
Endothelial Cell Apoptosis but Not Necrosis Is Inhibited by Ischemic Preconditioning

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Lactate Dehydrogenase Release
2.2. Endothelial Cell Viability in Double Staining with Propidium Iodide and Annexin V
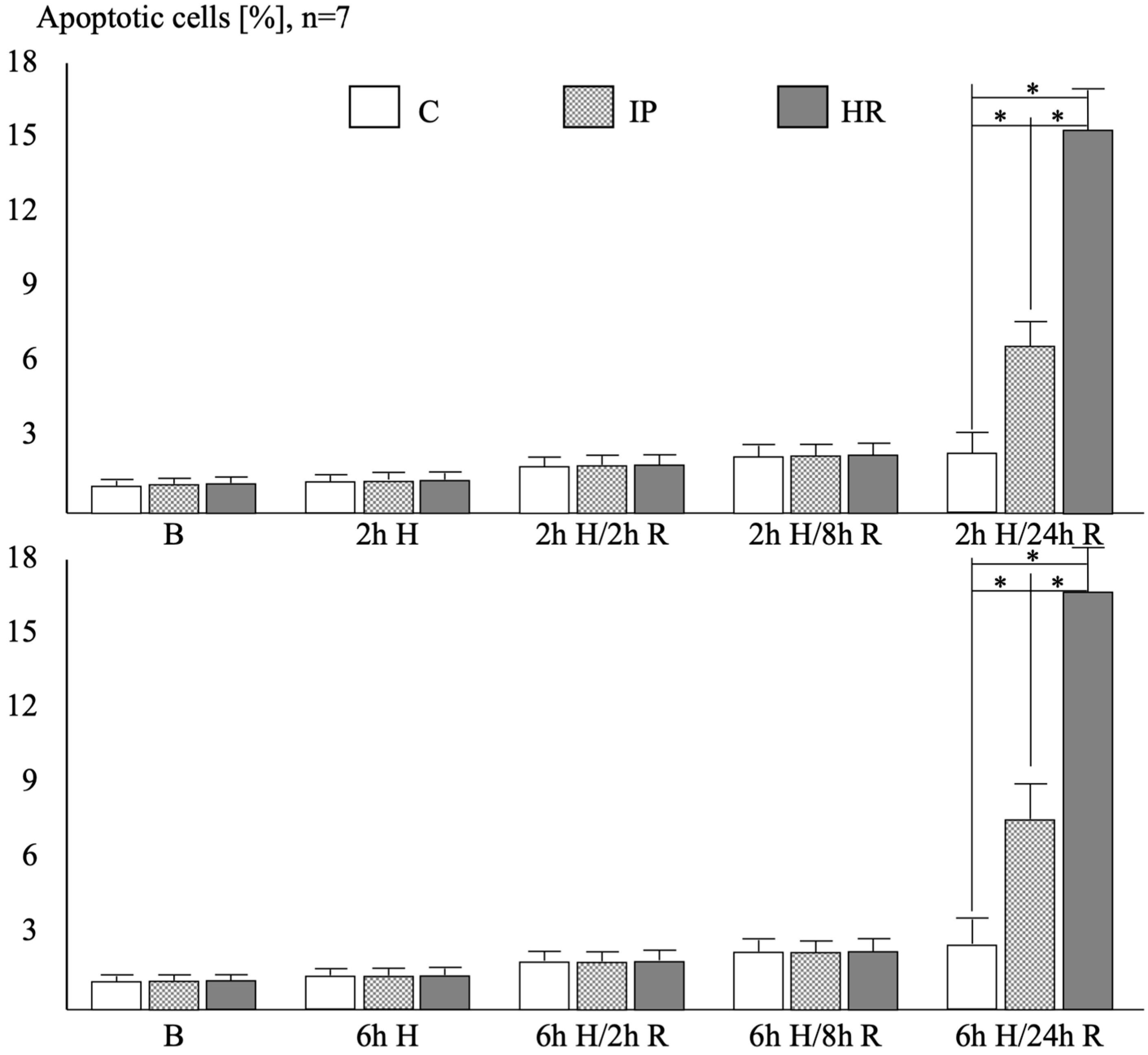
2.3. Endothelial Cell Apoptosis in the TUNEL Technique
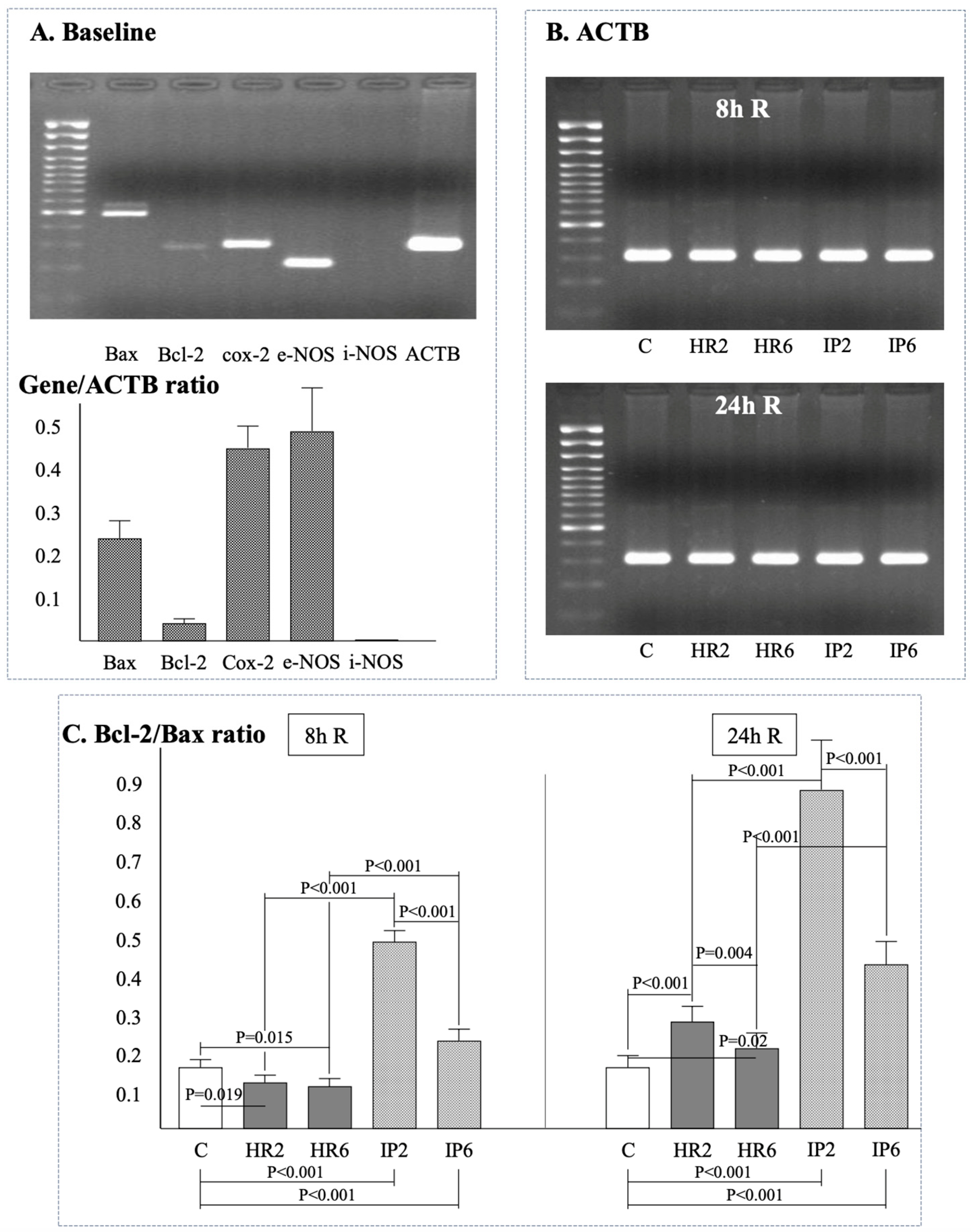
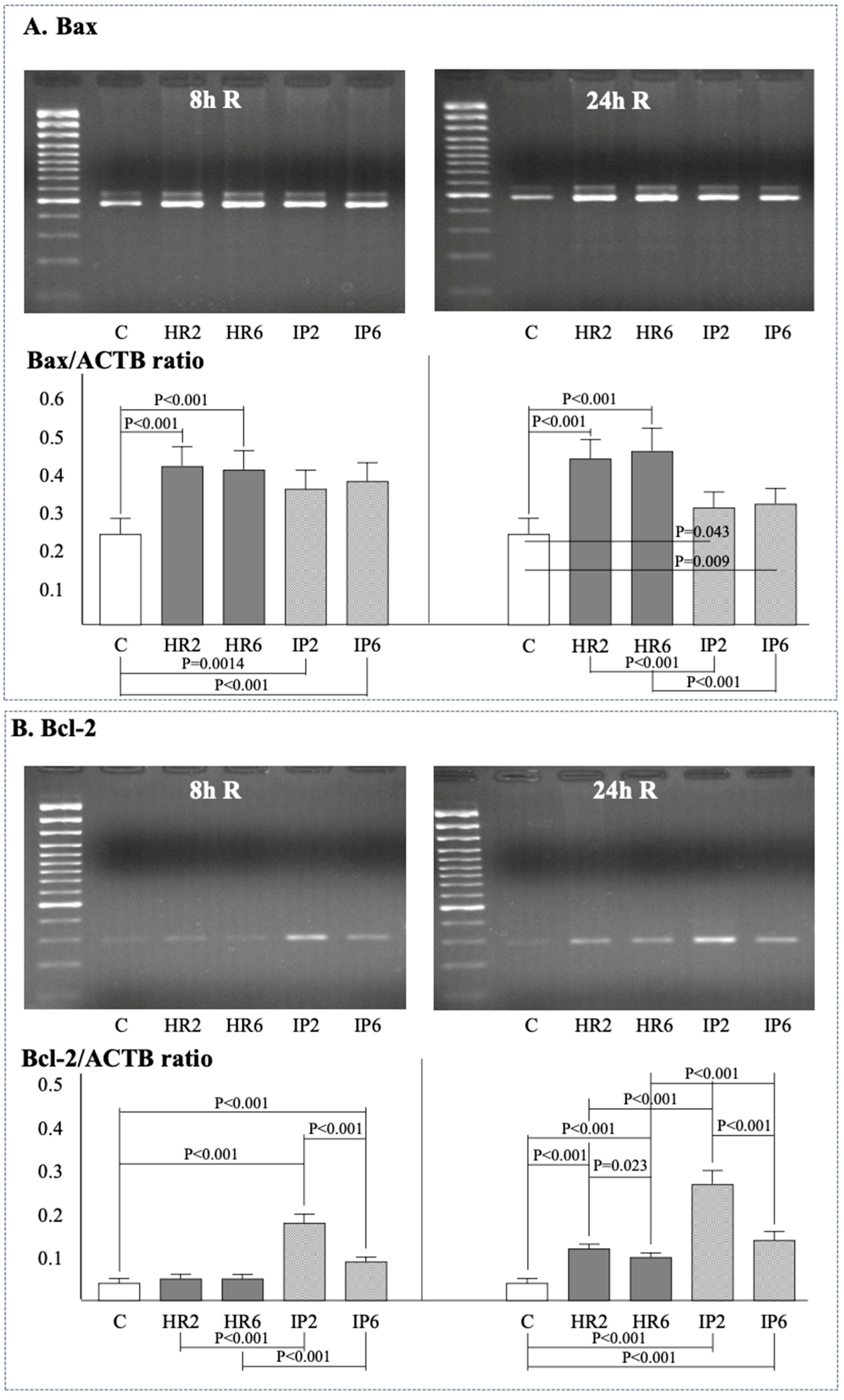
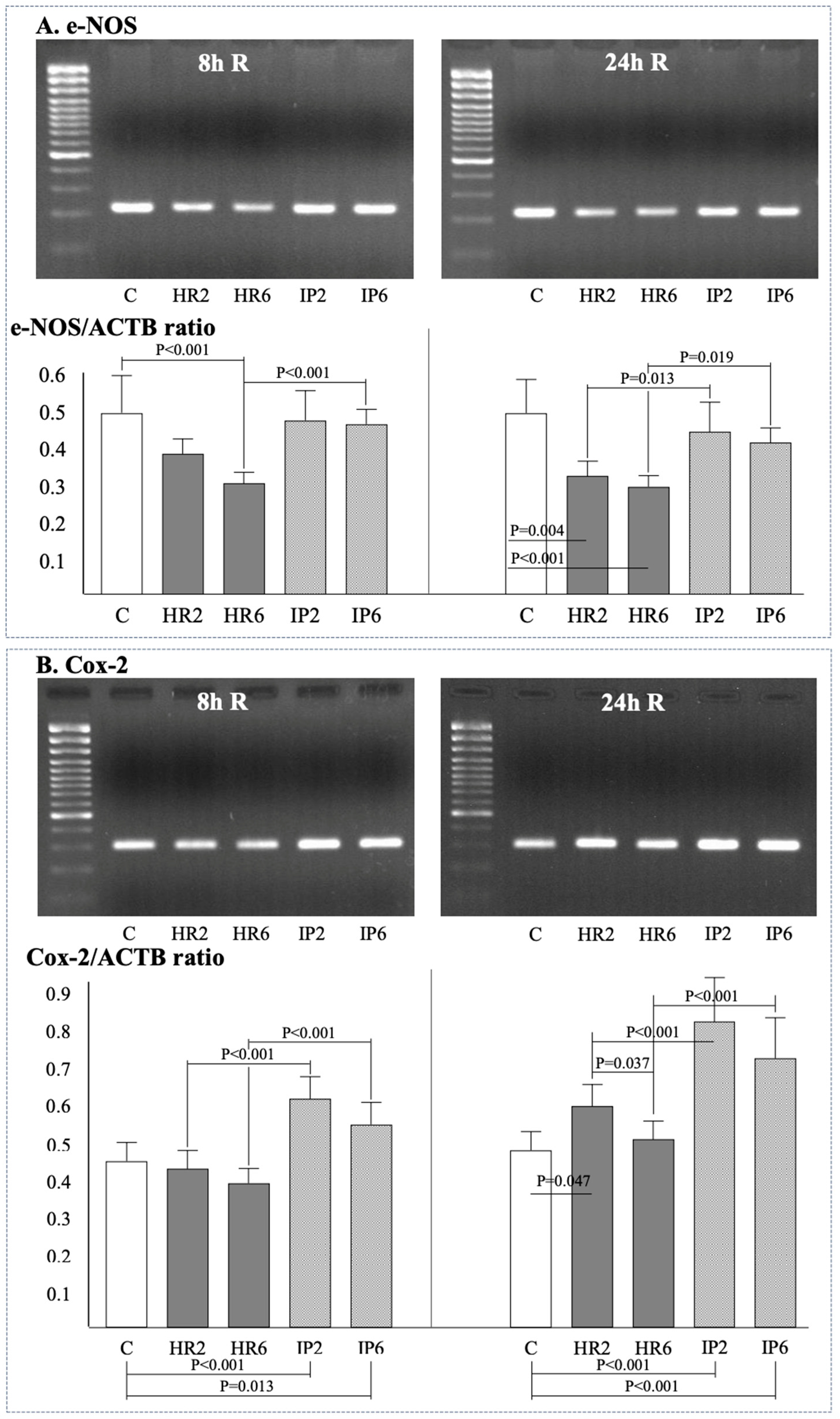
2.4. Expression of β-Actin, Bax, Bcl-2, e-NOS, i-NOS, and Cox-2
2.5. HUVEC Morphological Changes Following Hypoxia/Reoxygenation
3. Discussion
4. Materials and Methods
4.1. Isolation, Identification, and Culture of Endothelial Cells
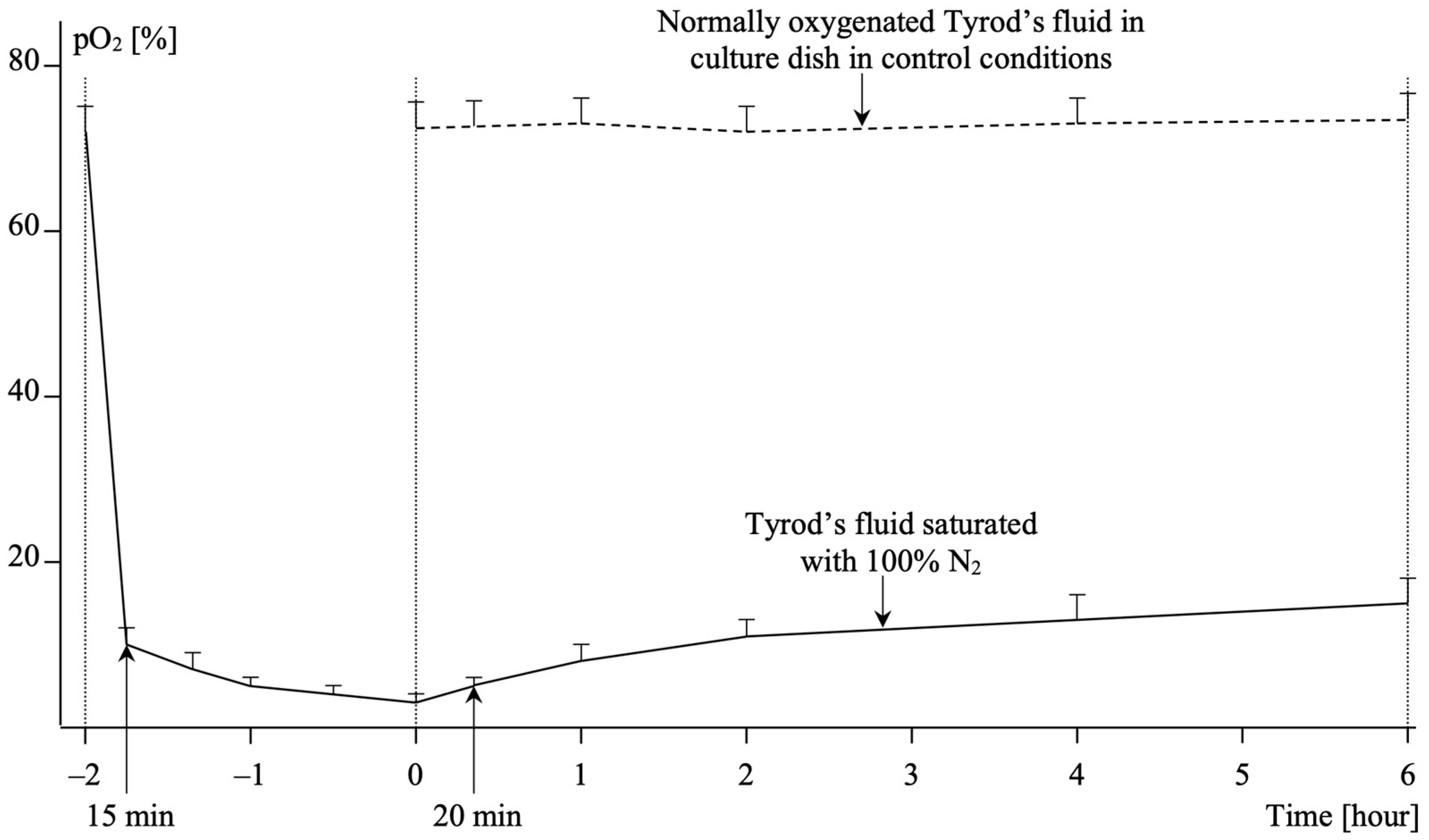
4.2. Hypoxia and Reoxygenation in HUVEC Culture
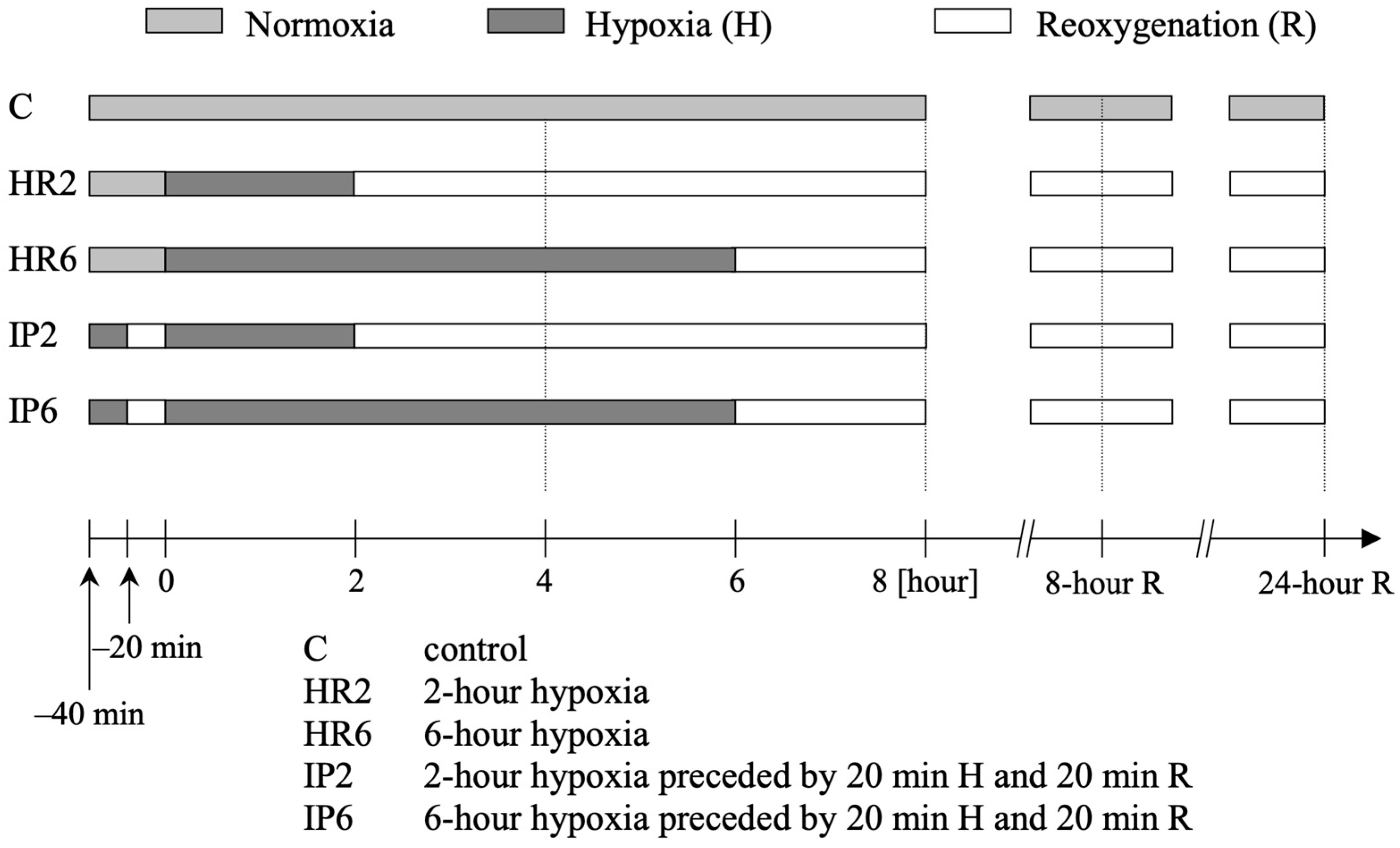
4.3. Study Protocol
4.4. Endothelial Cell Death Measurement by Lactate Dehydrogenase Release
4.5. Endothelial Cell Necrosis/Apoptosis in Double Staining with Propidium Iodide and Annexin V
4.6. Endothelial Cell Apoptosis in the TUNEL Technique
4.7. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
- -
- ACTB: s-5′-AGCGGGAAATCGTGCGTG-3′, a-5′-GGGTACATGGTGGTGCCTG-3′
- -
- Cox-2: s-5′-CCGGACAGGATTCTATGGAGA-3′, a-5′-CAATCATCAGGCGACAGGAGG-3′
- -
- Bax, Bcl-2, i-NOS, e-NOS: Purchased from R&D Systems Inc. (Minneapolis, MN, USA).
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- VanBenthuysen, K.M.; McMurtry, I.F.; Horwitz, L.D. Reperfusion after acute coronary occlusion in dogs impairs endothelium-dependent relaxation to acetylcholine and augments contractile reactivity in vitro. J. Clin. Investig. 1979, 87, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Tsao, P.; Lefer, A.M. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 1990, 82, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M. Injuries to the vascular endothelium: Vascular wall and endothelial dysfunction. Rev. Neurol. Dis. 2008, 5 (Suppl. 1), S4–S11. [Google Scholar] [PubMed]
- Daiber, A.; Andreadou, I.; Oelze, M.; Davidson, S.M.; Hausenloy, D.J. Discovery of new therapeutic redox targets for cardioprotection against ischemia/reperfusion injury and heart failure. Free Radic. Biol. Med. 2021, 163, 325–343. [Google Scholar] [CrossRef] [PubMed]
- DeFily, D.V.; Chillian, W.M. Preconditioning protects coronary arteriolar endothelium against ischemia-reperfusion injury. Am. J. Physiol. 1993, 265, H700–H706. [Google Scholar] [PubMed]
- Mehta, J.L.; Nichols, W.W.; Donnelly, W.H.; Lawson, D.L.; Saldeen, T.G. Impaired canine coronary vasodilator response to acetylcholine and bradykinin after occlusion-reperfusion. Circ. Res. 1989, 64, 43–54. [Google Scholar] [CrossRef]
- Dauber, I.M.; VanBenthuysen, K.M.; McMurtry, I.F.; Wheeler, G.S.; Lesnefsky, E.J.; Horwitz, L.D.; Weil, J.V. Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ. Res. 1990, 66, 986–998. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Humphrey, S.M.; Gavin, J.B.; Herdson, P.B. The relationship of ischemic contracture of vascular reperfusion in the isolated rat heart. J. Mol. Cell. Cardiol. 1980, 12, 1397–1406. [Google Scholar] [CrossRef]
- Inauen, W.; Granger, D.N.; Meininger, C.J.; Schelling, M.E.; Granger, H.J.; Kvietys, P.R.; Sasaki, M.; Elrod, J.W.; Jordan, P.; Itoh, M.; et al. An in vitro model of ischemia/reperfusion-induced microvascular injury. Am. J. Physiol. 1990, 259, G134–G139. [Google Scholar] [CrossRef]
- Szabó, P.L.; Dostal, C.; Pilz, P.M.; Hamza, O.; Acar, E.; Watzinger, S.; Mathew, S.; Kager, G.; Hallström, S.; Podesser, B.K.; et al. Remote ischemic perconditioning ameliorates myocardial ischemia and reperfusion-induced coronary endothelial dysfunction and aortic stiffness in rats. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Kaeffer, N.; Richard, V.; Francois, A.; Lallemand, F.; Henry, J.P.; Thuillez, C. Preconditioning prevents chronic reperfusion-induced coronary endothelial dysfunction in rats. Am. J. Physiol. 1996, 271, H842–H849. [Google Scholar] [CrossRef]
- Li, Q.; Guo, Z.; Wu, C.; Tu, Y.; Wu, Y.; Xie, E.; Yu, C.; Sun, W.; Li, X.; Zheng, J.; et al. Ischemia preconditioning alleviates ischemia/reperfusion injury-induced coronary no-reflow and contraction of microvascular pericytes in rats. Microvasc. Res. 2022, 142, 104349. [Google Scholar] [CrossRef] [PubMed]
- Thourani, V.H.; Nakamura, M.; Duarte, I.G.; Bufkin, B.L.; Zhao, Z.-Q.; Jordan, J.E.; Shearer, S.T.; Guyton, R.A.; Vinten-Johansen, J. Ischemic preconditioning attenuates postischemic coronary artery endothelial dysfunction in a model of minimally invasive direct coronary artery bypass. J. Thorac. Cardiovasc. Surg. 1999, 117, 383–389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beresewicz, A.; Czarnowska, E.; Maczewski, M. Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Mol. Cell. Biochem. 1998, 186, 87–97. [Google Scholar] [CrossRef]
- Bouchard, J.F.; Lamontagne, D. Mechanisms of protection afforded by preconditioning to endothelial function against ischemic injury. Am. J. Physiol. 1996, 271, H1801–H1806. [Google Scholar] [CrossRef]
- Maczewski, M.; Beresewicz, A. The role of adenosine and ATP-sensitive potassium channels in the protection afforded by ischemic preconditioning against the post-ischemic endothelial dysfunction in guinea-pig hearts. J. Mol. Cell. Cardiol. 1998, 30, 1735–1747. [Google Scholar] [CrossRef]
- Gesuete, R.; Stevens, S.L.; Stenzel-Poore, M.P. Role of circulating immune cells in stroke and preconditioning-induced protection. Acta Neurochir. Suppl. 2016, 121, 39–44. [Google Scholar]
- Akimitsu, T.; Gute, D.C.; Korthuis, R.J. Ischemic preconditioning attenuates postischemic leukocyte adhesion and emigration. Am. J. Physiol. 1996, 271, H2052–H2059. [Google Scholar] [CrossRef]
- Kubes, P.; Payne, D.; Ostrovsky, L. Preconditioning and adenosine in I/R-induced leukocyte-endothelial cell interactions. Am. J. Physiol. 1998, 274, H1230–H1238. [Google Scholar] [CrossRef]
- Kharbanda, R.K.; Peters, M.; Walton, B.; Kattenhorn, M.; Mullen, M.; Klein, N.; Vallance, P.; Deanfield, J.; MacAllister, R. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans in vivo. Circulation 2001, 103, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Simkhovich, B.Z.; Kloner, R.A.; Przyklenk, K. Does preconditioning protect the coronary vasculature from subsequent ischemia/reperfusion injury? Circulation 1993, 88, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 2–16. [Google Scholar]
- Bu, L.L.; Yuan, H.H.; Xie, L.L.; Guo, M.H.; Liao, D.F.; Zheng, X.L. New dawn for atherosclerosis: Vascular endothelial cell senescence and death. Int. J. Mol. Sci. 2023, 24, 15160. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, T.; Yanagi, S. Role of Mitochondrial Dynamics in Heart Diseases. Genes 2023, 14, 1876. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, A.; Suzuki, K.; Lorenz-Meyer, S.; Bode, C.; Schaper, J. The role of apoptosis in myocardial ischemia: A critical appraisal. Basic Res. Cardiol. 2001, 96, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kelland, E.; Patil, M.S.; Patel, S.; Cartland, S.P.; Kavurma, M.M. The prognostic, diagnostic, and therapeutic potential of TRAIL signalling in cardiovascular diseases. Int. J. Mol. Sci. 2023, 24, 6725. [Google Scholar] [CrossRef] [PubMed]
- Maulik, N.; Yoshida, T.; Engelman, R.M.; Deaton, D.; Flack, J.E.; Rousou, J.A.; Das, D.K. Ischemic preconditioning attenuates apoptotic cell death associated with ischemia/reperfusion. Mol. Cell. Biochem. 1998, 186, 139–145. [Google Scholar] [CrossRef]
- Nakamura, M.; Wang, N.-P.; Zhao, Z.-Q.; Wilcox, J.N.; Thourani, V.; Guyton, R.A.; Vinten-Johansen, J. Preconditioning decreases Bax expression, PMN accumulation and apoptosis in reperfused rat heart. Cardiovasc. Res. 2000, 45, 661–670. [Google Scholar] [CrossRef]
- Fliss, H.; Gattinger, D. Apoptosis in ischemic and reperfused rat myocardium. Circ. Res. 1996, 79, 949–956. [Google Scholar] [CrossRef]
- Maulik, N.; Engelman, R.M.; Rousou, J.A.; Flack, J.E., III; Deaton, D.; Das, D.K. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation 1999, 100, II369–II375. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.M.; Zhou, J. The role of PI3K/AKT signaling pathway in myocardial ischemia-reperfusion injury. Int. Immunopharmacol. 2023, 123, 110714. [Google Scholar] [CrossRef] [PubMed]
- Wal, P.; Aziz, N.; Singh, Y.K.; Wal, A.; Kosey, S.; Rai, A.K. Myocardial infarction as a consequence of mitochondrial dysfunction. Curr. Cardiol. Rev. 2023, 19, 23–30. [Google Scholar] [CrossRef]
- Popov, S.V.; Mukhomedzyanov, A.V.; Voronkov, N.S.; Derkachev, I.A.; Boshchenko, A.A.; Fu, F.; Sufianova, G.Z.; Khlestkina, M.S.; Maslov, L.N. Regulation of autophagy of the heart in ischemia and reperfusion. Apoptosis 2023, 28, 55–80. [Google Scholar] [CrossRef]
- Shirai, T.; Rao, V.; Weisel, R.D.; Ikonomidis, J.S.; Li, R.-K.; Tumiati, L.C.; Merante, F.; Mickle, D.A. Preconditioning human cardiomyocytes and endothelial cells. J. Thorac. Cardiovasc. Surg. 1998, 115, 210–219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.-I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for BcI-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Velez, D.A.; Wang, N.P.; Hewan-Lowe, K.O.; Nakamura, M.; Guyton, R.A.; Vinten-Johansen, J. Progressively developed myocardial apoptotic cell death during late phase of reperfusion. Apoptosis 2001, 6, 279–290. [Google Scholar] [CrossRef]
- Marber, M.S. Ischemic preconditioning in isolated cells. Circulation Res. 2000, 86, 926–931. [Google Scholar] [CrossRef]
- Kim, Y.D.; Fomsgaard, J.S.; Heim, K.F.; Ramwell, P.W.; Thomas, G.; Kagan, E.; Moore, S.P.; Coughlin, S.S.; Kuwahara, M.; Analouei, A. Brief ischemia-reperfusion induces stunning of endothelium in canine coronary artery. Circulation 1992, 85, 1473–1482. [Google Scholar] [CrossRef]
- Nicklas, J.M.; Gips, S.J. Decreased coronary flow reserve after transient myocardial ischemia in dogs. J. Am. Coll. Cardiol. 1989, 13, 195–199. [Google Scholar] [CrossRef]
- Zhou, X.; Zhai, X.; MAshraf, M. Preconditioning of bovine endothelial cells. The protective effect is mediated by an adenosine A2 receptor through a protein kinase C signaling pathway. Circ. Res. 1996, 78, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Richard, V.; Tamion, F.; Lallemand, F.; Lebreton, J.P.; Vaudry, H.; Daveau, M.; Thuillez, C. Protective effects of preconditioning in cultured rat endothelial cells: Effects on neutrophil adhesion and expression of ICAM-1 after anoxia and reoxygenation. Circulation 1999, 100, 541–546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maslov, L.N.; Popov, S.V.; Mukhomedzyanov, A.V.; Naryzhnaya, N.V.; Voronkov, N.S.; Ryabov, V.V.; Boshchenko, A.A.; Khaliulin, I.; Prasad, N.R.; Fu, F.; et al. Reperfusion cardiac injury: Receptors and the signaling mechanisms. Curr. Cardiol. Rev. 2022, 18, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Shinmura, K.; Tang, X.-L.; Kodani, E.; Xuan, Y.-T.; Guo, Y.; Dawn, B. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc. Res. 2002, 55, 506–519. [Google Scholar] [CrossRef] [PubMed]
- von Knethen, A.; Brune, B. Cyclooxygenase-2: An essential regulator of NO-mediated apoptosis. FASEB J. 1997, 11, 887–895. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, L.P.; Leung, G.K.; Kostyk, S.K.; Kourembanas, S.; Luchi, W.M.; Shimizu, M.H.M.; Canale, D.; Gois, P.H.F.; de Bragança, A.C.; Volpini, R.A.; et al. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am. J. Physiol. 1994, 267, H1921–H1927. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tomson, K.; Yang, B.; Mehta, P.; Croker, B.P.; Mehta, J.L. Modulation of constitutive nitric oxide synthase, bcl-2 and Fas expression in cultured human coronary endothelial cells exposed to anoxia-reoxygenation and angiotensin II: Role of AT1 receptor activation. Cardiovasc. Res. 1999, 41, 109–115. [Google Scholar] [CrossRef]
- Dominguez, A.; Iruela-Arispe, M.L. Integration of chemo-mechanical signaling in response to fluid shear stress by the endothelium. Curr. Opin. Cell. Biol. 2023, 85, 102232. [Google Scholar] [CrossRef]
- Zhou, H.L.; Jiang, X.Z.; Ventikos, Y. Role of blood flow in endothelial functionality: A review. Front. Cell. Dev. Biol. 2023, 11, 1259280. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n = 12 | Study Group | Baseline | H | 2 h R | 8 h R | 24 h R |
|---|---|---|---|---|---|---|
| 2 h hypoxia | C | 0.017 ± 0.010 | 0.019 ± 0.011 | 0.023 ± 0.028 | 0.043 ± 0.019 | 0.092 ± 0.016 |
| IP | 0.018 ± 0.011 | 0.020 ± 0.011 | 0.075 ± 0.021 * | 0.096 ± 0.028 *,** | 0.280 ± 0.052 * | |
| HR | 0.016 ± 0.009 | 0.019 ± 0.012 | 0.098 ± 0.027 * | 0.167 ± 0.033 * | 0.305 ± 0.048 * | |
| 6 h hypoxia | C | 0.017 ± 0.011 | 0.018 ± 0.011 | 0.035 ± 0.021 | 0.054 ± 0.027 | 0.110 ± 0.030 |
| IP | 0.017 ± 0.010 | 0.018 ± 0.012 | 0.117 ± 0.023 * | 0.134 ± 0.034 *,** | 0.330 ± 0.079 * | |
| HR | 0.018 ± 0.011 | 0.020 ± 0.011 | 0.146 ± 0.045 * | 0.193 ± 0.043 * | 0.370 ± 0.084 * |
| n = 6 | 2 h Hypoxia | 6 h Hypoxia | |||||
|---|---|---|---|---|---|---|---|
| C | IP | HR | C | IP | HR | ||
| 8 h R | Apoptotic | 2.3 ± 0.3 | 4.5 ± 0.5 *,** | 6.5 ± 0.85 * | 2.4 ± 0.3 | 5.4 ± 0.65 *,** | 7.9 ± 0.9 * |
| Necrotic | 5.8 ± 0.7 | 9.1 ± 1.0 **,*** | 17.5 ± 2.4 * | 6.2 ± 0.8 | 11.1 ± 1.6 **,*** | 20.2 ± 3.0 * | |
| Viable | 91.9 ± 1.5 | 86.4 ± 2.8 | 76.0 ± 4.8 | 91.4 ± 1.6 | 83.5 ± 2.7 | 72.1 ± 4.9 | |
| 24 h R | Apoptotic | 2.4 ± 0.35 | 7.4 ± 0.9 *,** | 15.6 ± 2.0 * | 2.6 ± 0.4 | 10.6 ± 1.3 | 20.7 ± 2.2 |
| Necrotic | 7.6 ± 0.91 | 26.9 ± 3.1 * | 30.2 ± 4.0 * | 8.8 ± 1.1 | 28.2 ± 3.3 * | 33.2 ± 4.3 * | |
| Viable | 90.0 ± 1.8 | 65.7 ± 3.8 | 54.2 ± 6.2 | 88.6 ± 1.8 | 61.2 ± 4.6 | 46.0 ± 6.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalewski, J.; Szajna, M.; Stępień, K.; Nowak, K.; Karcińska, A.; Yika, A.d.C.; Krawczyk, K.; Karwat, K.; Zalewska, M.; Pierzchalski, P. Endothelial Cell Apoptosis but Not Necrosis Is Inhibited by Ischemic Preconditioning. Int. J. Mol. Sci. 2024, 25, 1238. https://doi.org/10.3390/ijms25021238
Zalewski J, Szajna M, Stępień K, Nowak K, Karcińska A, Yika AdC, Krawczyk K, Karwat K, Zalewska M, Pierzchalski P. Endothelial Cell Apoptosis but Not Necrosis Is Inhibited by Ischemic Preconditioning. International Journal of Molecular Sciences. 2024; 25(2):1238. https://doi.org/10.3390/ijms25021238
Chicago/Turabian StyleZalewski, Jarosław, Marta Szajna, Konrad Stępień, Karol Nowak, Aleksandra Karcińska, Alicia del Carmen Yika, Kornelia Krawczyk, Krzysztof Karwat, Magdalena Zalewska, and Piotr Pierzchalski. 2024. "Endothelial Cell Apoptosis but Not Necrosis Is Inhibited by Ischemic Preconditioning" International Journal of Molecular Sciences 25, no. 2: 1238. https://doi.org/10.3390/ijms25021238
APA StyleZalewski, J., Szajna, M., Stępień, K., Nowak, K., Karcińska, A., Yika, A. d. C., Krawczyk, K., Karwat, K., Zalewska, M., & Pierzchalski, P. (2024). Endothelial Cell Apoptosis but Not Necrosis Is Inhibited by Ischemic Preconditioning. International Journal of Molecular Sciences, 25(2), 1238. https://doi.org/10.3390/ijms25021238