

Diagnostic Challenges during Inflammation and Cancer: Current Biomarkers and Future Perspectives in Navigating through the Minefield of Reactive versus Dysplastic and Cancerous Lesions in the Digestive System



 , , , ,
, , , ,  ,
,
Abstract
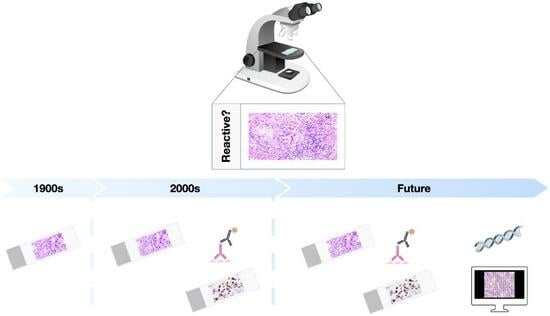
1. Introduction
2. Tissue Response during Chronic Inflammation and Diagnostic Dilemmas
2.1. Gastrointestinal Tract
2.1.1. Mouth
2.1.2. Esophagus
2.1.3. Stomach
2.1.4. Colon
2.2. Pancreas
2.3. Gallbladder and Extrahepatic Bile Ducts
2.4. Liver
3. Tissue-Based Biomarkers Differentiating Reactive from Neoplastic Lesions
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rocha e Silva, M. A brief survey of the history of inflammation. Agents Action 1978, 8, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.J.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 325. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Veronique Giroux, A.K.R. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Underwood, J.C. Lymphoreticular infiltration in human tumours: Prognostic and biological implications: A review. Br. J. Cancer 1974, 30, 538–548. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Georges, D.; Freddie Bray, J.F.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2019, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Michels, N.; van Aart, C.; Morisse, J.; Mullee, A.; Huybrechts, I. Chronic inflammation towards cancer incidence: A systematic review and meta-analysis of epidemiological studies. Crit. Rev. Oncol. Hematol. 2021, 157, 103177. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Schetter, A.J.; Heegaard, N.H.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2009, 31, 37–49. [Google Scholar] [CrossRef]
- Lynch, J.M. Understanding Pseudoepitheliomatous Hyperplasia. Pathol. Case Rev. 2004, 9, 36–45. [Google Scholar] [CrossRef]
- El-Khoury, J.; Kibbi, A.-G.; Abbas, O. Mucocutaneous pseudoepitheliomatous hyperplasia: A review. Am. J. Dermatopathol. 2012, 34, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Pascu, R.M.; Crăiţoiu, Ş.; Crăiţoiu, M.M.; Florescu, A.M.; Dăguci, L.; Petcu, I.C.; Pătru, C.L. The Role Played by Growth Factors TGF-β1, EGF and FGF7 in the Pathogeny of Oral Pseudoepitheliomatous Hyperplasia. Curr. Health Sci. J. 2017, 43, 246. [Google Scholar]
- Akilov, O.E.; Donovan, M.J.; Stepinac, T.; Carter, C.R.; Whitcomb, J.P.; Hasan, T.; McDowell, M.A. T helper type 1 cytokines and keratinocyte growth factor play a critical role in pseudoepitheliomatous hyperplasia initiation during cutaneous leishmaniasis. Arch. Dermatol. Res. 2007, 299, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Ra, S.H.; Su, A.; Li, X.; Binder, S. Molecularly enriched pathways and differentially expressed genes distinguishing cutaneous squamous cell carcinoma from pseudoepitheliomatous hyperplasia. Diagn. Mol. Pathol. 2013, 22, 41–47. [Google Scholar] [CrossRef]
- Que, J.; Garman, K.S.; Souza, R.F.; Spechler, S.J. Pathogenesis and Cells of Origin of Barrett’s Esophagus. Gastroenterology 2019, 157, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Spechler, S.J. Mechanisms and pathophysiology of Barrett oesophagus. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J. Barrett esophagus and risk of esophageal cancer: A clinical review. JAMA 2013, 310, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: Reparative lineages in the gastrointestinal mucosa. J. Pathol. 2018, 245, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hayakawa, Y.; Koike, K. Metaplasia in the Stomach—Precursor of Gastric Cancer? Int. J. Mol. Sci. 2017, 18, 2063. [Google Scholar] [CrossRef]
- Stairs, D.B.; Kong, J.; Lynch, J.P. Cdx genes, inflammation, and the pathogenesis of intestinal metaplasia. Prog. Mol. Biol. Transl. Sci. 2010, 96, 231. [Google Scholar]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef]
- Wada, Y.; Nakajima, S.; Kushima, R.; Takemura, S.; Mori, N.; Hasegawa, H.; Nakayama, T.; Mukaisho, K.-I.; Yoshida, A.; Umano, S.; et al. Pyloric, pseudopyloric, and spasmolytic polypeptide-expressing metaplasias in autoimmune gastritis: A case series of 22 Japanese patients. Virchows Arch. 2021, 479, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.R.; Goldenring, J.R. Injury, repair, inflammation and metaplasia in the stomach. J. Physiol. 2018, 596, 3861–3867. [Google Scholar] [CrossRef]
- Brown, J.W.; Cho, C.J.; Mills, J.C. Paligenosis: Cellular Remodeling During Tissue Repair. Annu. Rev. Physiol. 2022, 84, 461–483. [Google Scholar] [CrossRef]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. J. Tech. Methods Pathol. 1999, 79, 639–646. [Google Scholar]
- Fuchino, T.; Wada, Y.; Kodama, M.; Mukaisho, K.-I.; Mizukami, K.; Okimoto, T.; Kushima, R.; Murakami, K. Clinicopathological characteristics of pancreatic acinar cell metaplasia associated with Helicobacter pylori infection. BMC Gastroenterol. 2022, 22, 289. [Google Scholar] [CrossRef] [PubMed]
- El Hadad, J.; Schreiner, P.; Vavricka, S.R.; Greuter, T. The Genetics of Inflammatory Bowel Disease. Mol. Diagn. Ther. 2023, 28, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, L.; Riis, L.B. A close view on histopathological changes in inflammatory bowel disease, a narrative review. Dig. Med. Res. 2021, 4, 3. [Google Scholar] [CrossRef]
- Rubio, C.A. Corrupted Colonic Crypts Bordering Regenerating Mucosal Ulcers in Ulcerative Colitis. In Vivo 2017, 31, 669–671. [Google Scholar]
- Bankaitis, E.D.; Ha, A.; Kuo, C.J.; Magness, S.T. Reserve Stem Cells in Intestinal Homeostasis and Injury. Gastroenterology 2018, 155, 1346–1361. [Google Scholar] [CrossRef]
- Bradford, E.M.; Ryu, S.H.; Singh, A.P.; Lee, G.; Goretsky, T.; Sinh, P.; Williams, D.B.; Cloud, A.L.; Gounaris, E.; Patel, V.; et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef]
- Moparthi, L.; Koch, S. Wnt signaling in intestinal inflammation. Differentiation 2019, 108, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 2016, 143, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.d.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Lin, Q.; Gong, X.; Thomas, A.; Liu, Q. LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/beta-catenin signaling. Mol. Cell. Biol. 2012, 32, 2054–2064. [Google Scholar] [CrossRef]
- Pai, P.; Rachagani, S.; Dhawan, P.; Batra, S.K. Mucins and Wnt/β-catenin signaling in gastrointestinal cancers: An unholy nexus. Carcinogenesis 2016, 37, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Johnson, K.; Yin, J.; Lee, S.; Lin, R.; Yu, H.; In, J.; Foulke-Abel, J.; Zachos, N.C.; Donowitz, M.; et al. Chronic Inflammation in Ulcerative Colitis Causes Long-Term Changes in Goblet Cell Function. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 219–232. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Ungaro, F.; Noviello, D.; Lovisa, S.; Peyrin-Biroulet, L.; Danese, S. Revisiting fibrosis in inflammatory bowel disease: The gut thickens. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 169–184. [Google Scholar] [CrossRef]
- Ullman, T.; Odze, R.; Farraye, F.A. Diagnosis and management of dysplasia in patients with ulcerative colitis and Crohn’s disease of the colon. Inflamm. Bowel Dis. 2009, 15, 630–638. [Google Scholar] [CrossRef]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816.e1. [Google Scholar] [CrossRef]
- Rubin, C.E.; Haggitt, R.C.; Burmer, G.C.; Brentnall, T.A.; Stevens, A.C.; Levine, D.S.; Dean, P.J.; Kimmey, M.; Perera, D.R.; Rabinovitch, P.S. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992, 103, 1611–1620. [Google Scholar] [CrossRef]
- Mahmoud, R.; Shah, S.C.; Torres, J.; Castaneda, D.; Glass, J.; Elman, J.; Kumar, A.; Axelrad, J.; Harpaz, N.; Ullman, T.; et al. Association Between Indefinite Dysplasia and Advanced Neoplasia in Patients with Inflammatory Bowel Diseases Undergoing Surveillance. Clin. Gastroenterol. Hepatol. 2020, 18, 1518–1527.e3. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Detlefsen, S.; Feyerabend, B. Fibrosis of the pancreas: The initial tissue damage and the resulting pattern. Virchows Arch. Int. J. Pathol. 2004, 445, 1. [Google Scholar] [CrossRef]
- Esposito, I.; Hruban, R.H.; Verbeke, C.; Terris, B.; Zamboni, G.; Scarpa, A.; Morohoshi, T.; Suda, K.; Luchini, C.; Klimstra, D.S.; et al. Guidelines on the histopathology of chronic pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and the European Pancreatic Club. Pancreatology 2020, 20, 586–593. [Google Scholar] [PubMed]
- Hamada, S.; Matsumoto, R.; Masamune, A. Pancreatic Stellate Cells and Metabolic Alteration: Physiology and Pathophysiology. Front. Physiol. 2022, 13, 865105. [Google Scholar] [CrossRef] [PubMed]
- Ferdek, P.E.; Krzysztofik, D.; Stopa, K.B.; Kusiak, A.A.; Paw, M.; Wnuk, D.; Jakubowska, M.A. When healing turns into killing—The pathophysiology of pancreatic and hepatic fibrosis. J. Physiol. 2022, 600, 2579–2612. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Wen, H.-J.; Ruggeri, J.M.; Takeuchi, K.K.; Zhang, Y.; Magliano, M.P.d.; Crawford, H.C. Mitogen-activated Protein Kinase Kinase Activity Maintains Acinar-to-Ductal Metaplasia and Is Required for Organ Regeneration in Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2016, 3, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Satoh, M.; Sakai, Y.; Satoh, A.; Shimosegawa, T. Ligands of peroxisome proliferator-activated receptor-gamma block activation of pancreatic stellate cells. J. Biol. Chem. 2002, 277, 141–147. [Google Scholar] [CrossRef]
- Strobel, O.; Dor, Y.; Alsina, J.; Stirman, A.; Lauwers, G.; Trainor, A.; Castillo, C.F.-D.; Warshaw, A.L.; Thayer, S.P. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 2007, 133, 1999–2009. [Google Scholar] [CrossRef]
- Grimont, A.; Leach, S.D.; Chandwani, R. Uncertain Beginnings: Acinar and Ductal Cell Plasticity in the Development of Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 369–382. [Google Scholar] [CrossRef]
- Li, S.; Xie, K. Ductal metaplasia in pancreas. Biochim. Biophys. Acta BBA Rev. Cancer 2022, 1877, 188698. [Google Scholar] [CrossRef]
- Willet, S.G.; Lewis, M.A.; Miao, Z.-F.; Li, D.; Radyk, M.D.; Cunningham, R.L.; Burclaff, J.; Sibbel, G.; Lo, H.-Y.G.; Blanc, V.; et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 2018, 37, e98311. [Google Scholar] [CrossRef] [PubMed]
- Radyk, M.D.; Spatz, L.B.; Peña, B.L.; Brown, J.W.; Burclaff, J.; Cho, C.J.; Mills, J.C. ATF3 induces RAB7 to govern autodegradation in paligenosis, a conserved cell plasticity program. EMBO Rep. 2021, 22, e51806. [Google Scholar] [CrossRef] [PubMed]
- Chuvin, N.; Vincent, D.F.; Pommier, R.M.; Alcaraz, L.B.; Gout, J.; Caligaris, C.; Bartholin, L. Acinar-to-Ductal Metaplasia Induced by Transforming Growth Factor Beta Facilitates KRASG12D-driven Pancreatic Tumorigenesis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Liou, G.Y.; Bastea, L.; Fleming, A.; Doppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Bledsoe, J.R.; Shinagare, S.A.; Deshpande, V. Difficult Diagnostic Problems in Pancreatobiliary Neoplasia. Arch. Pathol. Lab. Med. 2015, 139, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.D.; Zhang, M.L.; VandenBussche, C.J. The Diagnostic Challenge of Evaluating Small Biopsies from the Pancreatobiliary System. Surg. Pathol. Clin. 2022, 15, 435–453. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Tomita, H.; Hara, A. Development of extrahepatic bile ducts and mechanisms of tumorigenesis: Lessons from mouse models. Pathol. Int. 2022, 72, 589–605. [Google Scholar] [CrossRef]
- Zimmermann, A. Tumors and Tumor-Like Lesions of the Hepatobiliary Tract; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Landas, S.K. Putative precursors of gallbladder dysplasia: A review of 400 routinely resected specimens. Arch. Pathol. Lab. Med. 2005, 129, 386–390. [Google Scholar] [CrossRef]
- Roa, J.C.; Basturk, O.; Adsay, V. Dysplasia and carcinoma of the gallbladder: Pathological evaluation, sampling, differential diagnosis and clinical implications. Histopathology 2021, 79, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Katabi, N. Neoplasia of gallbladder and biliary epithelium. Arch. Pathol. Lab. Med. 2010, 134, 1621–1627. [Google Scholar] [CrossRef]
- Robert, D.; Odze, J.R.G. Odze & Goldblum Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Oyasiji, T.; Zhang, J.; Kuvshinoff, B.; Iyer, R.; Hochwald, S.N. Molecular Targets in Biliary Carcinogenesis and Implications for Therapy. Oncologist 2015, 20, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, H.; Henson, R.; Meng, F.; Mize-Berge, J.; Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006, 66, 10517–10524. [Google Scholar] [CrossRef]
- Braconi, C.; Huang, N.; Patel, T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010, 51, 881–890. [Google Scholar] [CrossRef]
- Komori, J.; Marusawa, H.; Machimoto, T.; Endo, Y.; Kinoshita, K.; Kou, T.; Haga, H.; Ikai, I.; Uemoto, S.; Chiba, T. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology 2008, 47, 888–896. [Google Scholar] [CrossRef]
- Zhang, Z.; Lai, G.H.; Sirica, A.E. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology 2004, 39, 1028–1037. [Google Scholar] [CrossRef]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: Evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Kakuda, Y.; Sugino, T.; Sato, Y.; Fukumura, Y. Pathologies of Precursor Lesions of Biliary Tract Carcinoma. Cancers 2022, 14, 5358. [Google Scholar] [CrossRef]
- Hsu, M.; Sasaki, M.; Igarashi, S.; Sato, Y.; Nakanuma, Y. KRAS and GNAS mutations and p53 overexpression in biliary intraepithelial neoplasia and intrahepatic cholangiocarcinomas. Cancer 2013, 119, 1669–1674. [Google Scholar] [CrossRef]
- Sasaki, M.; Nitta, T.; Sato, Y.; Nakanuma, Y. Autophagy may occur at an early stage of cholangiocarcinogenesis via biliary intraepithelial neoplasia. Hum. Pathol. 2015, 46, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Khizar, H.; Hu, Y.; Wu, Y.; Yang, J. The role and implication of autophagy in cholangiocarcinoma. Cell Death Discov. 2023, 9, 332. [Google Scholar] [CrossRef]
- Gill, R.M.; Theise, N.D. Rappaport, Glisson, Hering, and Mall—Champions of Liver Microanatomy: Microscopic and Ultramicroscopic Anatomy of the Liver Into the Modern Age. Clin. Liver Dis. 2021, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Cain, O.; Chauhan, A.; Webb, G.J. Medical liver biopsy: Background, indications, procedure and histopathology. Frontline Gastroenterol. 2020, 11, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Lackner, C. Hepatocellular ballooning in nonalcoholic steatohepatitis: The pathologist’s perspective. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 223–231. [Google Scholar] [CrossRef]
- Zatloukal, K.; French, S.W.; Stumptner, C.; Strnad, P.; Harada, M.; Toivola, D.M.; Cadrin, M.; Omary, M.B. From Mallory to Mallory–Denk bodies: What, how and why. Exp. Cell Res. 2007, 313, 2033–2049. [Google Scholar] [CrossRef]
- Harada, M.; Hanada, S.; Toivola, D.M.; Ghori, N.; Omary, M.B. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008, 47, 2026–2035. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Katsarou, A.; Moustakas, I.I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Panagiotis Lembessis, Michael Koutsilieris, Antonios Chatzigeorgiou. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Chavakis, T. Immune Cells and Metabolism. Handb. Exp. Pharmacol. 2016, 233, 221–249. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 391–411. [Google Scholar] [CrossRef]
- Buchholz, M.; Kestler, H.A.; Holzmann, K.; Ellenrieder, V.; Schneiderhan, W.; Siech, M.; Adler, G.; Bachem, M.G.; Gress, T.M. Transcriptome analysis of human hepatic and pancreatic stellate cells: Organ-specific variations of a common transcriptional phenotype. J. Mol. Med. 2005, 83, 795–805. [Google Scholar] [CrossRef]
- Moustakas, I.I.; Katsarou, A.; Legaki, A.I.; Pyrina, I.; Ntostoglou, K.; Papatheodoridi, A.M.; Gercken, B.; Pateras, I.S.; Gorgoulis, V.G.; Koutsilieris, M.; et al. Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity. Int. J. Mol. Sci. 2021, 22, 3446. [Google Scholar] [CrossRef]
- Papatheodoridi, A.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Jurk, D. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Burel-Vandenbos, F.; Benchetrit, M.; Miquel, C.; Fontaine, D.; Auvergne, R.; Lebrun-Frenay, C.; Cardot-Leccia, N.; Michiels, J.-F.; Paquis-Flucklinger, V.; Virolle, T. EGFR immunolabeling pattern may discriminate low-grade gliomas from gliosis. J. Neuro-Oncol. 2011, 102, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Burel-Vandenbos, F.; Turchi, L.; Benchetrit, M.; Fontas, E.; Pedeutour, Z.; Rigau, V.; Almairac, F.; Ambrosetti, D.; Michiels, J.F.; Virolle, T. Cells with intense EGFR staining and a high nuclear to cytoplasmic ratio are specific for infiltrative glioma: A useful marker in neuropathological practice. Neuro-Oncology 2013, 15, 1278–1288. [Google Scholar] [CrossRef]
- Camelo-Piragua, S.; Jansen, M.; Ganguly, A.; Kim, J.C.; Cosper, A.K.; Dias-Santagata, D.; Nutt, C.L.; Iafrate, A.J.; Louis, D.N. A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: Chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J. Neuropathol. Exp. Neurol. 2011, 70, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Weissert, S.; Balss, J.; Habel, A.; Meyer, J.; Jager, D.; Ackermann, U.; Tessmer, C.; Korshunov, A.; Zentgraf, H.; et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010, 20, 245–254. [Google Scholar] [CrossRef]
- Capper, D.; Zentgraf, H.; Balss, J.; Hartmann, C.; von Deimling, A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009, 118, 599–601. [Google Scholar] [CrossRef]
- Sato, F.; Bhawal, U.K.; Osaki, S.; Sugiyama, N.; Oikawa, K.; Muragaki, Y. Differential immunohistochemical expression of DEC1, CK-1epsilon, and CD44 in oral atypical squamous epithelium and carcinoma in situ. Mol. Med. Rep. 2022, 25, 159. [Google Scholar] [CrossRef]
- Zarovnaya, E.; Black, C. Distinguishing pseudoepitheliomatous hyperplasia from squamous cell carcinoma in mucosal biopsy specimens from the head and neck. Arch. Pathol. Lab. Med. 2005, 129, 1032–1036. [Google Scholar] [CrossRef]
- Gologan, O.; Barnes, E.L.; Hunt, J.L. Potential diagnostic use of p16INK4A, a new marker that correlates with dysplasia in oral squamoproliferative lesions. Am. J. Surg. Pathol. 2005, 29, 792–796. [Google Scholar] [CrossRef]
- Coltrera, M.D.; Zarbo, R.J.; Sakr, W.A.; Gown, A.M. Markers for dysplasia of the upper aerodigestive tract. Suprabasal expression of PCNA, p53, and CK19 in alcohol-fixed, embedded tissue. Am. J. Pathol. 1992, 141, 817–825. [Google Scholar] [PubMed]
- Ohbu, M.; Kobayashi, N.; Okayasu, I. Expression of cell cycle regulatory proteins in the multistep process of oesophageal carcinogenesis: Stepwise over-expression of cyclin E and p53, reduction of p21(WAF1/CIP1) and dysregulation of cyclin D1 and p27(KIP1). Histopathology 2001, 39, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Dorer, R.; Odze, R.D. AMACR immunostaining is useful in detecting dysplastic epithelium in Barrett’s esophagus, ulcerative colitis, and Crohn’s disease. Am. J. Surg. Pathol. 2006, 30, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Lisovsky, M.; Falkowski, O.; Bhuiya, T. Expression of alpha-methylacyl-coenzyme A racemase in dysplastic Barrett’s epithelium. Hum. Pathol. 2006, 37, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Scheil-Bertram, S.; Lorenz, D.; Ell, C.; Sheremet, E.; Fisseler-Eckhoff, A. Expression of alpha-methylacyl coenzyme A racemase in the dysplasia carcinoma sequence associated with Barrett’s esophagus. Mod. Pathol. 2008, 21, 961–967. [Google Scholar] [CrossRef]
- Gadara, M.R.; Gonzalez, M.; Cartun, R.W.; Ligato, S. IMP3 Immunoreactivity is More Sensitive Than AMACR in Detecting Dysplastic Epithelium and Early Adenocarcinoma in Barrett Esophagus. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, H.; Hayat, U.; Manivel, J.; Iwamoto, C.; Peltola, J.; Hanson, B.; Larson, W.; Dachel, S.; Gravely, A.; Mesa, H. Surface Ki-67 Expression Improves Reproducibility of Dysplasia Diagnosis in Barrett’s Esophagus. Am. J. Clin. Pathol. 2020, 153, 695–704. [Google Scholar] [CrossRef]
- Rice, T.W.; Goldblum, J.R.; Falk, G.W.; Tubbs, R.R.; Kirby, T.J.; Casey, G. p53 immunoreactivity in Barrett’s metaplasia, dysplasia, and carcinoma. J. Thorac. Cardiovasc. Surg. 1994, 108, 1132–1137. [Google Scholar] [CrossRef]
- Lee, W.A. Alpha-methylacyl-CoA-racemase expression in adenocarcinoma, dysplasia and non-neoplastic epithelium of the stomach. Oncology 2006, 71, 246–250. [Google Scholar] [CrossRef]
- Sampalean, D.S.; Turcu, M.; Fetyko, A.; Bartha, J.R.; BaTaga, S.M.; Turdean, S.G. Immunohistochemical expression of Ki-67 and p53 along with their digitalized evaluation in the discriminatory analysis of reactive atypia and dysplastic lesions in gastrointestinal biopsies of the stomach. Rom. J. Morphol. Embryol. 2017, 58, 139–144. [Google Scholar]
- Dong, B.; Xie, Y.Q.; Chen, K.; Wang, T.; Tang, W.; You, W.C.; Li, J.Y. Differences in biological features of gastric dysplasia, indefinite dysplasia, reactive hyperplasia and discriminant analysis of these lesions. World J. Gastroenterol. 2005, 11, 3595–3600. [Google Scholar] [CrossRef] [PubMed]
- Strehl, J.D.; Hoegel, J.; Hornicek, I.; Hartmann, A.; Riener, M.O. Immunohistochemical expression of IMP3 and p53 in inflammatory lesions and neoplastic lesions of the gastric mucosa. Int. J. Clin. Exp. Pathol. 2014, 7, 2091–2101. [Google Scholar] [PubMed]
- Noffsinger, A.; Belli, J.; Miller, M.; Fenoglio-Preiser, C. A unique basal pattern of p53 expression in ulcerative colitis is associated with mutation in the p53 gene. Histopathology 2001, 39, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Machinami, R. p53 immunohistochemistry of ulcerative colitis-associated with dysplasia and carcinoma. Pathol. Int. 1999, 49, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Fujimori, T.; Mitomi, H.; Tomita, S.; Ichikawa, K.; Imura, J.; Fujii, S.; Itabashi, M.; Kameoka, S.; Igarashi, Y. Immunohistochemical assessment of a unique basal pattern of p53 expression in ulcerative-colitis-associated neoplasia using computer-assisted cytometry. Diagn. Pathol. 2014, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Tretiakova, M.; Antic, T.; Westerhoff, M.; Mueller, J.; Himmelfarb, E.A.; Wang, H.L.; Xiao, S.-Y. Diagnostic utility of CD10 in benign and malignant extrahepatic bile duct lesions. Am. J. Surg. Pathol. 2012, 36, 101–108. [Google Scholar] [CrossRef]
- Riener, M.-O.; Vogetseder, A.; Pestalozzi, B.C.; Clavien, P.-A.; Probst-Hensch, N.; Kristiansen, G.; Jochum, W. Cell adhesion molecules P-cadherin and CD24 are markers for carcinoma and dysplasia in the biliary tract. Hum. Pathol. 2010, 41, 1558–1565. [Google Scholar] [CrossRef]
- Zakharov, V.; Ren, B.; Ryan, C.; Cao, W. Diagnostic value of HMGA s, p53 and β-catenin in discriminating adenocarcinoma from adenoma or reactive atypia in ampulla and common bile duct biopsies. Histopathology 2013, 62, 778–787. [Google Scholar] [CrossRef]
- Zhao, H.; Davydova, L.; Mandich, D.; Cartun, R.W.; Ligato, S. S-100A4 protein and mesothelin expression in dysplasia and carcinoma of the extrahepatic bile duct. Am. J. Clin. Pathol. 2007, 127, 374–379. [Google Scholar] [CrossRef]
- Baumhoer, D.; Riener, M.-O.; Zlobec, I.; Tornillo, L.; Vogetseder, A.; Kristiansen, G.; Dietmaier, W.; Hartmann, A.; Wuensch, P.H.; Sessa, F. Expression of CD24, P-cadherin and S100A4 in tumors of the ampulla of Vater. Mod. Pathol. 2009, 22, 306–313. [Google Scholar] [CrossRef]
- Aishima, S.; Fujita, N.; Mano, Y.; Kubo, Y.; Tanaka, Y.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Oda, Y. Different roles of S100P overexpression in intrahepatic cholangiocarcinoma: Carcinogenesis of perihilar type and aggressive behavior of peripheral type. Am. J. Surg. Pathol. 2011, 35, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Harada, K.; Sasaki, M.; Nakanuma, Y. Clinicopathological significance of S 100 protein expression in cholangiocarcinoma. J. Gastroenterol. Hepatol. 2013, 28, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Lynch, B.C.; Lathrop, S.L.; Ye, D.; Ma, T.Y.; Cerilli, L.A. Expression of the p16 (INK4a) gene product in premalignant and malignant epithelial lesions of the gallbladder. Ann. Diagn. Pathol. 2008, 12, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Wilentz, R.E.; Su, G.H.; Le Dai, J.; Sparks, A.B.; Argani, P.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: A new marker of DPC4 inactivation. Am. J. Pathol. 2000, 156, 37–43. [Google Scholar] [CrossRef]
- Wachter, D.L.; Schlabrakowski, A.; Hoegel, J.; Kristiansen, G.; Hartmann, A.; Riener, M.-O. Diagnostic value of immunohistochemical IMP3 expression in core needle biopsies of pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2011, 35, 873–877. [Google Scholar] [CrossRef]
- Mikata, R.; Yasui, S.; Kishimoto, T.; Kouchi, Y.; Shingyoji, A.; Senoo, J.; Takahashi, K.; Nagashima, H.; Kusakabe, Y.; Ohyama, H. Diagnostic value of IMP3 and p53 immunohistochemical staining in EUS-guided fine-needle aspiration for solid pancreatic tumors. Sci. Rep. 2021, 11, 17257. [Google Scholar] [CrossRef] [PubMed]
- Senoo, J.; Mikata, R.; Kishimoto, T.; Hayashi, M.; Kusakabe, Y.; Yasui, S.; Yamato, M.; Ohyama, H.; Sugiyama, H.; Tsuyuguchi, T. Immunohistochemical analysis of IMP3 and p53 expression in endoscopic ultrasound-guided fine needle aspiration and resected specimens of pancreatic diseases. Pancreatology 2018, 18, 176–183. [Google Scholar] [CrossRef]
- Ibrahim, D.A.; Abouhashem, N.S. Diagnostic value of IMP3 and mesothelin in differentiating pancreatic ductal adenocarcinoma from chronic pancreatitis. Pathol. Res. Pract. 2016, 212, 288–293. [Google Scholar] [CrossRef]
- Burnett, A.S.; Quinn, P.L.; Ajibade, D.V.; Peters, S.R.; Ahlawat, S.K.; Mahmoud, O.M.; Chokshi, R.J. Design of an immunohistochemistry biomarker panel for diagnosis of pancreatic adenocarcinoma. Pancreatology 2019, 19, 842–849. [Google Scholar] [CrossRef]
- Liu, H.; Shi, J.; Anandan, V.; Wang, H.L.; Diehl, D.; Blansfield, J.; Gerhard, G.; Lin, F. Reevaluation and identification of the best immunohistochemical panel (pVHL, Maspin, S100P, IMP-3) for ductal adenocarcinoma of the pancreas. Arch. Pathol. Lab. Med. 2012, 136, 601–609. [Google Scholar] [CrossRef]
- Witzke, K.E.; Großerueschkamp, F.; Jütte, H.; Horn, M.; Roghmann, F.; von Landenberg, N.; Bracht, T.; Kallenbach-Thieltges, A.; Käfferlein, H.; Brüning, T. Integrated Fourier transform infrared imaging and proteomics for identification of a candidate histochemical biomarker in bladder cancer. Am. J. Pathol. 2019, 189, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Neal, D.J.; Amin, M.B.; Smith, S.C. CK20 versus AMACR and p53 immunostains in evaluation of Urothelial Carcinoma in Situ and Reactive Atypia. Diagn. Pathol. 2020, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Alston, E.L.; Zynger, D.L. Does the addition of AMACR to CK20 help to diagnose challenging cases of urothelial carcinoma in situ? Diagn. Pathol. 2019, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Aron, M.; Luthringer, D.J.; McKenney, J.K.; Hansel, D.E.; Westfall, D.E.; Parakh, R.; Mohanty, S.K.; Balzer, B.; Amin, M.B. Utility of a triple antibody cocktail intraurothelial neoplasm-3 (IUN-3-CK20/CD44s/p53) and α-methylacyl-CoA racemase (AMACR) in the distinction of urothelial carcinoma in situ (CIS) and reactive urothelial atypia. Am. J. Surg. Pathol. 2013, 37, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.K.; Desai, S.; Cohen, C.; Amin, M.B. Discriminatory immunohistochemical staining of urothelial carcinoma in situ and non-neoplastic urothelium: An analysis of cytokeratin 20, p53, and CD44 antigens. Am. J. Surg. Pathol. 2001, 25, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.K.; Przybycin, C.G.; McKenney, J.K.; Magi-Galluzzi, C. Immunohistochemical staining patterns of Ki-67 and p53 in florid reactive urothelial atypia and urothelial carcinoma in situ demonstrate significant overlap. Hum. Pathol. 2020, 98, 81–88. [Google Scholar] [CrossRef]
- Edgecombe, A.; Nguyen, B.N.; Djordjevic, B.; Belanger, E.C.; Mai, K.T. Utility of cytokeratin 5/6, cytokeratin 20, and p16 in the diagnosis of reactive urothelial atypia and noninvasive component of urothelial neoplasia. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 264–271. [Google Scholar] [CrossRef]
- Di Sciascio, L.; Ambrosi, F.; Franceschini, T.; Giunchi, F.; Franchini, E.; Massari, F.; Bianchi, F.M.; Colecchia, M.; Fiorentino, M.; Ricci, C. Could double stain for p53/CK20 be a useful diagnostic tool for the appropriate classification of flat urothelial lesions? Pathol. Res. Pract. 2022, 234, 153937. [Google Scholar] [CrossRef]
- Jung, S.; Wu, C.; Eslami, Z.; Tanguay, S.; Aprikian, A.; Kassouf, W.; Brimo, F. The role of immunohistochemistry in the diagnosis of flat urothelial lesions: A study using CK20, CK5/6, P53, Cd138, and Her2/Neu. Ann. Diagn. Pathol. 2014, 18, 27–32. [Google Scholar] [CrossRef]
- Schwarz, S.; Rechenmacher, M.; Filbeck, T.; Knuechel, R.; Blaszyk, H.; Hartmann, A.; Brockhoff, G. Value of multicolour fluorescence in situ hybridisation (UroVysion) in the differential diagnosis of flat urothelial lesions. J. Clin. Pathol. 2008, 61, 272–277. [Google Scholar] [CrossRef]
- Mallofré, C.; Castillo, M.; Morente, V.; Solé, M. Immunohistochemical expression of CK20, p53, and Ki-67 as objective markers of urothelial dysplasia. Mod. Pathol. 2003, 16, 187–191. [Google Scholar] [CrossRef]
- Harnden, P.; Eardley, I.; Joyce, A.; Southgate, J. Cytokeratin 20 as an objective marker of urothelial dysplasia. Br. J. Urol. 1996, 78, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Gunia, S.; Kakies, C.; May, M.; Koch, S.; Erbersdobler, A. Lewisy antigen (blood group 8, BG8) is a useful marker in the histopathological differential diagnosis of flat urothelial lesions of the urinary bladder. J. Clin. Pathol. 2011, 64, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Bastacky, S.; Parwani, A.V.; McHale, T.; Dhir, R. p16ink4 immunoreactivity is a reliable marker for urothelial carcinoma in situ. Hum. Pathol. 2008, 39, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.T.; Cviko, A.; Riethdorf, S.; Riethdorf, L.; Quade, B.J.; Sun, D.; Duensing, S.; Sheets, E.E.; Munger, K.; Crum, C.P. Ki-67, cyclin E, and p16 INK4 are complimentary surrogate biomarkers for human papilloma virus-related cervical neoplasia. Am. J. Surg. Pathol. 2001, 25, 884–891. [Google Scholar] [CrossRef]
- Qiao, X.; Bhuiya, T.A.; Spitzer, M. Differentiating high-grade cervical intraepithelial lesion from atrophy in postmenopausal women using Ki-67, cyclin E, and p16 immunohistochemical analysis. J. Low. Genit. Tract Dis. 2005, 9, 100–107. [Google Scholar] [CrossRef]
- Iaconis, L.; Hyjek, E.; Ellenson, L.H.; Pirog, E.C. p16 and Ki-67 immunostaining in atypical immature squamous metaplasia of the uterine cervix: Correlation with human papillomavirus detection. Arch. Pathol. Lab. Med. 2007, 131, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Rock, K.L.; Woda, B.A.; Jiang, Z.; Fraire, A.E.; Dresser, K. IMP3 is a novel biomarker for adenocarcinoma in situ of the uterine cervix: An immunohistochemical study in comparison with p16INK4a expression. Mod. Pathol. 2007, 20, 242–247. [Google Scholar] [CrossRef]
- Mittal, K.; Mesia, A.; Demopoulos, R.I. MIB-1 expression is useful in distinguishing dysplasia from atrophy in elderly women. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 1999, 18, 122–124. [Google Scholar] [CrossRef]
- Bulten, J.; de Wilde, P.C.; Schijf, C.; van der Laak, J.A.; Wienk, S.; Poddighe, P.J.; Hanselaar, A.G. Decreased expression of Ki-67 in atrophic cervical epithelium of post-menopausal women. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2000, 190, 545–553. [Google Scholar] [CrossRef]
- Simon, R.A.; Peng, S.-L.; Liu, F.; Quddus, M.R.; Zhang, C.; Steinhoff, M.M.; Lawrence, W.D.; Sung, C.J. Tubal metaplasia of the endometrium with cytologic atypia: Analysis of p53, Ki-67, TERT, and long-term follow-up. Mod. Pathol. 2011, 24, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Brustmann, H. Immunohistochemical detection of human telomerase reverse transcriptase (hTERT), topoisomerase IIα expression, and apoptosis in endometrial adenocarcinoma and atypical hyperplasia. Int. J. Gynecol. Pathol. 2005, 24, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Isacson, C.; Levine, R.; Kurman, R.J.; Cho, K.R.; Hedrick, L. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. Am. J. Pathol. 1997, 150, 177. [Google Scholar]
- Negri, G.; Egarter-Vigl, E.; Kasal, A.; Romano, F.; Haitel, A.; Mian, C. p16INK4a is a useful marker for the diagnosis of adenocarcinoma of the cervix uteri and its precursors: An immunohistochemical study with immunocytochemical correlations. Am. J. Surg. Pathol. 2003, 27, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Regauer, S.; Reich, O. CK17 and p16 expression patterns distinguish (atypical) immature squamous metaplasia from high-grade cervical intraepithelial neoplasia (CIN III). Histopathology 2007, 50, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Skapa, P.; Robova, H.; Rob, L.; Zamecnik, J. p16 INK4a Immunoprofiles of Squamous Lesions of the Uterine Cervix–Implications for the Reclassification of Atypical Immature Squamous Metaplasia. Pathol. Oncol. Res. 2013, 19, 707–714. [Google Scholar] [CrossRef]
- van der Marel, J.; van Baars, R.; Alonso, I.; del Pino, M.; van de Sandt, M.; Lindeman, J.; ter Harmsel, B.; Boon, M.; Smedts, F.; Ordi, J. Oncogenic human papillomavirus–infected immature Metaplastic cells and cervical Neoplasia. Am. J. Surg. Pathol. 2014, 38, 470–479. [Google Scholar] [CrossRef]
- McMullen-Tabry, E.R.; Schechter, S.A.; Wang, G.Y.; Sciallis, A.P.; Hrycaj, S.M.; Chan, M.P.; Skala, S.L. p53/CK17 dual stain improves accuracy of distinction between differentiated vulvar intraepithelial neoplasia and its mimics. Int. J. Gynecol. Pathol. 2022, 41, 298–306. [Google Scholar] [CrossRef]
- Dasgupta, S.; Koljenović, S.; van den Bosch, T.P.; Swagemakers, S.M.; van der Hoeven, N.M.; van Marion, R.; van der Spek, P.J.; van Doorn, H.C.; van Kemenade, F.J.; Ewing-Graham, P.C. Evaluation of immunohistochemical markers, CK17 and SOX2, as adjuncts to p53 for the diagnosis of differentiated vulvar intraepithelial neoplasia (dVIN). Pharmaceuticals 2021, 14, 324. [Google Scholar] [CrossRef]
- Brustmann, H.; Brunner, A. Immunohistochemical expression of SOX2 in vulvar intraepithelial neoplasia and squamous cell carcinoma. Int. J. Gynecol. Pathol. 2013, 32, 323–328. [Google Scholar] [CrossRef]
- Cigognetti, M.; Lonardi, S.; Fisogni, S.; Balzarini, P.; Pellegrini, V.; Tironi, A.; Bercich, L.; Bugatti, M.; Rossi, G.; Murer, B. BAP1 (BRCA1-associated protein 1) is a highly specific marker for differentiating mesothelioma from reactive mesothelial proliferations. Mod. Pathol. 2015, 28, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Hamasaki, M.; Matsumoto, S.; Sato, A.; Tsujimura, T.; Kawahara, K.; Iwasaki, A.; Okamoto, T.; Oda, Y.; Honda, H. Immunohistochemical detection of MTAP and BAP1 protein loss for mesothelioma diagnosis: Comparison with 9p21 FISH and BAP1 immunohistochemistry. Lung Cancer 2017, 104, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.B.; Dacic, S.; Miller, C.; Cheung, S.; Churg, A. Utility of methylthioadenosine phosphorylase compared with BAP1 immunohistochemistry, and CDKN2A and NF2 fluorescence in situ hybridization in separating reactive mesothelial proliferations from epithelioid malignant mesotheliomas. Arch. Pathol. Lab. Med. 2018, 142, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Hida, T.; Hamasaki, M.; Matsumoto, S.; Sato, A.; Tsujimura, T.; Kawahara, K.; Hiroshima, K.; Oda, Y.; Nabeshima, K. A combination of MTAP and BAP1 immunohistochemistry in pleural effusion cytology for the diagnosis of mesothelioma. Cancer Cytopathol. 2018, 126, 54–63. [Google Scholar] [CrossRef]
- Yoshimura, M.; Kinoshita, Y.; Hamasaki, M.; Matsumoto, S.; Hida, T.; Oda, Y.; Iwasaki, A.; Nabeshima, K. Highly expressed EZH2 in combination with BAP1 and MTAP loss, as detected by immunohistochemistry, is useful for differentiating malignant pleural mesothelioma from reactive mesothelial hyperplasia. Lung Cancer 2019, 130, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Chapel, D.B.; Schulte, J.J.; Berg, K.; Churg, A.; Dacic, S.; Fitzpatrick, C.; Galateau-Salle, F.; Hiroshima, K.; Krausz, T.; Le Stang, N. MTAP immunohistochemistry is an accurate and reproducible surrogate for CDKN2A fluorescence in situ hybridization in diagnosis of malignant pleural mesothelioma. Mod. Pathol. 2020, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hasteh, F.; Lin, G.Y.; Weidner, N.; Michael, C.W. The use of immunohistochemistry to distinguish reactive mesothelial cells from malignant mesothelioma in cytologic effusions. Cancer Cytopathol. J. Am. Cancer Soc. 2010, 118, 90–96. [Google Scholar] [CrossRef]
- Attanoos, R.; Griffin, A.; Gibbs, A. The use of immunohistochemistry in distinguishing reactive from neoplastic mesothelium. A novel use for desmin and comparative evaluation with epithelial membrane antigen, p53, platelet-derived growth factor-receptor, P-glycoprotein and Bcl-2. Histopathology 2003, 43, 231–238. [Google Scholar] [CrossRef]
- McKelvie, P.A.; Chan, F.; Yu, Y.; Waring, P.; Gresshoff, I.; Farrell, S.; Williams, R.A. The prognostic significance of the BRAFV600E mutation in papillary thyroid carcinoma detected by mutation-specific immunohistochemistry. Pathology 2013, 45, 637–644. [Google Scholar] [CrossRef]
- Ilie, M.I.; Lassalle, S.; Long-Mira, E.; Bonnetaud, C.; Bordone, O.; Lespinet, V.; Lamy, A.; Sabourin, J.-C.; Haudebourg, J.; Butori, C. Diagnostic value of immunohistochemistry for the detection of the BRAFV600E mutation in papillary thyroid carcinoma: Comparative analysis with three DNA-based assays. Thyroid 2014, 24, 858–866. [Google Scholar] [CrossRef]
- Chui, M.H.; Cassol, C.A.; Asa, S.L.; Mete, O. Follicular epithelial dysplasia of the thyroid: Morphological and immunohistochemical characterization of a putative preneoplastic lesion to papillary thyroid carcinoma in chronic lymphocytic thyroiditis. Virchows Arch. 2013, 462, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yan, J.; Zhang, C.; Qin, S.; Qin, L.; Liu, L.; Wang, X.; Li, N. Expression of papillary thyroid carcinoma-associated molecular markers and their significance in follicular epithelial dysplasia with papillary thyroid carcinoma-like nuclear alterations in Hashimoto’s thyroiditis. Int. J. Clin. Exp. Pathol. 2014, 7, 7999. [Google Scholar] [PubMed]
- Liu, H.; Lin, F. Application of immunohistochemistry in thyroid pathology. Arch. Pathol. Lab. Med. 2015, 139, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Pellegata, N.S.; Huang, Y.; Nagaraja, H.N.; de la Chapelle, A.; Kloos, R.T. Galectin-3, fibronectin-1, CITED-1, HBME1 and cytokeratin-19 immunohistochemistry is useful for the differential diagnosis of thyroid tumors. Mod. Pathol. 2005, 18, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Morreau, H.; Kievit, J.; Romijn, J.A.; Carrasco, N.; Smit, J.W. Combined immunostaining with galectin-3, fibronectin-1, CITED-1, Hector Battifora mesothelial-1, cytokeratin-19, peroxisome proliferator-activated receptor-γ, and sodium/iodide symporter antibodies for the differential diagnosis of non-medullary thyroid carcinoma. Eur. J. Endocrinol. 2008, 158, 375–384. [Google Scholar]
- Saggiorato, E.; De Pompa, R.; Volante, M.; Cappia, S.; Arecco, F.; Dei Tos, A.; Orlandi, F.; Papotti, M. Characterization of thyroid’follicular neoplasms’ in fine-needle aspiration cytological specimens using a panel of immunohistochemical markers: A proposal for clinical application. Endocr. Relat. Cancer 2005, 12, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Slosar, M.; Vohra, P.; Prasad, M.; Fischer, A.; Quinlan, R.; Khan, A. Insulin-like growth factor mRNA binding protein 3 (IMP3) is differentially expressed in benign and malignant follicular patterned thyroid tumors. Endocr. Pathol. 2009, 20, 149–157. [Google Scholar] [CrossRef]
- Bian, Y.S.; Osterheld, M.C.; Bosman, F.T.; Benhattar, J.; Fontolliet, C. p53 gene mutation and protein accumulation during neoplastic progression in Barrett’s esophagus. Mod. Pathol. 2001, 14, 397–403. [Google Scholar] [CrossRef][Green Version]
- Kaminagakura, E.; Bonan, P.R.; Lopes, M.A.; Almeida, O.P. Cell proliferation and p53 expression in pseudoepitheliomatous hyperplasia of oral paracoccidioidomycosis. Mycoses 2006, 49, 393–396. [Google Scholar] [CrossRef]
- Lu, D.; Vohra, P.; Chu, P.G.; Woda, B.; Rock, K.L.; Jiang, Z. An oncofetal protein IMP3: A new molecular marker for the detection of esophageal adenocarcinoma and high-grade dysplasia. Am. J. Surg. Pathol. 2009, 33, 521–525. [Google Scholar] [CrossRef]
- Feng, W.; Zhou, Z.; Peters, J.H.; Khoury, T.; Zhai, Q.; Wei, Q.; Truong, C.D.; Song, S.W.; Tan, D. Expression of insulin-like growth factor II mRNA-binding protein 3 in human esophageal adenocarcinoma and its precursor lesions. Arch. Pathol. Lab. Med. 2011, 135, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.T.; Budnick, S.D.; Logani, S. Immunohistochemical detection of p16INK4a in dysplastic lesions of the oral cavity. Mod. Pathol. 2006, 19, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Legan, M.; Luzar, B.; Marolt, V.F.; Cör, A. Expression of cyclooxygenase-2 is associated with p53 accumulation in premalignant and malignant gallbladder lesions. World J. Gastroenterol. 2006, 12, 3425. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, A.; Yamauchi, Y.; Hirohashi, S. p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int. J. Cancer 1996, 69, 28–33. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Castresana, J.S.; Rubio, M.P.; Vázquez, J.J.; Idoate, M.; Sober, A.J.; Seizinger, B.R.; Barnhill, R.L. Lack of allelic deletion and point mutation as mechanisms of p53 activation in human malignant melanoma. Int. J. Cancer 1993, 55, 562–565. [Google Scholar] [CrossRef]
- Rubio, M.P.; von Deimling, A.; Yandell, D.W.; Wiestler, O.D.; Gusella, J.F.; Louis, D.N. Accumulation of wild type p53 protein in human astrocytomas. Cancer Res. 1993, 53, 3465–3467. [Google Scholar]
- Noffsinger, A.; Unger, B.; Fenoglio-Preiser, C.M. Increased cell proliferation characterizes Crohn’s disease. Mod. Pathol. 1998, 11, 1198–1203. [Google Scholar] [PubMed]
- Degrauwe, N.; Suva, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Woda, B.A.; Jiang, Z. Oncofetal protein IMP3, a new cancer biomarker. Adv. Anat. Pathol. 2014, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Wang, X.; Liu, M.; Zeng, Z.; Lin, C.; Xu, W.; Ma, W.; Wang, J.; Xiang, Q.; Johnston, R.N.; et al. The Oncogenic Functions of Insulin-like Growth Factor 2 mRNA-Binding Protein 3 in Human Carcinomas. Curr. Pharm. Des. 2020, 26, 3939–3954. [Google Scholar] [CrossRef] [PubMed]
- Chapel, D.B.; Schulte, J.J.; Husain, A.N.; Krausz, T. Application of immunohistochemistry in diagnosis and management of malignant mesothelioma. Transl. Lung Cancer Res. 2020, 9, S3–S27. [Google Scholar] [CrossRef] [PubMed]
- Raffone, A.; Travaglino, A.; D’Antonio, A.; De Marco, M.; Caccese, M.; Mascolo, M.; Insabato, L.; Zeppa, P.; Rosati, A.; Mollo, A.; et al. BAG3 expression correlates with the grade of dysplasia in squamous intraepithelial lesions of the uterine cervix. Acta Obstet. Gynecol. Scand. 2020, 99, 99–104. [Google Scholar] [CrossRef] [PubMed]
- De Marco, M.; Falco, A.; Iaccarino, R.; Raffone, A.; Mollo, A.; Guida, M.; Rosati, A.; Chetta, M.; Genovese, G.; De Caro, F.; et al. An emerging role for BAG3 in gynaecological malignancies. Br. J. Cancer 2021, 125, 789–797. [Google Scholar] [CrossRef]
- Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Genetically engineered mouse models in cancer research. Adv. Cancer Res. 2010, 106, 113–164. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Zheng, Y.; Sefik, E.; Astle, J.; Karatepe, K.; Oz, H.H.; Solis, A.G.; Jackson, R.; Luo, H.R.; Bruscia, E.M.; Halene, S.; et al. Human neutrophil development and functionality are enabled in a humanized mouse model. Proc. Natl. Acad. Sci. USA 2022, 119, e2121077119. [Google Scholar] [CrossRef]
- Hayden, P.J.; Harbell, J.W. Special review series on 3D organotypic culture models: Introduction and historical perspective. In Vitro Cell. Dev. Biol. Anim. 2021, 57, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Laczko, D.; Wang, F.; Johnson, F.B.; Jhala, N.; Rosztoczy, A.; Ginsberg, G.G.; Falk, G.W.; Rustgi, A.K.; Lynch, J.P. Modeling Esophagitis Using Human Three-Dimensional Organotypic Culture System. Am. J. Pathol. 2017, 187, 1787–1799. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fleming Martinez, A.K.; Storz, P. Mimicking and Manipulating Pancreatic Acinar-to-Ductal Metaplasia in 3-dimensional Cell Culture. J. Vis. Exp. 2019, 144, e59096. [Google Scholar] [CrossRef]
- DeHaan, R.K.; Sarvestani, S.K.; Huang, E.H. Organoid Models of Colorectal Pathology: Do They Hold the Key to Personalized Medicine? A Systematic Review. Dis. Colon Rectum 2020, 63, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Van de Laar, E.; Cabanero, M.; Tarumi, S.; Hasenoeder, S.; Wang, D.; Virtanen, C.; Suzuki, T.; Bandarchi, B.; Sakashita, S.; et al. SOX2 and PI3K Cooperate to Induce and Stabilize a Squamous-Committed Stem Cell Injury State during Lung Squamous Cell Carcinoma Pathogenesis. PLoS Biol. 2016, 14, e1002581. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.M.; De Haan, P.; Ronaldson-Bouchard, K.; Kim, G.A.; Ko, J.; Rho, H.S.; Toh, Y.C. A guide to the organ-on-a-chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Cooks, T.; Theodorou, S.D.; Paparouna, E.; Rizou, S.V.; Myrianthopoulos, V.; Gorgoulis, V.G.; Pateras, I.S. Immunohisto(cyto)chemistry: An old time classic tool driving modern oncological therapies. Histol. Histopathol. 2019, 34, 335–352. [Google Scholar]
- Hwang, L.A.; Phang, B.H.; Liew, O.W.; Iqbal, J.; Koh, X.H.; Koh, X.Y.; Othman, R.; Xue, Y.; Richards, A.M.; Lane, D.P.; et al. Monoclonal Antibodies against Specific p53 Hotspot Mutants as Potential Tools for Precision Medicine. Cell Rep. 2018, 22, 299–312. [Google Scholar] [CrossRef]
- Danks, M.K.; Whipple, D.O.; McPake, C.R.; Lu, D.; Harris, L.C. Differences in epitope accessibility of p53 monoclonal antibodies suggest at least three conformations or states of protein binding of p53 protein in human tumor cell lines. Cell Death Differ. 1998, 5, 678–686. [Google Scholar] [CrossRef][Green Version]
- Wei, W.J.; Shen, C.T.; Song, H.J.; Qiu, Z.L.; Luo, Q.Y. MicroRNAs as a potential tool in the differential diagnosis of thyroid cancer: A systematic review and meta-analysis. Clin. Endocrinol. 2016, 84, 127–133. [Google Scholar] [CrossRef]
- Turai, P.I.; Herold, Z.; Nyiro, G.; Borka, K.; Micsik, T.; Toke, J.; Szucs, N.; Toth, M.; Patocs, A.; Igaz, P. Tissue miRNA Combinations for the Differential Diagnosis of Adrenocortical Carcinoma and Adenoma Established by Artificial Intelligence. Cancers 2022, 14, 895. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Mitwally, N.; Soliman, A.S.; Yousef, E. Potential Diagnostic and Prognostic Utility of miR-141, miR-181b1, and miR-23b in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8589. [Google Scholar] [CrossRef] [PubMed]
- Quiohilag, K.; Caie, P.; Oniscu, A.; Brenn, T.; Harrison, D. The differential expression of micro-RNAs 21, 200c, 204, 205, and 211 in benign, dysplastic and malignant melanocytic lesions and critical evaluation of their role as diagnostic biomarkers. Virchows Arch. Int. J. Pathol. 2020, 477, 121–130. [Google Scholar] [CrossRef] [PubMed]
- James, J.P.; Riis, L.B.; Malham, M.; Hogdall, E.; Langholz, E.; Nielsen, B.S. MicroRNA Biomarkers in IBD-Differential Diagnosis and Prediction of Colitis-Associated Cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; Ra, S.; Li, X.; Zhou, J.; Binder, S. Differentiating cutaneous squamous cell carcinoma and pseudoepitheliomatous hyperplasia by multiplex qRT-PCR. Mod. Pathol. 2013, 26, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Niazi, M.K.K.; Parwani, A.V.; Gurcan, M.N. Digital pathology and artificial intelligence. Lancet Oncol. 2019, 20, e253–e261. [Google Scholar] [CrossRef]
- Baxi, V.; Edwards, R.; Montalto, M.; Saha, S. Digital pathology and artificial intelligence in translational medicine and clinical practice. Mod. Pathol. 2022, 35, 23–32. [Google Scholar] [CrossRef]
- Srinidhi, C.L.; Ciga, O.; Martel, A.L. Deep neural network models for computational histopathology: A survey. Med. Image Anal. 2021, 67, 101813. [Google Scholar] [CrossRef]
- Alam, M.R.; Abdul-Ghafar, J.; Yim, K.; Thakur, N.; Lee, S.H.; Jang, H.J.; Jung, C.K.; Chong, Y. Recent Applications of Artificial Intelligence from Histopathologic Image-Based Prediction of Microsatellite Instability in Solid Cancers: A Systematic Review. Cancers 2022, 14, 2590. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyo, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Hong, R.; Liu, W.; DeLair, D.; Razavian, N.; Fenyo, D. Predicting endometrial cancer subtypes and molecular features from histopathology images using multi-resolution deep learning models. Cell Rep. Med. 2021, 2, 100400. [Google Scholar] [CrossRef] [PubMed]
- Ash, J.T.; Darnell, G.; Munro, D.; Engelhardt, B.E. Joint analysis of expression levels and histological images identifies genes associated with tissue morphology. Nat. Commun. 2021, 12, 1609. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Nelson, M.S.; Savari, O.; Loeffler, A.G.; Eliceiri, K.W. Differentiation of pancreatic ductal adenocarcinoma and chronic pancreatitis using graph neural networks on histopathology and collagen fiber features. J. Pathol. Inform. 2022, 13, 100158. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.C.B.; Bergonzini, A.; Lopez Chiloeches, M.; Paparouna, E.; Butter, D.; Theodorou, S.D.P.; Haykal, M.M.; Boutet-Robinet, E.; Tebaldi, T.; Wakeham, A.; et al. Influence of the microenvironment on modulation of the host response by typhoid toxin. Cell Rep. 2021, 35, 108931. [Google Scholar] [CrossRef]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.P.; Namini, M.R.J.; Hansen, M.B.; Martin, O.C.B.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunostaining Pattern | ||||
|---|---|---|---|---|
| Anatomical Position | Protein (s) | Reactive Lesions | Precancerous–Cancerous Lesions | Reference |
| CNS | EGFR | Gliosis: (+) weak membranous | Gliomas: (+) strong membranous | [105,106] |
| IDH1 p.R132H | Gliosis: (−) | Gliomas: usually (+) diffused and strong cytoplasmic and weak nuclear | [107,108,109] | |
| P53 | Gliosis: (−) | Gliomas: occasionally (+) diffused and strong nuclear | [107,108,109] | |
| Oral cavity | CK-1ε | Atypical squamous epithelium: (+) weak nuclear | Carcinoma in situ: (+) strong nuclear | [110] |
| CD44 | Atypical squamous epithelium: (+) weak membranous | Carcinoma in situ: (+) strong membranous | [110] | |
| E-Cadherin | Pseudoepitheliomatous hyperplasia: (+) membranous | Squamous cell carcinoma: decreased (+) membranous in the invasive front | [111] | |
| DEC1 | Atypical squamous epithelium: (+) strong nuclear | Carcinoma in situ: (+) weak nuclear | [110] | |
| Ki67 | Pseudoepitheliomatous hyperplasia: (+) nuclear restricted in basal and parabasal cells | Dysplasia: often (+) extended to the spinous layer | [191] | |
| MMP-1 | Pseudoepitheliomatous hyperplasia: (+) cytoplasmic with a basal cell pattern | Squamous cell carcinoma: (+) diffused cytoplasmic | [19] | |
| PCNA | Inflammatory lesion: (+) nuclear in the basal layer | Dysplasia: consistently (+) nuclear in the suprabasal layer | [112,113] | |
| P16INK4a | Inflammatory lesion: (−) or minimal (+) cytoplasmic/nuclear restricted in the basal cells | Dysplasia: (−) or often (+) strong and diffused cytoplasmic/nuclear in the middle and upper thirds or (−) | [112,194] | |
| P53 | Pseudoepitheliomatosis hyperplasia: occasionally (+) moderate-intensity nuclear with a basal-cell layer pattern | Dysplasia/squamous cell carcinoma: often (+) intense and diffused nuclear | [19,111,191] | |
| Esophagus | Ki67 | Normal/RA: focal (+) nuclear, usually restricted to the lower third | HGD/carcinoma: (+) diffused nuclear | [114] |
| P53 | Normal/RA: usually (−) and to a lesser extent focal (+) weak nuclear | HGD/carcinoma: (+) diffused and intense nuclear and rarely (−) (null pattern) | [114] | |
| Esophagus (Barrett’s) | AMACR (P504S) | IND: usually (−) and to a lesser extent (+) with focal cytoplasmic | LGD: often (+) diffused and, to a lesser extent, focal cytoplasmic; HGD/ACC: usually (+) diffused and, to a lesser extent, focal cytoplasmic | [115,116,117] |
| IMP3 | IND: rarely (+) with cytoplasmic and membranous | LGD: occasionally (+) with cytoplasmic and membranous; HGD: often (+) with cytoplasmic and membranous | [118,192,193] | |
| Ki67 | BE: (+) nuclear at the base of the crypt | Dysplasia: (+) diffused nuclear | [119] | |
| P53 | BE: (−) | LGD: usually (+) diffused nuclear; HGD: regularly (+) diffused nuclear | [120,190] | |
| Stomach | AMACR (P504S) | Non-neoplastic epithelium: (−) and rarely (+) weak cytoplasmic | Dysplasia/adenocarcinoma: usually (+) moderate and strong cytoplasmic | [121] |
| Ki67 | RA: (+) nuclear with a limited expression pattern | LGD/HGD: (+) often diffused nuclear (with an expansion of the proliferating zone) | [122,123] | |
| IMP3 | RA: often (+) with focal cytoplasmic and membranous (in the basal part of the cell) | LGD: often (+) weak cytoplasmic and membranous; HGD: often (+) diffused moderate/intense cytoplasmic and membranous | [124] | |
| P53 | RA: (−) or (+) focal and rarely diffused nuclear | LGD: rarely (+) weak-to-moderate nuclear; HGD: often (+) moderate/strong nuclear | [122,123] | |
| Colon | AMACR (P504S) | IND: rarely (+) focal cytoplasmic | LGD/HGD/ACC: (+) often diffused cytoplasmic | [115] |
| P21WAF1 | Regenerative atypia and indefinite for dysplasia: (+) strong nuclear mainly located in the superficial portion of colonic glands that are p53 (−) | Dysplasia and ACA: (−) in areas with (+) diffused P53 status | [125,126,127] | |
| P53 | Regenerative atypia and indefinite for dysplasia: (+) mainly few isolated cells with weak and moderate and to a lesser extent basal/nested nuclear | Dysplasia and ACA: (+) strong and diffused, basal/nested, and to a lesser extent few isolated cells nuclear | [125,126,127] | |
| Biliary tract | CD10 | Normal/RA: (+) strong membranous with continuous apical pattern | HGD/ECC: (−) and rarely (+) focal moderate membranous | [128] |
| CD24 | Normal/RA: (−) or (+) focally membranous/cytoplasmic | Dysplastic epithelium/ECC/ICC/GBC: (+) strong membranous/cytoplasmic | [129] | |
| P-Cadherin | Normal/RA: (−) or rarely (+) focal membranous | Dysplastic epithelium/ECC/ICC/GBC: often (+) focal/diffused membranous | [129] | |
| HMGA1, HMGA2 | RA: (+) weak/moderate nuclear | Carcinoma: (+) intense nuclear | [130] | |
| Mesothelin | RA: (−) | High-grade BillN and EHBDCa: often (+) diffused cytoplasmic and membranous | [131] | |
| P53 | Normal/RA: (−) or (+) focal weak/moderate nuclear | Dysplastic epithelium/ECC/ICC/GBC: often (+) diffused and intense nuclear | [129,130] | |
| S100A, S100A4 | Normal/RA: (−) or rare (+) cytoplasmic and nuclear | Dysplasia (including high-grade BillN)/carcinomas arising in periampullary duodenal mucosa/EHBD: usually (+) diffused membranous and cytoplasmic | [131,132] | |
| S100P | RA: (−) or rarely (+) nuclear, weak cytoplasmic | High-grade BillN and ICC: occasionally (+) diffused and intense nuclear and cytoplasmic | [133,134] | |
| Gallbladder | P16INK4a | Normal/RA: (−) and rarely (+) nuclear | Dysplasia/carcinoma: often (+) diffused and intense nuclear | [135] |
| P53 | Normal: (−) | Dysplasia/carcinoma: often (+) diffused and intense nuclear | [195] | |
| COX2 | Normal: (−) and rarely (+) | Dysplasia/carcinoma: often (+) diffused cytoplasmic/nuclear | [195] | |
| Pancreas | DPC4 (SMAD4) | Benign: (+) diffused cytoplasmic and occasionally nuclear | PDAC: usually (−), occasionally (+) diffused cytoplasmic and nuclear | [136] |
| IMP3 | Normal/pancreatitis: (−) and rarely (+) focal membranous and cytoplasmic | PDAC: usually (+) diffused membranous and cytoplasmic | [137,138,139,140,141,142] | |
| Maspin | Normal: usually (−), rarely (+) focal nuclear and cytoplasmic | PDAC: usually (+) diffused nuclear and cytoplasmic | [142] | |
| Mesothelin | Pancreatitis: (−) and rarely (+) with focal membranous and cytoplasmic | PDAC: usually (+) diffused membranous and cytoplasmic | [140] | |
| P53 | Pancreatitis: (−), rarely (+) | PDAC: often (+) diffused intense nuclear | [138,139] | |
| S100A4 | Normal: (+) focal membranous and cytoplasmic | PDAC: often (+) diffused membranous and cytoplasmic | [141] | |
| S100P | Normal: usually (−), rarely (+) focal nuclear and cytoplasmic | PDAC: usually (+) diffused nuclear and cytoplasmic | [142] | |
| VHL | Normal: (+) diffused cytoplasmic | PDAC: (+) focal cytoplasmic | [141,142] | |
| Urinary Bladder | AHNAK2 | RUA: (−) | Urothelial CIS: (+) diffused cytoplasmic | [143] |
| AMACR (P504s) | RUA: (−) | Urothelial CIS: often (+) diffused and intense cytoplasmic | [144,145] | |
| CD44 | RUA: usually (+) membranous with a basal-to-full-thickness pattern | Urothelial CIS: often (−) or (+) focal membranous with a basal pattern | [146,147,148] | |
| CK5/6 | RUA: (+) diffused and intense membranous with full-thickness pattern | Urothelial CIS: (−) and rarely (+) membranous with a basal pattern | [149] | |
| CK20 | RUA: (+) membranous limited to umbrella cells | Urothelial CIS: usually (+) full-thickness membranous | [144,145,146,147,148,150,151,152,153,154] | |
| HER2/Neu | RUA: usually (−) or (+) faint membranous limited to umbrella cells | Urothelial CIS: often (+) moderate-to-intense full-thickness membranous | [151,152,155] | |
| Lewis(y) antigen | RUA: (+) patchy membranous | Urothelial CIS: (+) intense full-thickness membranous | [155] | |
| P16INK4a | RUA: occasionally (−) or (+) weak nuclear and cytoplasmic | Urothelial CIS/HGUC: (+) diffused and intense nuclear and cytoplasmic | [156] | |
| P53 | RUA: (−) or (+) patchy and weak nuclear | Urothelial CIS:often (+) diffused and intense nuclear or rarely (−) | [148,150,151,152,153] | |
| Uterine Cervix | Cyclin E | RC and atrophic cervical epithelium: mainly (−), rarely (+) nuclear | HSIL: occasionally (+) diffused full-thickness nuclear | [157,158,159] |
| IMP3 | Normal: (−); tubular metaplasia: (−) | In situ adenocarcinoma: (+) diffused and intense nuclear and cytoplasmic | [160] | |
| Ki67 | RC and atrophic cervical epithelium: few (+) scattered basal and parabasal nuclei, rarely (+) in the upper two thirds | HSIL: (+) diffuse full-thickness nuclear | [157,158,159,161,162,163,164,165] | |
| P16INK4a | Normal, RC, and atrophic cervical: mainly (−) and occasionally (+) weak, in the lower half of the epithelium nuclear and cytoplasmic | LSIL: (+) varying intensity, mainly in the lower half of the epithelium nuclear and cytoplasmic; HSIL: (+) diffuse and intense full-thickness nuclear and cytoplasmic | [157,158,159,160,166,167,168,169] | |
| P53 | Atypical tubal metaplasia: (−) and often focal weak (+) | Uterine serous carcinoma: frequently (+) diffused and moderate-to-intense nuclear; rarely moderate (+) nuclear or (−) | [163,164,165] | |
| TERT | Atypical tubal metaplasia: (−) | Uterine serous carcinoma: (+) weak, moderate, and intense nuclear | [163,164,165] | |
| Vulva | CK17 | Normal/reactive entity: usually (−); to a lesser extent (+) patchy and weak suprabasal and rarely (+) moderate–intense suprabasal membranous | VIN: usually (+) moderate–strong full-thickness or suprabasal membranous and, to a lesser extent, patchy moderate–intense suprabasal membranous | [170,171] |
| P53 | Reactive entity: (+) patchy and weak nuclear | VIN: often (+) diffused and intense nuclear | [170,171] | |
| SOX2 | Normal/lichen sclerosus: usually (+) scattered faint or moderate/intense basal and suprabasal nuclear | VIN: usually (+) moderate/intense and full-thickness nuclear | [172] | |
| Pleura | BAP1 | Reactive mesothelial hyperplasia: (+) diffused nuclear | Malignant mesothelioma: frequent (−) | [173,174,175,176,177,178] |
| Desmin | Reactive mesothelial hyperplasia: usually intense and diffused (+) cytoplasmic | Malignant mesothelioma: usually (−), occasionally focal, and rarely diffused (+) cytoplasmic with faint/moderate intensity | [179,180] | |
| EMA | Reactive mesothelial hyperplasia: usually (−), occasionally (+) focal membranous, and rarely (+) diffused membranous | Malignant mesothelioma: usually (+) intense and diffused membranous | [179,180] | |
| MTAP | Reactive mesothelial hyperplasia: (+) diffused cytoplasmic | Malignant mesothelioma: frequent (−) | [173,174,175,176,177,178] | |
| P53 | Reactive mesothelial hyperplasia: usually (−) and rarely (+) intense nuclear | Malignant mesothelioma: often diffused and intense (+) nuclear | [179,180] | |
| Thyroid gland | BRAF p.V600E | Normal: (−) | PTC: (+) diffused cytoplasmic | [181,182] |
| CITED1 | Normal/RA: (−) | PTC: (+) diffused cytoplasmic and nuclear | [181,182,183,184,185,186,187,188] | |
| CK19 | Normal/RA: mainly (−) and to a lesser extent (+) focal weak/moderate membranous | PTC: frequently (+) moderate/intense membranous | ||
| CD56 | Normal/RA: (+) intense membranous | PTC: mainly (–) and, to a lesser extent, (+) weak membranous | ||
| FN1 | Normal/RA: (−) | PTC: (+) cytoplasmic and membranous | ||
| Galectin-3 | Normal/RA: (−) | PTC: frequently (+) diffused cytoplasmic | ||
| HBME-1 | Normal/RA: (−) | PTC: frequently (+) diffuse and intense membranous | ||
| IMP3 | Thyroiditis Hashimoto: (−) | FVPC, FC: often (+) with diffused membranous and cytoplasmic | [189] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pateras, I.S.; Igea, A.; Nikas, I.P.; Leventakou, D.; Koufopoulos, N.I.; Ieronimaki, A.I.; Bergonzini, A.; Ryu, H.S.; Chatzigeorgiou, A.; Frisan, T.; et al. Diagnostic Challenges during Inflammation and Cancer: Current Biomarkers and Future Perspectives in Navigating through the Minefield of Reactive versus Dysplastic and Cancerous Lesions in the Digestive System. Int. J. Mol. Sci. 2024, 25, 1251. https://doi.org/10.3390/ijms25021251
Pateras IS, Igea A, Nikas IP, Leventakou D, Koufopoulos NI, Ieronimaki AI, Bergonzini A, Ryu HS, Chatzigeorgiou A, Frisan T, et al. Diagnostic Challenges during Inflammation and Cancer: Current Biomarkers and Future Perspectives in Navigating through the Minefield of Reactive versus Dysplastic and Cancerous Lesions in the Digestive System. International Journal of Molecular Sciences. 2024; 25(2):1251. https://doi.org/10.3390/ijms25021251
Chicago/Turabian StylePateras, Ioannis S., Ana Igea, Ilias P. Nikas, Danai Leventakou, Nektarios I. Koufopoulos, Argyro Ioanna Ieronimaki, Anna Bergonzini, Han Suk Ryu, Antonios Chatzigeorgiou, Teresa Frisan, and et al. 2024. "Diagnostic Challenges during Inflammation and Cancer: Current Biomarkers and Future Perspectives in Navigating through the Minefield of Reactive versus Dysplastic and Cancerous Lesions in the Digestive System" International Journal of Molecular Sciences 25, no. 2: 1251. https://doi.org/10.3390/ijms25021251
APA StylePateras, I. S., Igea, A., Nikas, I. P., Leventakou, D., Koufopoulos, N. I., Ieronimaki, A. I., Bergonzini, A., Ryu, H. S., Chatzigeorgiou, A., Frisan, T., Kittas, C., & Panayiotides, I. G. (2024). Diagnostic Challenges during Inflammation and Cancer: Current Biomarkers and Future Perspectives in Navigating through the Minefield of Reactive versus Dysplastic and Cancerous Lesions in the Digestive System. International Journal of Molecular Sciences, 25(2), 1251. https://doi.org/10.3390/ijms25021251