CrossRef]
- Sorra, K.E.; Fiala, J.C.; Harris, K.M. Critical assessment of the involvement of perforations, spinules, and spine branching in hippocampal synapse formation. J. Comp. Neurol. 1998, 398, 225–240. [Google Scholar] [CrossRef]
- Sun, Y.; Smirnov, M.; Kamasawa, N.; Yasuda, R. Rapid Ultrastructural Changes in the PSD and Surrounding Membrane after Induction of Structural LTP in Single Dendritic Spines. J. Neurosci. 2021, 41, 7003–7014. [Google Scholar] [CrossRef]
- Fortin, D.A.; Davare, M.A.; Srivastava, T.; Brady, J.D.; Nygaard, S.; Derkach, V.A.; Soderling, T.R. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J. Neurosci. 2010, 30, 11565–11575. [Google Scholar] [CrossRef]
- Araya, R.; Vogels, T.P.; Yuste, R. Activity-dependent dendritic spine neck changes are correlated with synaptic strength. Proc. Natl. Acad. Sci. USA 2014, 111, E2895–E2904. [Google Scholar] [CrossRef]
- Chubykin, A.A.; Liu, X.; Comoletti, D.; Tsigelny, I.; Taylor, P.; Sudhof, T.C. Dissection of synapse induction by neuroligins: Effect of a neuroligin mutation associated with autism. J. Biol. Chem. 2005, 280, 22365–22374. [Google Scholar] [CrossRef] [PubMed]
- Dean, C.; Scholl, F.G.; Choih, J.; DeMaria, S.; Berger, J.; Isacoff, E.; Scheiffele, P. Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 2003, 6, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.R.; Zhang, X.; Jin, S.X.; Linhoff, M.W.; Craig, A.M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 2004, 119, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovska, V.; Ge, Y.; Xu, Q.; Li, S.; Zhang, P.; Craig, A.M. Neurexin-beta Mediates the Synaptogenic Activity of Amyloid Precursor Protein. J. Neurosci. 2022, 42, 8936–8947. [Google Scholar] [CrossRef] [PubMed]
- Nimchinsky, E.A.; Sabatini, B.L.; Svoboda, K. Structure and function of dendritic spines. Annu. Rev. Physiol. 2002, 64, 313–353. [Google Scholar] [CrossRef] [PubMed]
- Yuste, R.; Bonhoeffer, T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci. 2001, 24, 1071–1089. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Okabe, S. Imaging of spine synapses using super-resolution microscopy. Anat. Sci. Int. 2021, 96, 343–358. [Google Scholar] [CrossRef]
- Borczyk, M.; Radwanska, K.; Giese, K.P. The importance of ultrastructural analysis of memory. Brain Res. Bull. 2021, 173, 28–36. [Google Scholar] [CrossRef]
- Schreiner, D.; Simicevic, J.; Ahrne, E.; Schmidt, A.; Scheiffele, P. Quantitative isoform-profiling of highly diversified recognition molecules. eLife 2015, 4, e07794. [Google Scholar] [CrossRef]
- Fuccillo, M.V.; Foldy, C.; Gokce, O.; Rothwell, P.E.; Sun, G.L.; Malenka, R.C.; Sudhof, T.C. Single-Cell mRNA Profiling Reveals Cell-Type-Specific Expression of Neurexin Isoforms. Neuron 2015, 87, 326–340. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, Z.J. Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc. Natl. Acad. Sci. USA 2010, 107, 22699–22704. [Google Scholar] [CrossRef] [PubMed]
- Neupert, C.; Schneider, R.; Klatt, O.; Reissner, C.; Repetto, D.; Biermann, B.; Niesmann, K.; Missler, M.; Heine, M. Regulated Dynamic Trafficking of Neurexins Inside and Outside of Synaptic Terminals. J. Neurosci. 2015, 35, 13629–13647. [Google Scholar] [CrossRef] [PubMed]
- Yuste, R.; Bonhoeffer, T. Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 2004, 5, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Sigler, A.; Oh, W.C.; Imig, C.; Altas, B.; Kawabe, H.; Cooper, B.H.; Kwon, H.B.; Rhee, J.S.; Brose, N. Formation and Maintenance of Functional Spines in the Absence of Presynaptic Glutamate Release. Neuron 2017, 94, 304–311.e4. [Google Scholar] [CrossRef]
- Verhage, M.; Maia, A.S.; Plomp, J.J.; Brussaard, A.B.; Heeroma, J.H.; Vermeer, H.; Toonen, R.F.; Hammer, R.E.; van den Berg, T.K.; Missler, M.; et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000, 287, 864–869. [Google Scholar] [CrossRef]
- Sando, R.; Bushong, E.; Zhu, Y.; Huang, M.; Considine, C.; Phan, S.; Ju, S.; Uytiepo, M.; Ellisman, M.; Maximov, A. Assembly of Excitatory Synapses in the Absence of Glutamatergic Neurotransmission. Neuron 2017, 94, 312–321.e3. [Google Scholar] [CrossRef]
- Tyler, W.J.; Pozzo-Miller, L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J. Physiol. 2003, 553, 497–509. [Google Scholar] [CrossRef]
- Gottmann, K.; Mittmann, T.; Lessmann, V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 2009, 199, 203–234. [Google Scholar] [CrossRef]
- Ramsey, A.M.; Tang, A.H.; LeGates, T.A.; Gou, X.Z.; Carbone, B.E.; Thompson, S.M.; Biederer, T.; Blanpied, T.A. Subsynaptic positioning of AMPARs by LRRTM2 controls synaptic strength. Sci. Adv. 2021, 7, eabf3126. [Google Scholar] [CrossRef]
- Goncalves, J.; Bartol, T.M.; Camus, C.; Levet, F.; Menegolla, A.P.; Sejnowski, T.J.; Sibarita, J.B.; Vivaudou, M.; Choquet, D.; Hosy, E. Nanoscale co-organization and coactivation of AMPAR, NMDAR, and mGluR at excitatory synapses. Proc. Natl. Acad. Sci. USA 2020, 117, 14503–14511. [Google Scholar] [CrossRef]
- Li, S.; Raychaudhuri, S.; Lee, S.A.; Brockmann, M.M.; Wang, J.; Kusick, G.; Prater, C.; Syed, S.; Falahati, H.; Ramos, R.; et al. Asynchronous release sites align with NMDA receptors in mouse hippocampal synapses. Nat. Commun. 2021, 12, 677. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Aoto, J.; Sudhof, T.C. Alternative Splicing of Presynaptic Neurexins Differentially Controls Postsynaptic NMDA and AMPA Receptor Responses. Neuron 2019, 102, 993–1008.e5. [Google Scholar] [CrossRef] [PubMed]
- Aoto, J.; Foldy, C.; Ilcus, S.M.; Tabuchi, K.; Sudhof, T.C. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat. Neurosci. 2015, 18, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Patzke, C.; Liakath-Ali, K.; Seigneur, E.; Sudhof, T.C. GluD1 is a signal transduction device disguised as an ionotropic receptor. Nature 2021, 595, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.P.; Nedivi, E. Spine Dynamics: Are They All the Same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Matsuzaki, M.; Noguchi, J.; Yasumatsu, N.; Nakahara, H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003, 26, 360–368. [Google Scholar] [CrossRef]
- Bhouri, M.; Morishita, W.; Temkin, P.; Goswami, D.; Kawabe, H.; Brose, N.; Sudhof, T.C.; Craig, A.M.; Siddiqui, T.J.; Malenka, R. Deletion of LRRTM1 and LRRTM2 in adult mice impairs basal AMPA receptor transmission and LTP in hippocampal CA1 pyramidal neurons. Proc. Natl. Acad. Sci. USA 2018, 115, E5382–E5389. [Google Scholar] [CrossRef]
- Jiang, M.; Polepalli, J.; Chen, L.Y.; Zhang, B.; Sudhof, T.C.; Malenka, R.C. Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Mol. Psychiatry 2017, 22, 375–383. [Google Scholar] [CrossRef][Green Version]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Sudhof, T.C.; Brose, N. Neuroligins determine synapse maturation and function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.J.; Gokce, O.; Sudhof, T.C. Neuroligins Sculpt Cerebellar Purkinje-Cell Circuits by Differential Control of Distinct Classes of Synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef]
- de Arce, K.P.; Ribic, A.; Chowdhury, D.; Watters, K.; Thompson, G.J.; Sanganahalli, B.G.; Lippard, E.T.C.; Rohlmann, A.; Strittmatter, S.M.; Missler, M.; et al. Concerted roles of LRRTM1 and SynCAM 1 in organizing prefrontal cortex synapses and cognitive functions. Nat. Commun. 2023, 14, 459. [Google Scholar] [CrossRef] [PubMed]
- Takashima, N.; Odaka, Y.S.; Sakoori, K.; Akagi, T.; Hashikawa, T.; Morimura, N.; Yamada, K.; Aruga, J. Impaired cognitive function and altered hippocampal synapse morphology in mice lacking Lrrtm1, a gene associated with schizophrenia. PLoS ONE 2011, 6, e22716. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.B.; Sabatini, B.L. Glutamate induces de novo growth of functional spines in developing cortex. Nature 2011, 474, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Holtmaat, A.; Wilbrecht, L.; Knott, G.W.; Welker, E.; Svoboda, K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 2006, 441, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Rujescu, D.; Ingason, A.; Collier, D.A.; O’Donovan, M.C.; Owen, M.J. Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr. Bull. 2009, 35, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Kasem, E.; Kurihara, T.; Tabuchi, K. Neurexins and neuropsychiatric disorders. Neurosci. Res. 2018, 127, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef]
- Tromp, A.; Mowry, B.; Giacomotto, J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 2021, 26, 747–760. [Google Scholar] [CrossRef]
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef]
- Doherty, J.L.; O’Donovan, M.C.; Owen, M.J. Recent genomic advances in schizophrenia. Clin. Genet. 2012, 81, 103–109. [Google Scholar] [CrossRef]
- Larsen, E.; Menashe, I.; Ziats, M.N.; Pereanu, W.; Packer, A.; Banerjee-Basu, S. A systematic variant annotation approach for ranking genes associated with autism spectrum disorders. Mol. Autism 2016, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Duda, M.; Zhang, H.; Li, H.D.; Wall, D.P.; Burmeister, M.; Guan, Y. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl. Psychiatry 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Kishikawa, S.; Higgins, A.W.; Seong, I.S.; Donovan, D.J.; Shen, Y.; Lally, E.; Weiss, L.A.; Najm, J.; Kutsche, K.; et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Bucan, M.; Abrahams, B.S.; Wang, K.; Glessner, J.T.; Herman, E.I.; Sonnenblick, L.I.; Alvarez Retuerto, A.I.; Imielinski, M.; Hadley, D.; Bradfield, J.P.; et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Lowther, C.; Speevak, M.; Armour, C.M.; Goh, E.S.; Graham, G.E.; Li, C.; Zeesman, S.; Nowaczyk, M.J.; Schultz, L.A.; Morra, A.; et al. Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet. Med. 2017, 19, 53–61. [Google Scholar] [CrossRef]
- Flaherty, E.; Zhu, S.; Barretto, N.; Cheng, E.; Deans, P.J.M.; Fernando, M.B.; Schrode, N.; Francoeur, N.; Antoine, A.; Alganem, K.; et al. Neuronal impact of patient-specific aberrant NRXN1alpha splicing. Nat. Genet. 2019, 51, 1679–1690. [Google Scholar] [CrossRef]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 409, 10–13. [Google Scholar] [CrossRef]
- Camacho-Garcia, R.J.; Planelles, M.I.; Margalef, M.; Pecero, M.L.; Martinez-Leal, R.; Aguilera, F.; Vilella, E.; Martinez-Mir, A.; Scholl, F.G. Mutations affecting synaptic levels of neurexin-1beta in autism and mental retardation. Neurobiol. Dis. 2012, 47, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Preisel, M.; Brugger, K.; Wortmann, S.B.; Mayr, J.A. Case Report-An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions. Genes 2023, 14, 1217. [Google Scholar] [CrossRef] [PubMed]
- Rabaneda, L.G.; Robles-Lanuza, E.; Nieto-Gonzalez, J.L.; Scholl, F.G. Neurexin dysfunction in adult neurons results in autistic-like behavior in mice. Cell Rep. 2014, 8, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Blanpied, T.A.; Ehlers, M.D. Microanatomy of dendritic spines: Emerging principles of synaptic pathology in psychiatric and neurological disease. Biol. Psychiatry 2004, 55, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Baltan, S.; Jawaid, S.S.; Chomyk, A.M.; Kidd, G.J.; Chen, J.; Battapady, H.D.; Chan, R.; Dutta, R.; Trapp, B.D. Neuronal hibernation following hippocampal demyelination. Acta Neuropathol. Commun. 2021, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, I.; Rico, B.; Marin, O. Neural circuit dysfunction in mouse models of neurodevelopmental disorders. Curr. Opin. Neurobiol. 2018, 48, 174–182. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes. Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef]
- Gray, E.G. Electron microscopy of synaptic contacts on dendrite spines of the cerebral cortex. Nature 1959, 183, 1592–1593. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohrmann, L.; Seebach, J.; Missler, M.; Rohlmann, A. Distinct Alterations in Dendritic Spine Morphology in the Absence of β-Neurexins. Int. J. Mol. Sci. 2024, 25, 1285. https://doi.org/10.3390/ijms25021285
Mohrmann L, Seebach J, Missler M, Rohlmann A. Distinct Alterations in Dendritic Spine Morphology in the Absence of β-Neurexins. International Journal of Molecular Sciences. 2024; 25(2):1285. https://doi.org/10.3390/ijms25021285
Chicago/Turabian StyleMohrmann, Leonie, Jochen Seebach, Markus Missler, and Astrid Rohlmann. 2024. "Distinct Alterations in Dendritic Spine Morphology in the Absence of β-Neurexins" International Journal of Molecular Sciences 25, no. 2: 1285. https://doi.org/10.3390/ijms25021285
APA StyleMohrmann, L., Seebach, J., Missler, M., & Rohlmann, A. (2024). Distinct Alterations in Dendritic Spine Morphology in the Absence of β-Neurexins. International Journal of Molecular Sciences, 25(2), 1285. https://doi.org/10.3390/ijms25021285