
Effect of β-Estradiol on Adipogenesis in a 3T3-L1 Cell Model of Prelamin A Accumulation





Abstract
1. Introduction
2. Results
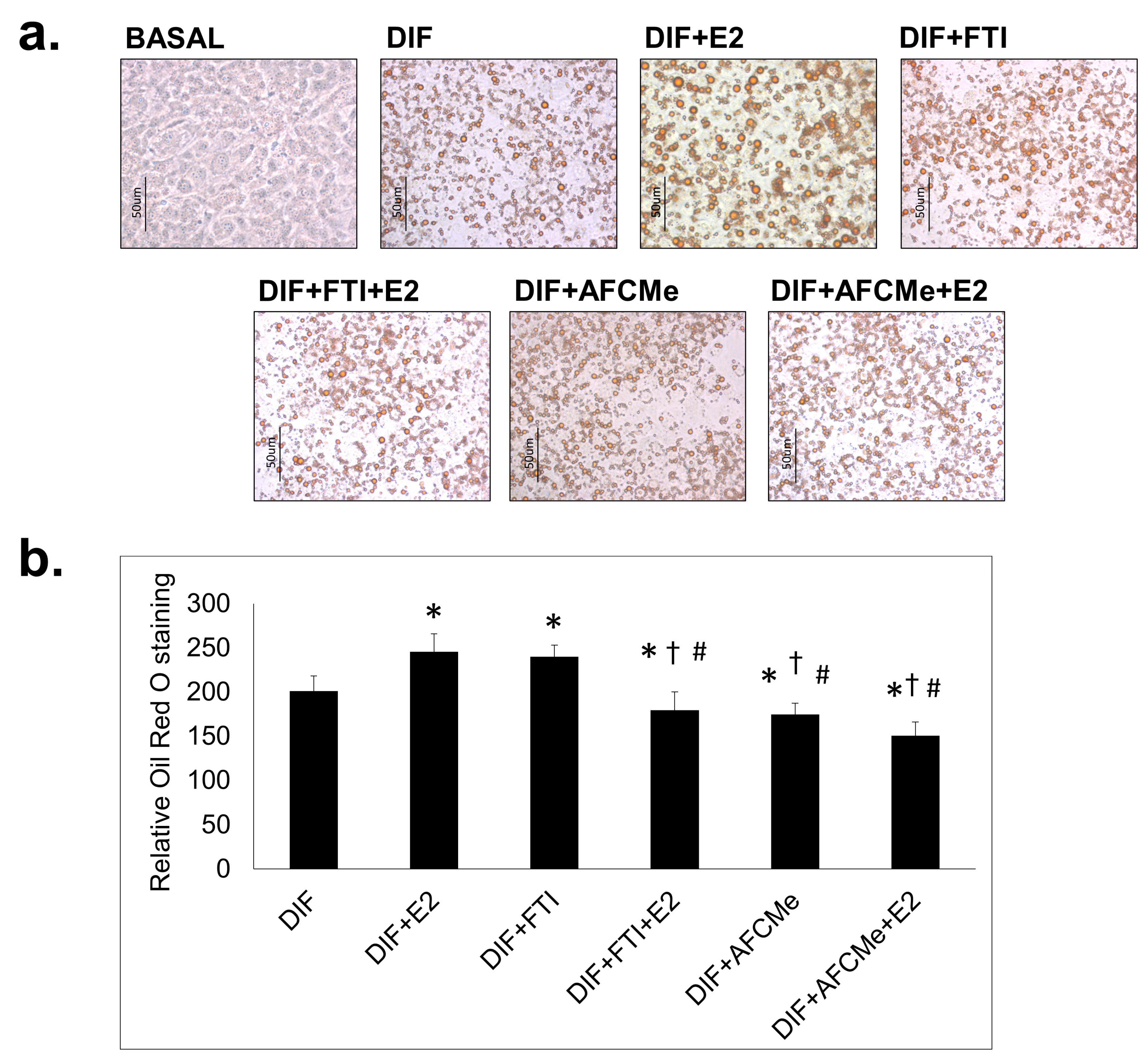
2.1. Assessment of Adipogenic Differentiation
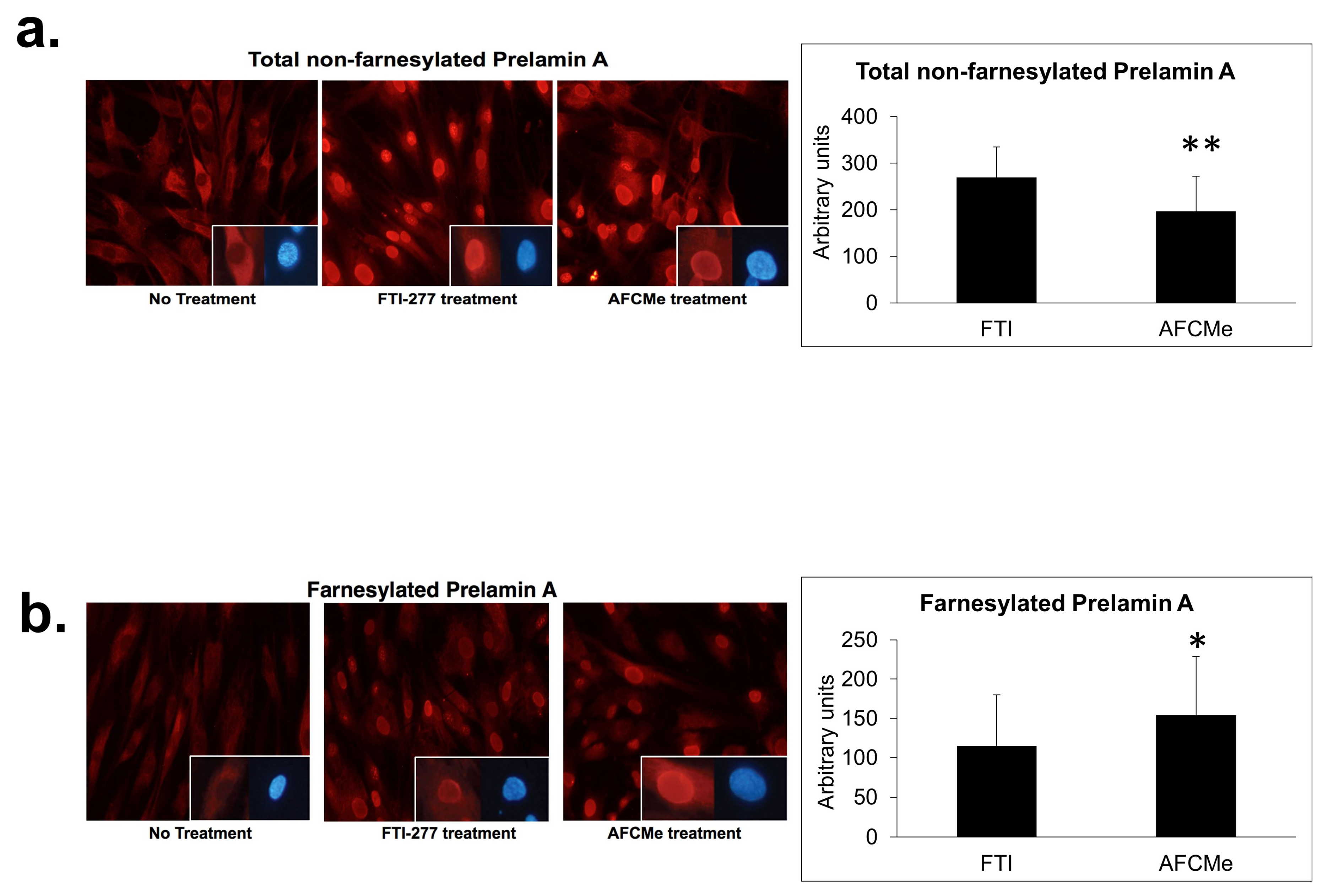
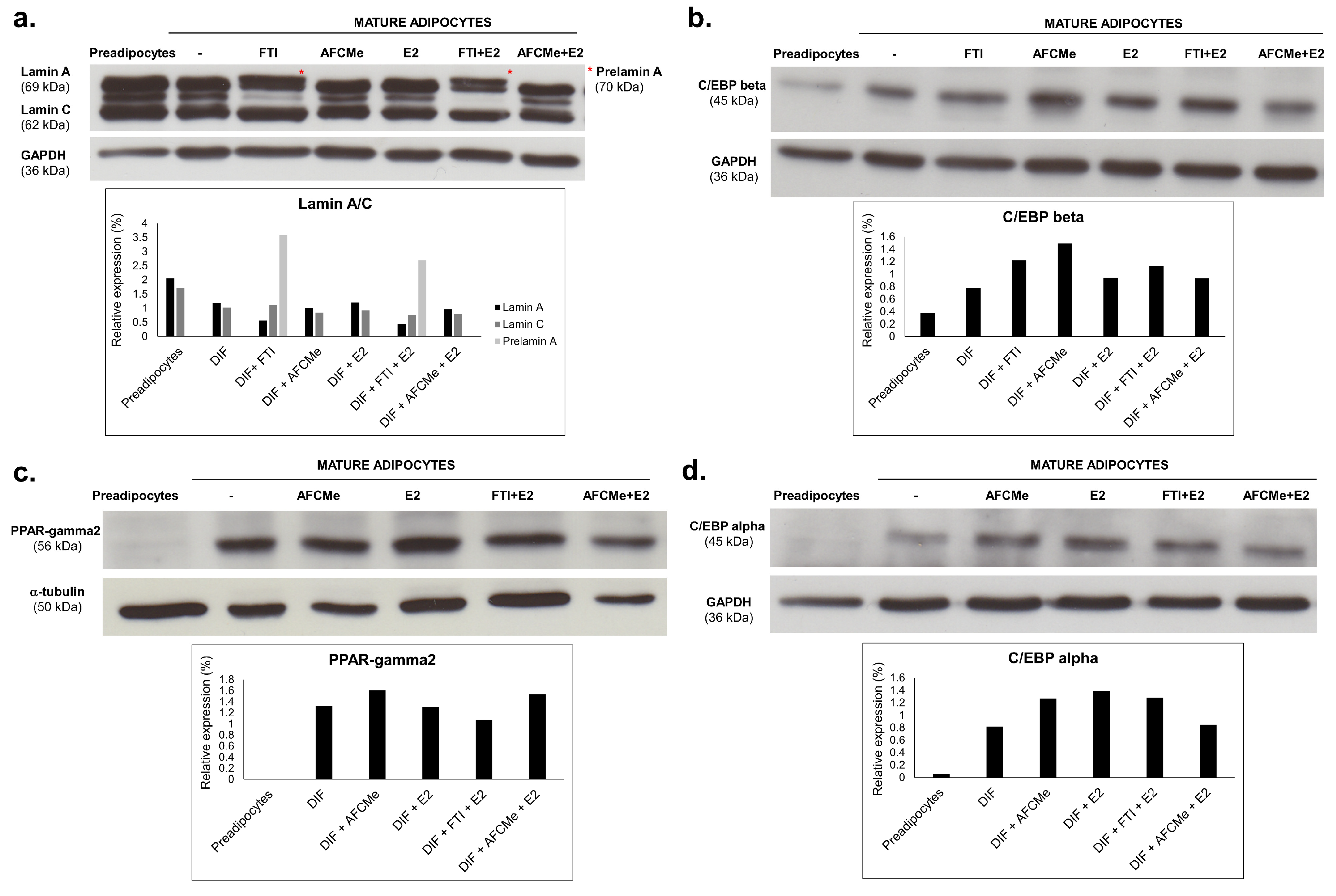
2.2. Prelamin A Accumulation after Drug Treatment
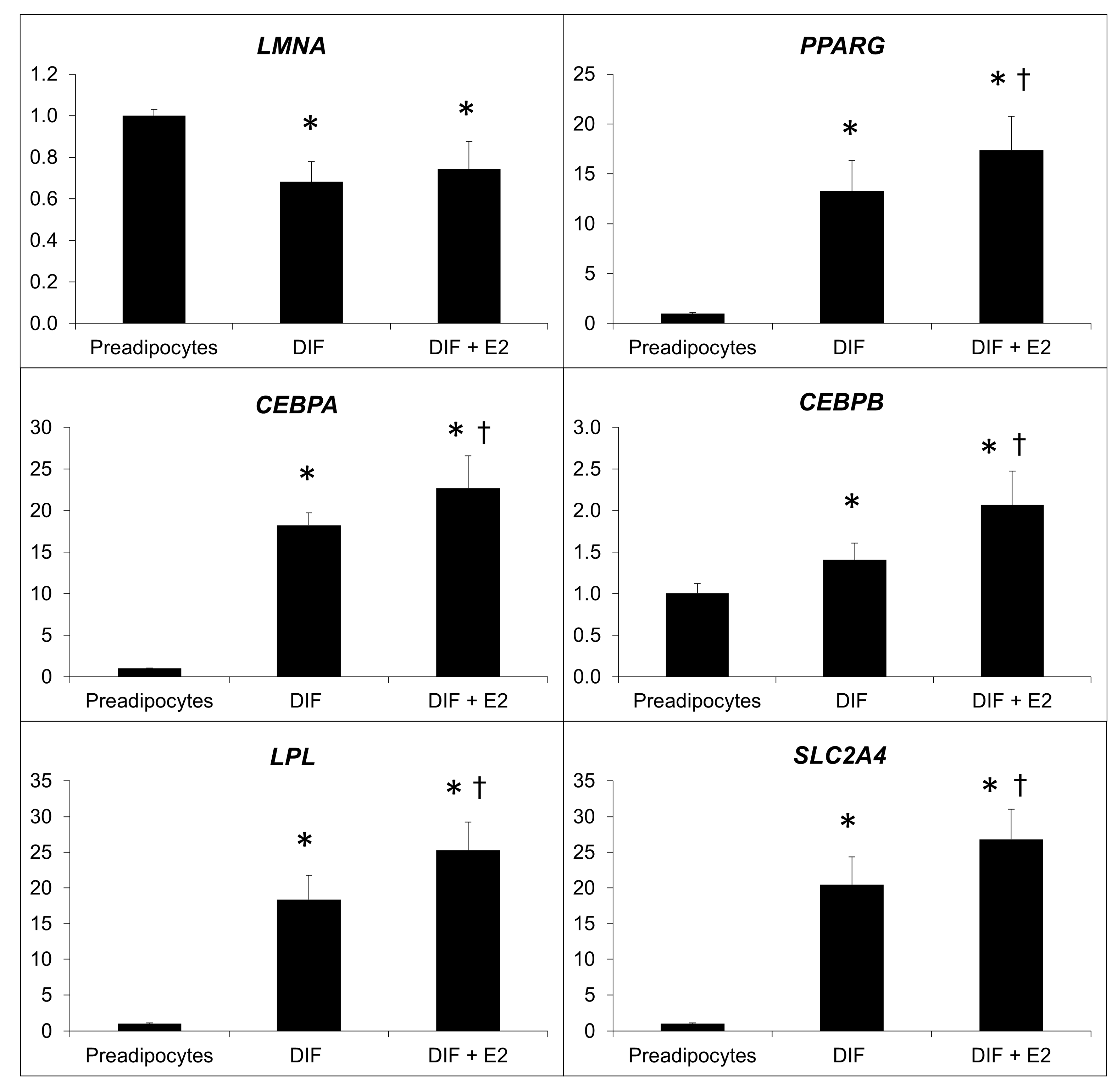
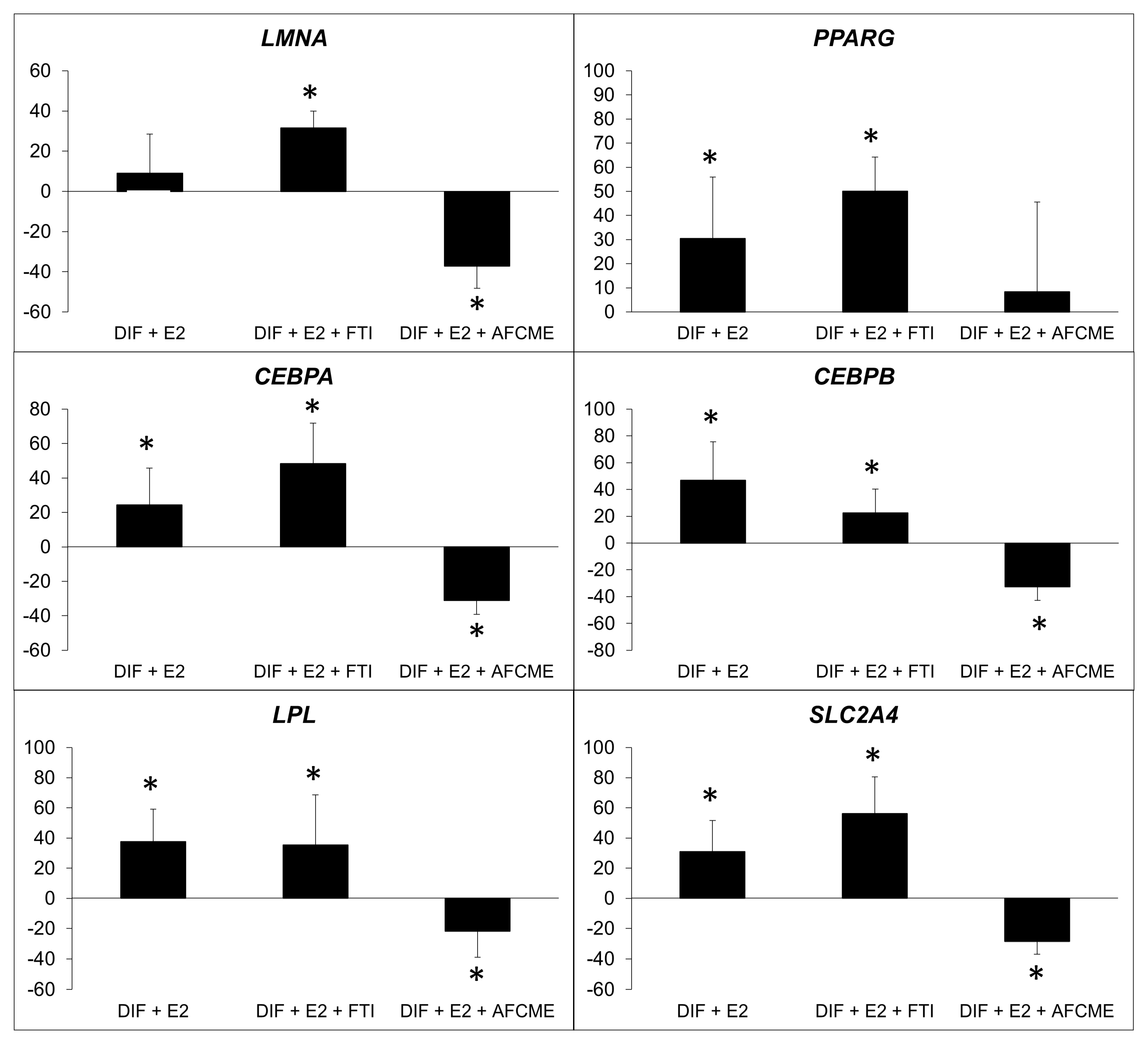
2.3. Effect of β-Estradiol and/or Inhibitors of Prelamin A Maturation in Adipogenic Gene Expression
2.4. Effects of Processing Inhibitors in Adipogenic Protein Evaluation
3. Discussion
4. Materials and Methods
4.1. Adipose Tissue Biopsy and Cell Cultures
4.2. Adipocyte Differentiation
4.3. 17-β-Estradiol Treatment and Inhibition of Lamin A/C Maturation
4.4. Oil Red O Staining
4.5. RNA Extraction and Retrotranscription
4.6. Real-Time PCR
4.7. Western Blotting
4.8. Immunofluorescence
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garg, A. Lipodystrophies. Am. J. Med. 2000, 108, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Vilar, D.; Loidi, L.; Dominguez, F.; Cabezas-Cerrato, J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm. Metab. Res. 2003, 35, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Hegele, R.A. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Rusinol, A.E.; Sinensky, M.S. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J. Cell Sci. 2006, 119, 3265–3272. [Google Scholar] [CrossRef]
- Candelario, J.; Borrego, S.; Reddy, S.; Comai, L. Accumulation of distinct prelamin A variants in human diploid fibroblasts differentially affects cell homeostasis. Exp. Cell Res. 2011, 317, 319–329. [Google Scholar] [CrossRef]
- Dominici, S.; Fiori, V.; Magnani, M.; Schena, E.; Capanni, C.; Camozzi, D.; D’Apice, M.R.; Le Dour, C.; Auclair, M.; Caron, M.; et al. Different prelamin A forms accumulate in human fibroblasts: A study in experimental models and progeria. Eur. J. Histochem. EJH 2009, 53, e6. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Lattanzi, G.; Gonzalez-Mendez, B.; Costa-Freitas, A.T.; Prieto, D.; Columbaro, M.; Mattioli, E.; Victoria, B.; Martinez-Sanchez, N.; Ramazanova, A.; et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009, 46, 40–48. [Google Scholar] [CrossRef]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Victoria, B.; González-Méndez, B.; Barreiro, F.; Fernández-Rodríguez, B.; Cereijo, R.; Gallego-Escuredo, J.M.; Villarroya, F.; Pañeda-Menéndez, A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clin. Endocrinol. 2012, 76, 816–824. [Google Scholar] [CrossRef]
- Bjorntorp, P. Hormonal control of regional fat distribution. Hum. Reprod. 1997, 12 (Suppl. S1), 21–25. [Google Scholar] [CrossRef]
- Lemieux, S.; Prud’homme, D.; Bouchard, C.; Tremblay, A.; Despres, J.P. Sex differences in the relation of visceral adipose tissue accumulation to total body fatness. Am. J. Clin. Nutr. 1993, 58, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Tokunaga, K.; Fujioka, S.; Kobatake, T.; Keno, Y.; Yoshida, S.; Shimomura, I.; Tarui, S.; Matsuzawa, Y. Sexual dimorphism of age-related changes in whole-body fat distribution in the obese. Int. J. Obes. Relat. Metab. Disord. 1994, 18, 207-202. [Google Scholar]
- Meseguer, A.; Puche, C.; Cabero, A. Sex steroid biosynthesis in white adipose tissue. Horm. Metab. Res. 2002, 34, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Auclair, M.; Donadille, B.; Bereziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef]
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef]
- Casasola, A.; Scalzo, D.; Nandakumar, V.; Halow, J.; Recillas-Targa, F.; Groudine, M.; Rincon-Arano, H. Prelamin A processing, accumulation and distribution in normal cells and laminopathy disorders. Nucleus 2016, 7, 84–102. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.S.; Barnes, R.H., 2nd; Tu, Y.; Ren, S.; Andres, D.A.; Spielmann, H.P.; Lammerding, J.; Wang, Y.; Young, S.G.; Fong, L.G. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum. Mol. Genet. 2010, 19, 2682–2694. [Google Scholar] [CrossRef]
- Rebuffe-Scrive, M.; Enk, L.; Crona, N.; Lonnroth, P.; Abrahamsson, L.; Smith, U.; Bjorntorp, P. Fat cell metabolism in different regions in women. Effect of menstrual cycle, pregnancy, and lactation. J. Clin. Investig. 1985, 75, 1973–1976. [Google Scholar] [CrossRef]
- White, U.A.; Stephens, J.M. Transcriptional factors that promote formation of white adipose tissue. Mol. Cell Endocrinol. 2010, 318, 10–14. [Google Scholar] [CrossRef]
- Gomez-Ambrosi, J.; Fruhbeck, G. Evidence for the involvement of resistin in inflammation and cardiovascular disease. Curr. Diabetes Rev. 2005, 1, 227–234. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, P.; Kaku, H.; Zhang, K.; Watanabe, M.; Saika, T.; Nasu, Y.; Kumon, H. A comparison of proteomic profiles changes during 17beta-estradiol treatment in human prostate cancer PC-3 cell line. Cancer Genom. Proteom. 2009, 6, 331–335. [Google Scholar]
- Tsukune, N.; Naito, M.; Kubota, T.; Ozawa, Y.; Nagao, M.; Ohashi, A.; Sato, S.; Takahashi, T. Lamin A overexpression promotes osteoblast differentiation and calcification in the MC3T3-E1 preosteoblastic cell line. Biochem. Biophys. Res. Commun. 2017, 488, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Schena, E.; Mattioli, E.; Peres, C.; Zanotti, L.; Morselli, P.; Iozzo, P.; Guzzardi, M.A.; Bernardini, C.; Forni, M.; Nesci, S.; et al. Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction. Cells 2023, 12, 2586. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, W.; Qiu, Z.; Zhou, Z. G protein-coupled estrogen receptor: A promising therapeutic target for aldosterone-induced hypertension. Front. Endocrinol. 2023, 14, 1226458. [Google Scholar] [CrossRef] [PubMed]
- Fatima, L.A.; Campello, R.S.; Barreto-Andrade, J.N.; Passarelli, M.; Santos, R.S.; Clegg, D.J.; Machado, U.F. Estradiol stimulates adipogenesis and Slc2a4/GLUT4 expression via ESR1-mediated activation of CEBPA. Mol. Cell Endocrinol. 2019, 498, 110447. [Google Scholar] [CrossRef]
- Jeong, S.; Yoon, M. 17beta-Estradiol inhibition of PPARgamma-induced adipogenesis and adipocyte-specific gene expression. Acta Pharmacol. Sin. 2011, 32, 230–238. [Google Scholar] [CrossRef]
- Tu, Y.; Sanchez-Iglesias, S.; Araujo-Vilar, D.; Fong, L.G.; Young, S.G. LMNA missense mutations causing familial partial lipodystrophy do not lead to an accumulation of prelamin A. Nucleus 2016, 7, 512–521. [Google Scholar] [CrossRef]
- Oldenburg, A.; Briand, N.; Sorensen, A.L.; Cahyani, I.; Shah, A.; Moskaug, J.O.; Collas, P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 2017, 216, 2731–2743. [Google Scholar] [CrossRef]
- Xiao, C.; Yu, M.; Liu, J.; Wu, H.; Deng, M.; Zhang, Q.; Xiao, X. Generation of an integration-free induced pluripotent stem cell line (PUMCHi001-A) from a patient with familial partial lipodystrophy type 2 (FPLD2) carrying a heterozygous p.R349W (c.1045C > T) mutation in the LMNA gene. Stem. Cell Res. 2020, 42, 101651. [Google Scholar] [CrossRef]
- Savage, D.B. Mouse models of inherited lipodystrophy. Dis. Model. Mech. 2009, 2, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Magre, J.; Prieur, X. Not Enough Fat: Mouse Models of Inherited Lipodystrophy. Front. Endocrinol. 2022, 13, 785819. [Google Scholar] [CrossRef] [PubMed]
- Wojtanik, K.M.; Edgemon, K.; Viswanadha, S.; Lindsey, B.; Haluzik, M.; Chen, W.; Poy, G.; Reitman, M.; Londos, C. The role of LMNA in adipose: A novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation. J. Lipid Res. 2009, 50, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Santi, S.; Maraldi, N.M.; D’Apice, M.R.; Novelli, G.; Riccio, M.; Squarzoni, S.; Foisner, R.; et al. Drugs affecting prelamin A processing: Effects on heterochromatin organization. Exp. Cell Res. 2008, 314, 453–462. [Google Scholar] [CrossRef]
- Kilic, F.; Dalton, M.B.; Burrell, S.K.; Mayer, J.P.; Patterson, S.D.; Sinensky, M. In Vitro Assay and Characterization of the Farnesylation-dependent Prelamin A Endoprotease. J. Biol. Chem. 1997, 272, 5298–5304. [Google Scholar] [CrossRef] [PubMed]
- Zini, N.; Avnet, S.; Ghisu, S.; Maraldi, N.M.; Squarzoni, S.; Baldini, N.; Lattanzi, G. Effects of prelamin A processing inhibitors on the differentiation and activity of human osteoclasts. J. Cell. Biochem. 2008, 105, 34–40. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Chen, L.-c.; Maynard-Smith, L.A.; Lisa Ooi, A.G.; Wandless, T.J. A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 2006, 126, 995–1004. [Google Scholar] [CrossRef]
- Sanchez-Iglesias, S.; Unruh-Pinheiro, A.; Guillin-Amarelle, C.; Gonzalez-Mendez, B.; Ruiz-Riquelme, A.; Rodriguez-Canete, B.L.; Rodriguez-Garcia, S.; Guillen-Navarro, E.; Domingo-Jimenez, R.; Araujo-Vilar, D. Skipped BSCL2 Transcript in Celia’s Encephalopathy (PELD): New Insights on Fatty Acids Involvement, Senescence and Adipogenesis. PLoS ONE 2016, 11, e0158874. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number NCBI | Forward Primer | Reverse Primer | Probe Number | Probe Sequence | |
|---|---|---|---|---|---|
| LMNA | NM_019390 | ATCCGCATTGACAGCCTCT | TCCAGGTCACGCAGCTTT | 102 | CTCAGCCA |
| PPARG | NM_011146.3 | GAAAGACAACGGACAAATCACC | GGGGGTGATATGTTTGAACTTG | 7 | CTTCTCCC |
| LPL | NM_008509.2 | GCTGGTGGGAAATGATGTG | TGGACGTTGTCTAGGGGGTA | 25 | CTCCTCCA |
| GLUT4 (SLC2A4) | NM_009204.2 | GACGGACACTCCATCTGTTG | GCCACGATGGAGACATAGC | 5 | TGTGGCTG |
| Rn18s | NR_003278.2 | AAACGGCTACCACATCCAAG | TACAGGGCCTCGAAAGAGTC | 74 | CTGCTGCC |
| C/EBPβ | NM_009883.3 | TGATGCAATCCGGATCAA | CACGTGTGTTGCGTCAGTC | 102 | CTCAGCCA |
| C/EBPα | NM_007678.3 | AAACAACGCAACGTGGAGA | GCGGTCATTGTCACTGGTC | 67 | TGCTGGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobelo-Gómez, S.; Sánchez-Iglesias, S.; Fernández-Pombo, A.; Araújo-Vilar, D. Effect of β-Estradiol on Adipogenesis in a 3T3-L1 Cell Model of Prelamin A Accumulation. Int. J. Mol. Sci. 2024, 25, 1282. https://doi.org/10.3390/ijms25021282
Cobelo-Gómez S, Sánchez-Iglesias S, Fernández-Pombo A, Araújo-Vilar D. Effect of β-Estradiol on Adipogenesis in a 3T3-L1 Cell Model of Prelamin A Accumulation. International Journal of Molecular Sciences. 2024; 25(2):1282. https://doi.org/10.3390/ijms25021282
Chicago/Turabian StyleCobelo-Gómez, Silvia, Sofía Sánchez-Iglesias, Antía Fernández-Pombo, and David Araújo-Vilar. 2024. "Effect of β-Estradiol on Adipogenesis in a 3T3-L1 Cell Model of Prelamin A Accumulation" International Journal of Molecular Sciences 25, no. 2: 1282. https://doi.org/10.3390/ijms25021282
APA StyleCobelo-Gómez, S., Sánchez-Iglesias, S., Fernández-Pombo, A., & Araújo-Vilar, D. (2024). Effect of β-Estradiol on Adipogenesis in a 3T3-L1 Cell Model of Prelamin A Accumulation. International Journal of Molecular Sciences, 25(2), 1282. https://doi.org/10.3390/ijms25021282