
Integrated Transcriptomics and Metabolomics Reveal Changes in Cell Homeostasis and Energy Metabolism in Trachinotus ovatus in Response to Acute Hypoxic Stress
 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Transcriptomics Analysis
2.1.1. Sequencing Quality and Sample Relationship
2.1.2. Identification of Differentially Expressed Genes (DEGs)
2.1.3. Gene Expression Trends
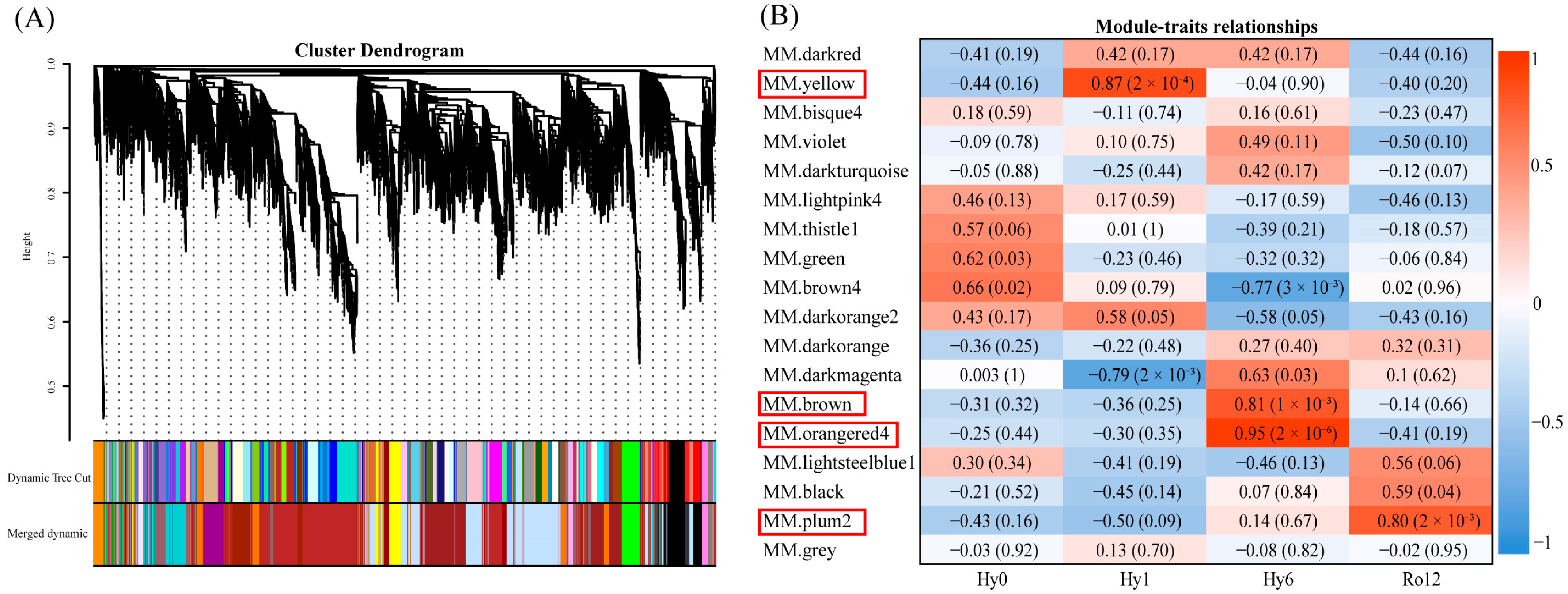
2.1.4. Gene Co-Expression Network Construction
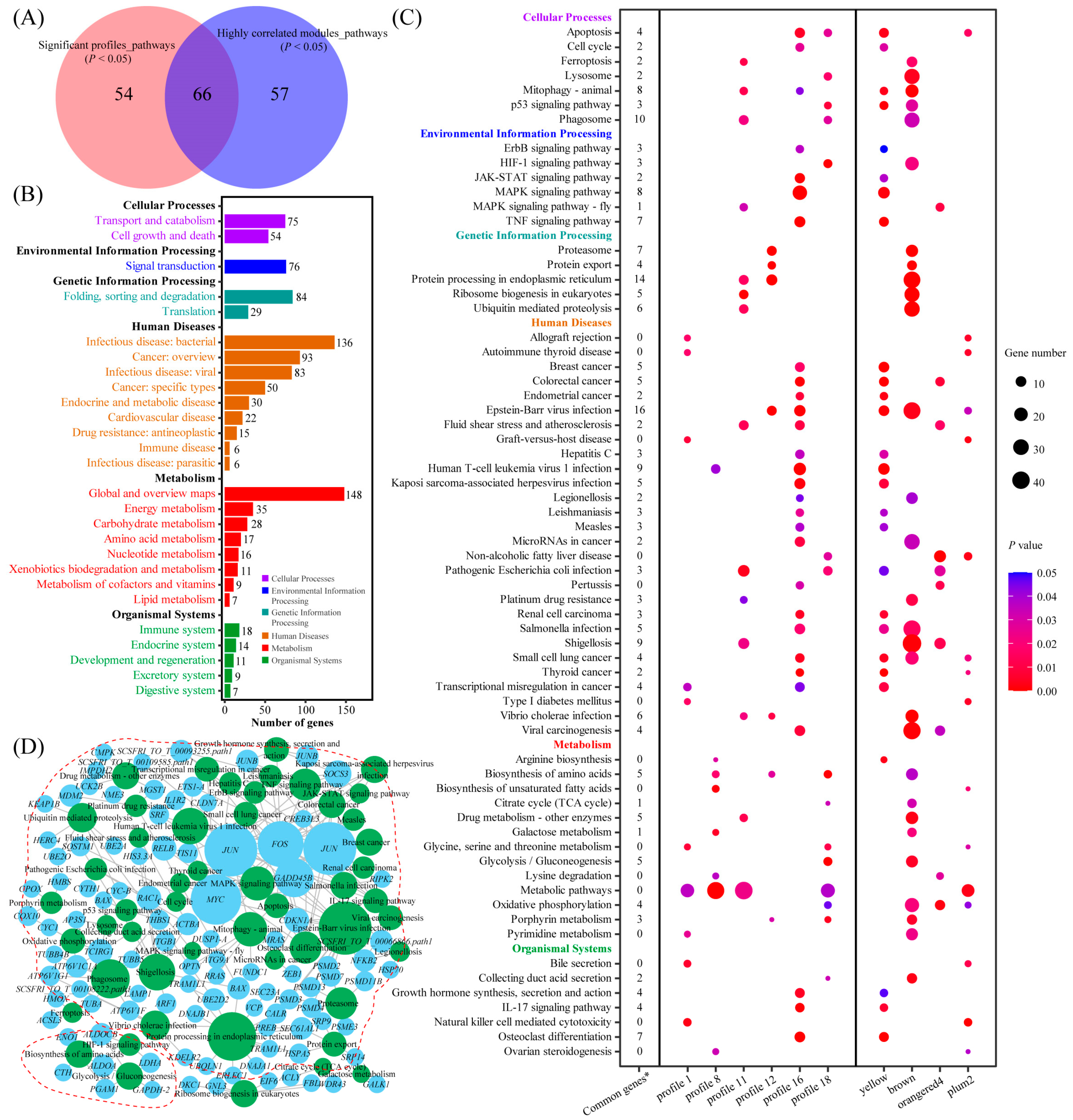
2.1.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
2.1.6. Gene Ontology (GO) Enrichment Analysis
2.1.7. Protein–Protein Interaction (PPI) Network Analysis
2.2. Metabolomics Analysis
2.2.1. Metabolite Identification and Multivariate Statistical Analysis
2.2.2. Identification of Differentially Expressed Metabolites (DEMs)
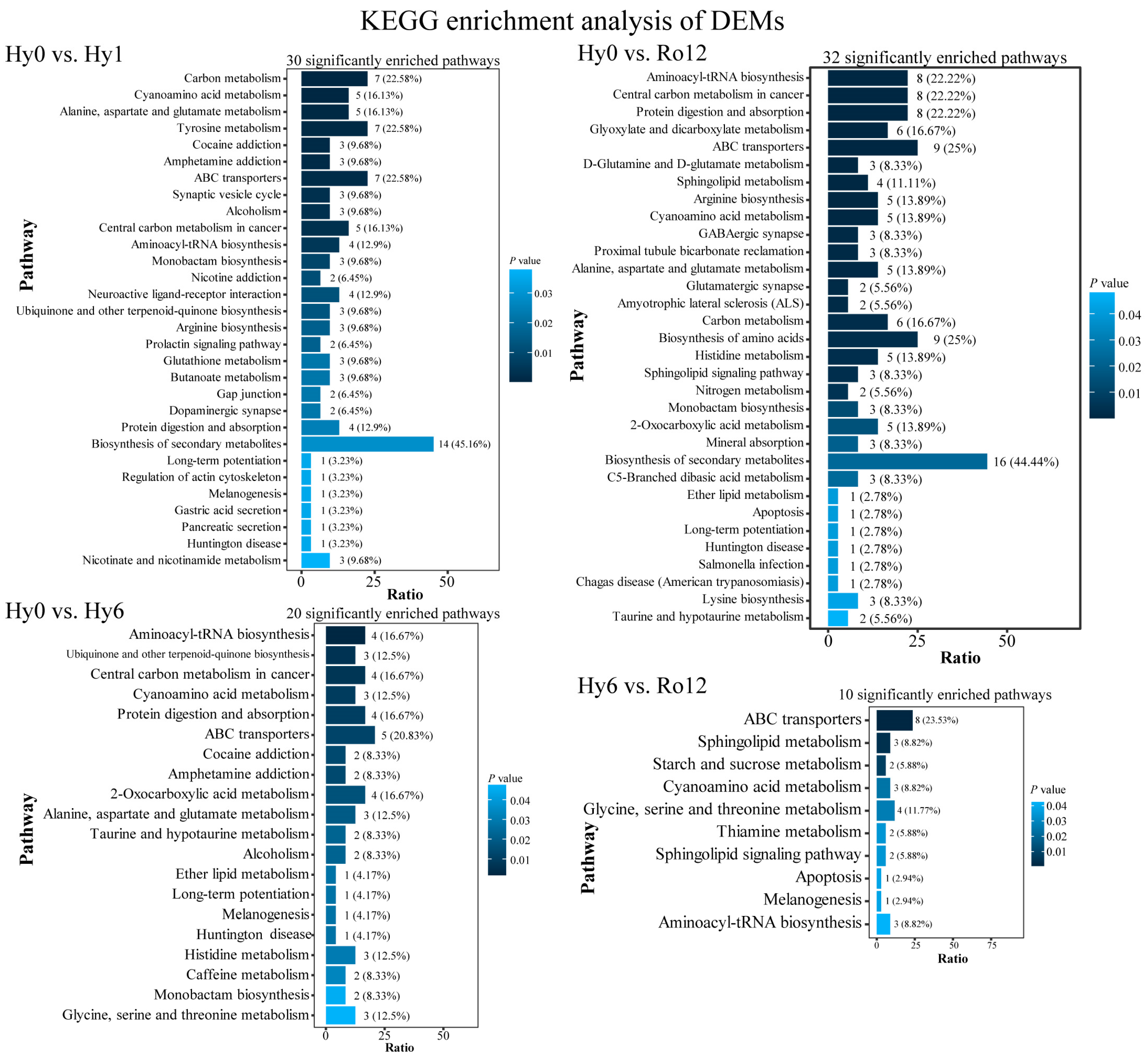
2.2.3. KEGG Enrichment Analysis of DEMs
2.3. Integrated KEGG Enrichment Analysis of Transcriptomics and Metabolomics
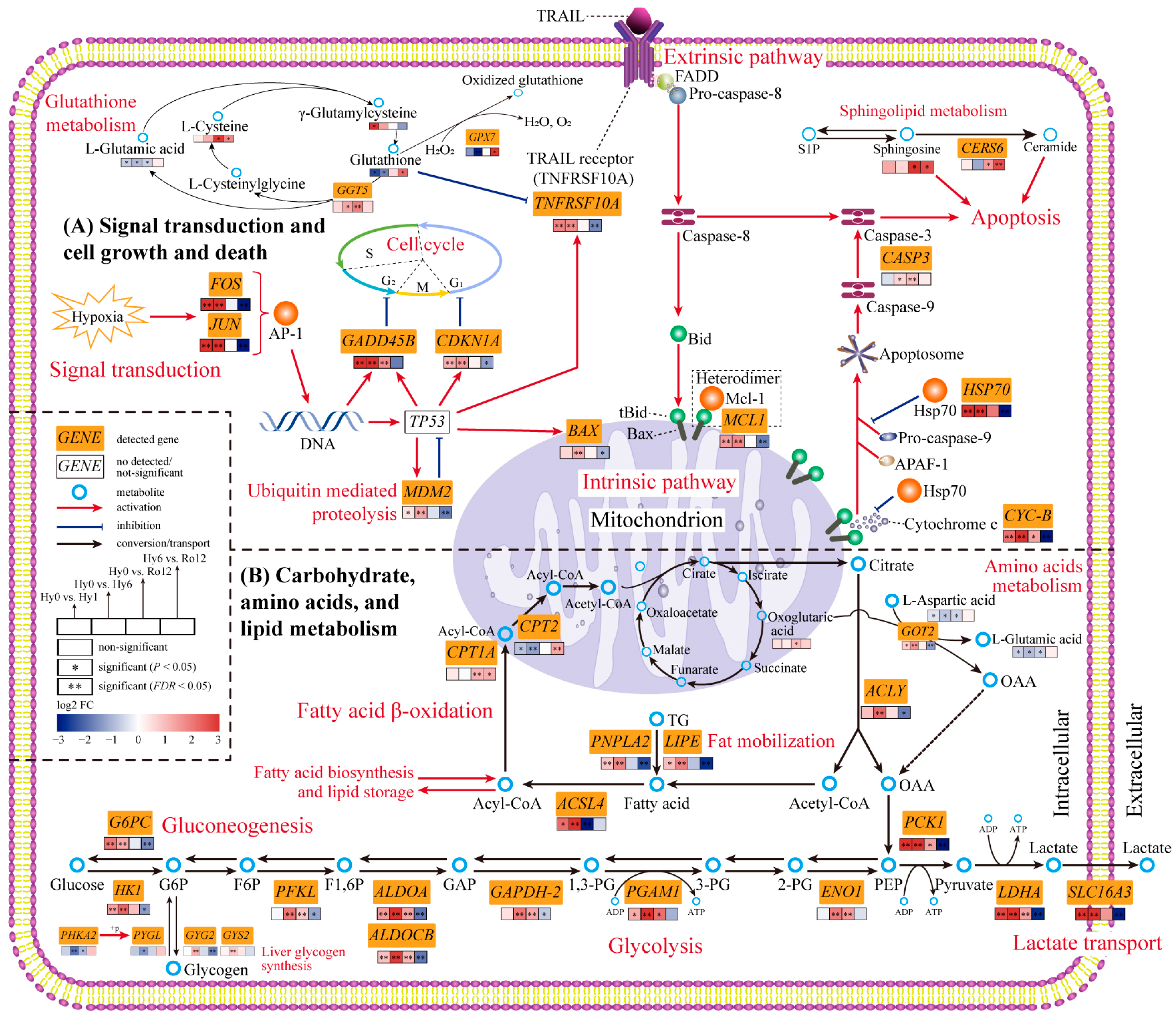
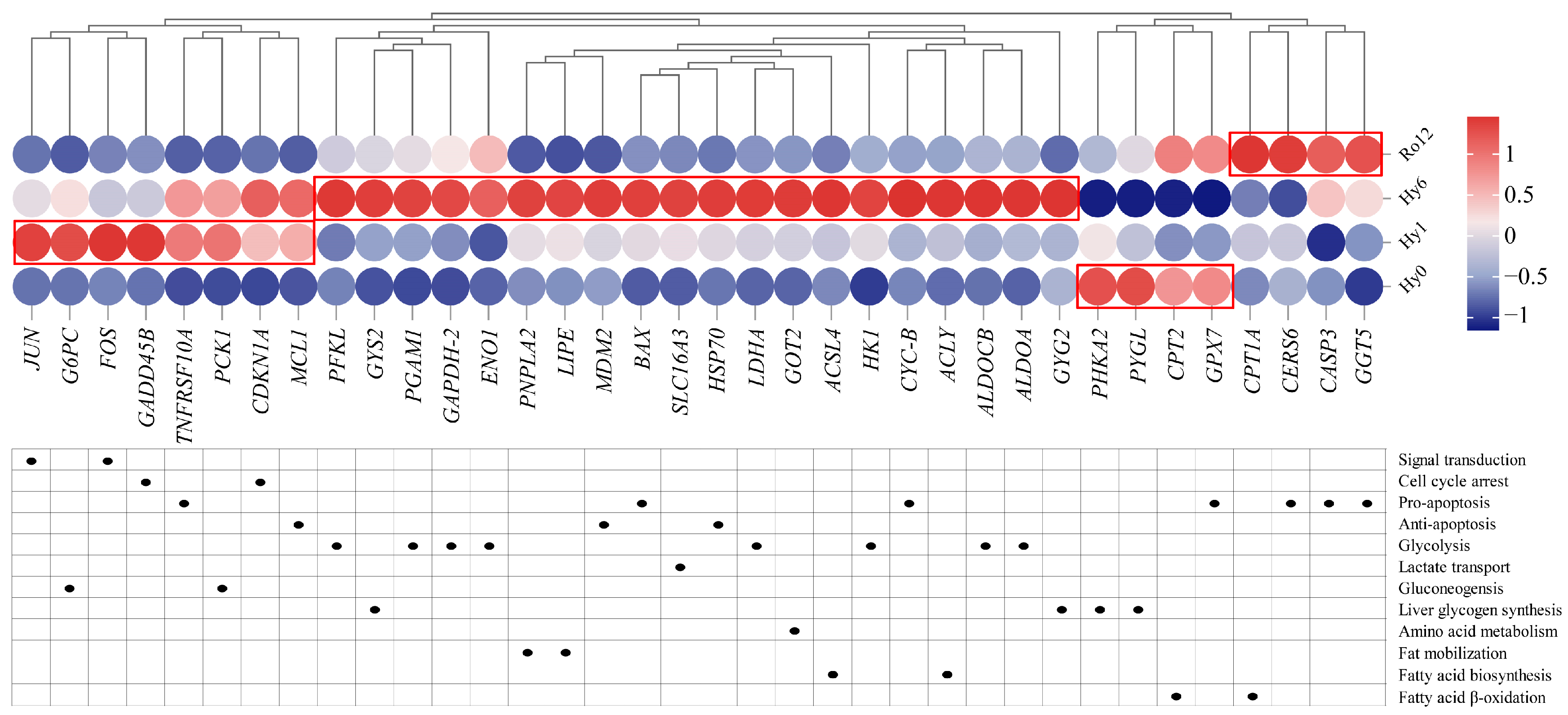
2.4. Key Genes, Metabolites, and Biological Pathways
2.5. Key Gene Expression Analysis during Acute Hypoxic Stress and Re-Oxygenation
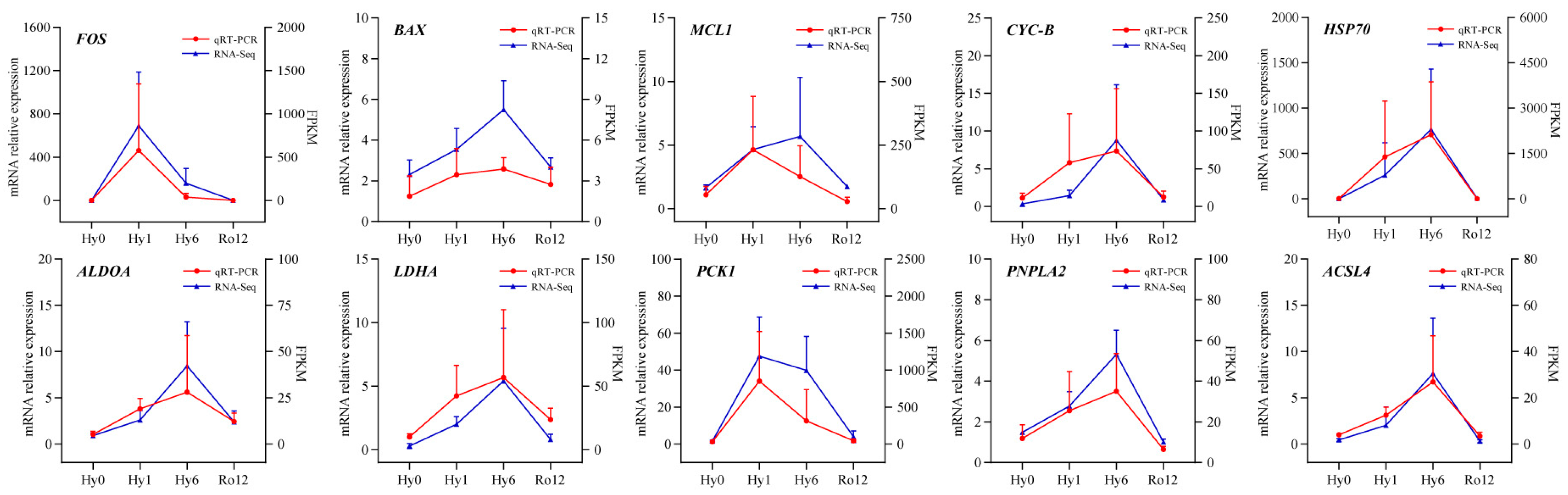
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation of Key Genes
3. Discussion
3.1. Hub Genes in Response to Hypoxia Adaptation
3.2. Cell Growth and Death in Response to Acute Hypoxic Stress and Re-Oxygenation
3.2.1. Cell Cycle Arrest during the Hypoxia Stage
3.2.2. Balance of Both Pro-Apoptosis and Anti-Apoptosis Processes under Acute Hypoxic Stress
3.2.3. Continuous Apoptosis after Re-Oxygenation
3.3. Carbohydrate Metabolism, Amino Acid Metabolism, and Lipid Metabolism in Response to Acute Hypoxic Stress and Re-Oxygenation
3.3.1. Enhancing Anaerobic Glycolysis and Lactate Transport for Hypoxia Adaptation
3.3.2. Enhancing Gluconeogenesis and Glycogen Synthesis for Hypoxia Adaptation
3.3.3. Enhancing Amino Acid Metabolism for Hypoxia Adaption
3.3.4. Enhancing Fat Mobilization and Fatty Acid Biosynthesis for Hypoxia Adaption
3.3.5. Activating Fatty Acid β-Oxidation and Aerobic Metabolism after Re-Oxygenation
3.4. Key Periods for Acute Hypoxic Stress and Re-Oxygenation in T. ovatus
4. Materials and Methods
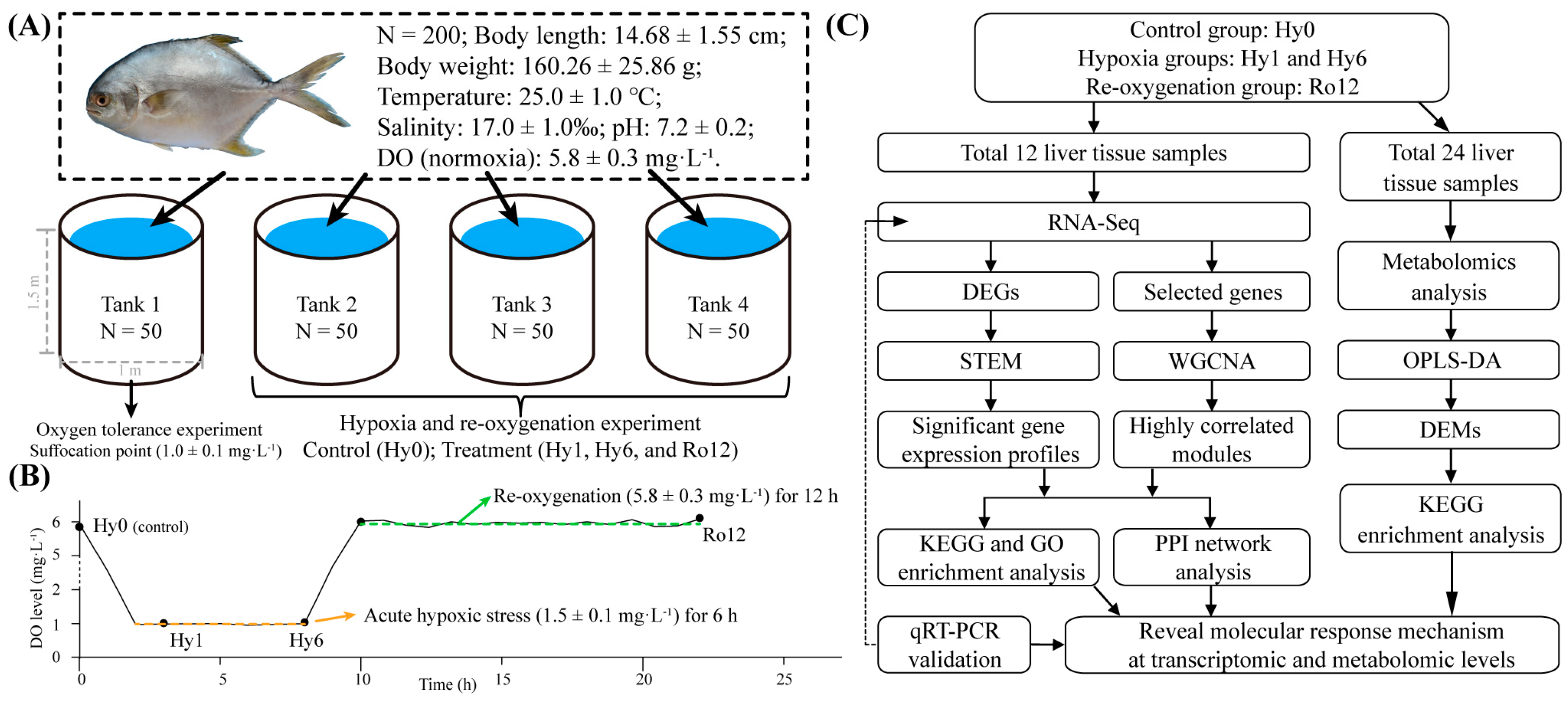
4.1. Ethics Statement and Fish Management
4.2. Experimental Design and Sample Collection
4.3. Transcriptomics Analysis
4.4. Metabolomics Analysis
4.5. Integrated Analysis of Transcriptomics and Metabolomics
4.6. qRT-PCR Validation for Key Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diaz, R.J. Overview of Hypoxia around the World. J. Environ. Qual. 2001, 30, 275–281. [Google Scholar] [CrossRef]
- Hughes, B.B.; Levey, M.D.; Fountain, M.C.; Carlisle, A.B.; Chavez, F.P.; Gleason, M.G. Climate mediates hypoxic stress on fish diversity and nursery function at the land–sea interface. Proc. Natl. Acad. Sci. USA 2015, 112, 8025–8030. [Google Scholar] [CrossRef] [PubMed]
- Thronson, A.; Quigg, A. Fifty-Five Years of Fish Kills in Coastal Texas. Estuaries Coasts 2008, 31, 802–813. [Google Scholar] [CrossRef]
- Breitburg, D.; Levin, L.A.; Oschlies, A.; Grégoire, M.; Chavez, F.P.; Conley, D.J.; Garçon, V.; Gilbert, D.; Gutiérrez, D.; Isensee, K.; et al. Declining oxygen in the global ocean and coastal waters. Science 2018, 359, eaam7240. [Google Scholar] [CrossRef] [PubMed]
- Dan, X.-M.; Yan, G.-J.; Zhang, A.-J.; Cao, Z.-D.; Fu, S.-J. Effects of stable and diel-cycling hypoxia on hypoxia tolerance, postprandial metabolic response, and growth performance in juvenile qingbo (Spinibarbus sinensis). Aquaculture 2014, 428–429, 21–28. [Google Scholar] [CrossRef]
- Zhang, G.; Yin, S.; Mao, J.; Liang, F.; Zhao, C.; Li, P.; Zhou, G.; Chen, S.; Tang, Z. Integrated analysis of mRNA-seq and miRNA-seq in the liver of Pelteobagrus vachelli in response to hypoxia. Sci. Rep. 2016, 6, 22907. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, G.; Yin, D.; Li, Y.; Zhang, Y.; Cheng, J.; Zhang, K.; Ji, J.; Wang, T.; Jia, Y.; et al. Integrated Application of Multiomics Strategies Provides Insights into the Environmental Hypoxia Response in Pelteobagrus vachelli Muscle. Mol. Cell. Proteom. 2022, 21, 100196. [Google Scholar] [CrossRef] [PubMed]
- Tzaneva, V.; Vadeboncoeur, C.; Ting, J.; Perry, S.F. Effects of hypoxia-induced gill remodelling on the innervation and distribution of ionocytes in the gill of goldfish, Carassius auratus. J. Comp. Neurol. 2013, 522, 118–130. [Google Scholar] [CrossRef]
- Cameron, J.S.; DeWitt, J.P.; Ngo, T.T.; Yajnik, T.; Chan, S.; Chung, E.; Kang, E. Cardiac KATP channel alterations associated with acclimation to hypoxia in goldfish (Carassius auratus L.). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2013, 164, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.W.; Brechin, J.G.; Nilsson, G.E.; Vaage, J.; Stensløkken, K.-O.; Hardy, K.M.; Burnett, K.G.; Burnett, L.E.; Sandvik, G.K.; Jensen, F.B.; et al. Tissue-specific changes in protein synthesis rates in vivo during anoxia in crucian carp. Am. J. Physiol. Integr. Comp. Physiol. 1996, 271, R897–R904. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.X.; Wang, P.F.; Ou, Y.J.; Wen, J.F.; Li, J.E.; Wang, W.; Xie, M.J. The effect of acute and chronic hypoxia stress on liver tissue structure and oxidation in juvenile golden pompano (Trachinotus ovatus). Chin. J. Zool. 2016, 51, 1049–1058. [Google Scholar]
- Wang, M.; Li, B.; Wang, J.; Xie, S.; Zhang, L. Skin transcriptome and physiological analyses reveal the metabolic and immune responses of yellow catfish (Pelteobagrus fulvidraco) to acute hypoxia. Aquaculture 2021, 546, 737277. [Google Scholar] [CrossRef]
- Zheng, X.; Fu, D.; Cheng, J.; Tang, R.; Chu, M.; Chu, P.; Wang, T.; Yin, S. Effects of hypoxic stress and recovery on oxidative stress, apoptosis, and intestinal microorganisms in Pelteobagrus vachelli. Aquaculture 2021, 543, 736945. [Google Scholar] [CrossRef]
- Turko, A.J.; Cisternino, B.; Wright, P.A. Calcified gill filaments increase respiratory function in fishes. Proc. R. Soc. B. 2020, 287, 20192796. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, G.E.; Randall, D. Adaptations to hypoxia in fishes. In Respiratory Physiology of Vertebrates: Life with and with-out Oxygen; Cambridge University Press: Cambridge, UK, 2010; pp. 131–173. [Google Scholar]
- Richards, J.G. Physiological, behavioral and biochemical adaptations of intertidal fishes to hypoxia. J. Exp. Biol. 2011, 214, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W. The hypoxia signaling pathway and hypoxic adaptation in fishes. Sci. China Life Sci. 2015, 58, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.L.; Zhao, L.L.; Wu, H.; Liu, Q.; Liao, L.; Luo, J.; Lian, W.Q.; Cui, C.; Jin, L.; Ma, J.D.; et al. Acute hypoxia changes the mode of glucose and lipid utilization in the liver of the largemouth bass (Micropterus salmoides). Sci. Total Environ. 2020, 713, 135157. [Google Scholar] [CrossRef]
- Yang, S.; Wu, H.; He, K.; Yan, T.; Zhou, J.; Zhao, L.; Sun, J.; Lian, W.; Zhang, D.; Du, Z.; et al. Response of AMP-activated protein kinase and lactate metabolism of largemouth bass (Micropterus salmoides) under acute hypoxic stress. Sci. Total Environ. 2019, 666, 1071–1079. [Google Scholar] [CrossRef]
- Ding, J.; Liu, C.; Luo, S.; Zhang, Y.; Gao, X.; Wu, X.; Shen, W.; Zhu, J. Transcriptome and physiology analysis identify key metabolic changes in the liver of the large yellow croaker (Larimichthys crocea) in response to acute hypoxia. Ecotoxicol. Environ. Saf. 2020, 189, 109957. [Google Scholar] [CrossRef]
- Gracey, A.Y.; Troll, J.V.; Somero, G.N. Hypoxia-induced gene expression profiling in the euryoxic fish Gillichthys mirabilis. Proc. Natl. Acad. Sci. USA 2001, 98, 1993–1998. [Google Scholar] [CrossRef]
- Zhao, L.L.; Sun, J.L.; Liang, J.; Liu, Q.; Luo, J.; Li, Z.Q.; Yan, T.M.; Zhou, J.; Yang, S. Enhancing lipid metabolism and inducing antioxidant and immune responses to adapt to acute hypoxic stress in Schizothorax prenanti. Aquaculture 2020, 519, 734933. [Google Scholar] [CrossRef]
- Yan, L.; Wang, P.; Zhao, C.; Fan, S.; Lin, H.; Guo, Y.; Ma, Z.; Qiu, L. Toxic responses of liver in Lateolabrax maculatus during hypoxia and re-oxygenation. Aquat. Toxicol. 2021, 236, 105841. [Google Scholar] [CrossRef]
- Wang, M.; Wu, F.; Xie, S.; Zhang, L. Acute hypoxia and reoxygenation: Effect on oxidative stress and hypoxia signal transduction in the juvenile yellow catfish (Pelteobagrus fulvidraco). Aquaculture 2020, 531, 735903. [Google Scholar] [CrossRef]
- Liang, Y.-S.; Wu, R.-X.; Niu, S.-F.; Miao, B.-B.; Liang, Z.-B.; Zhai, Y. Liver transcriptome analysis reveals changes in energy metabolism, oxidative stress, and apoptosis in pearl gentian grouper exposed to acute hypoxia. Aquaculture 2022, 561, 738635. [Google Scholar] [CrossRef]
- Ou, Y.J.; Fan, C.Y.; Li, J.E.; Yu, N.; Su, H. Acute hypoxia stress on blood biochemical indexes in selective group of Trachinotus ovatus. Acta. Oceanol. Sin. 2014, 36, 126–131. [Google Scholar]
- Ou, Y.J.; Chen, S.X.; Wang, P.F.; Li, J.E.; Wen, J.F.; Wang, W.; Xie, M.J. Study on oxidative stress response and physiological metabolism related indices of Trachinotus ovatus under hypoxia stress. South China Fish. Sci. 2017, 13, 120–124. [Google Scholar]
- Bureau of Fisheries, Ministry of Agriculture. Chinese Fishery Statistical Yearbook; China Agriculture Press: Beijing, China, 2023; Chapter 2; pp. 15–46. (In Chinese) [Google Scholar]
- San, L.; Liu, B.; Liu, B.; Guo, H.; Guo, L.; Zhang, N.; Zhu, K.; Jiang, S.; Zhang, D. Transcriptome Analysis of Gills Provides Insights Into Translation Changes Under Hypoxic Stress and Reoxygenation in Golden Pompano, Trachinotus ovatus (Linnaeus 1758). Front. Mar. Sci. 2021, 8, 763622. [Google Scholar] [CrossRef]
- Herrera, D.G.; Robertson, H.A. Activation of c-fos in the brain. Prog. Neurobiol. 1996, 50, 83–107. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Liu, Z.-G.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Hwang, H.-W.; Baxter, L.L.; Loftus, S.K.; Cronin, J.C.; Trivedi, N.S.; Borate, B.; Pavan, W.J. Distinct microRNA expression signatures are associated with melanoma subtypes and are regulated by HIF1A. Pigment. Cell Melanoma Res. 2014, 27, 777–787. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Hoffman, B. Gadd45 stress sensor genes. Adv. Exp. Med. Biol. 2013, 793, 1–134. [Google Scholar]
- Elkeles, A.; Juven-Gershon, T.; Israeli, D.; Wilder, S.; Zalcenstein, A.; Oren, M. The c-fos Proto-Oncogene Is a Target for Transactivation by the p53 Tumor Suppressor. Mol. Cell Biol. 1999, 19, 2594–2600. [Google Scholar] [CrossRef]
- Di, G.; Li, H.; Zhao, Y.; Lin, Y.; Lan, D.; Kong, X.; Chen, X. Comprehensive transcriptomic analysis reveals insights into the gill response to hypoxia and Poly I:C in Qihe crucian carp Carassius auratus. Aquac. Rep. 2022, 24, 101154. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, Y.; Liang, L.; Shen, L.; Zhu, J.; Wang, Y.; Zhang, J.; Zhang, Z. Knockdown Of TRIM31 Enhances Colorectal Cancer Radiosensitivity By Inducing DNA Damage And Activating Apoptosis. OncoTargets Ther. 2019, 12, 8179–8188. [Google Scholar] [CrossRef]
- Skovira, J.W.; Wu, J.; Matyas, J.J.; Kumar, A.; Hanscom, M.; Kabadi, S.V.; Fang, R.; Faden, A.I. Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J. Neuroinflamm. 2016, 13, 299. [Google Scholar] [CrossRef]
- Gao, Y.; Yin, H.; Zhang, Y.; Dong, Y.; Yang, F.; Wu, X.; Liu, H. Dexmedetomidine protects hippocampal neurons against hypoxia/reoxygenation-induced apoptosis through activation HIF-1α/p53 signaling. Life Sci. 2019, 232, 116611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Zhang, Y.; Herman, B. Caspases, apoptosis and aging. Ageing Res. Rev. 2003, 2, 357–366. [Google Scholar] [CrossRef]
- Taranukhin, A.G.; Taranukhina, E.Y.; Saransaari, P.; Podkletnova, I.M.; Pelto-Huikko, M.; Oja, S.S. Neuroprotection by taurine in ethanol-induced apoptosis in the developing cerebellum. J. Biomed. Sci. 2010, 17, S12. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, Y.; Li, B.; Gao, S. DBA-induced caspase-3-dependent apoptosis occurs through mitochondrial translocation of cyt-c in the rat hippocampus. Mol. Biosyst. 2017, 13, 1863–1873. [Google Scholar] [CrossRef]
- Schlegel, C.R.; Fonseca, A.-V.; Stöcker, S.; Georgiou, M.L.; Misterek, M.B.; E Munro, C.; Carmo, C.R.; Seckl, M.J.; Costa-Pereira, A.P. DAPK2 is a novel modulator of TRAIL-induced apoptosis. Cell Death Differ. 2014, 21, 1780–1791. [Google Scholar] [CrossRef]
- Matsumaru, K.; Ji, C.; Kaplowitz, N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 2003, 37, 1425–1434. [Google Scholar] [CrossRef]
- Liu, W.; Zhao, C.; Wang, P.; Wang, S.; Lin, H.; Qiu, L. The response of glutathione peroxidase 1 and glutathione peroxidase 7 under different oxidative stresses in black tiger shrimp, Penaeus monodon. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2018, 217, 1–13. [Google Scholar] [CrossRef]
- Wickham, S.; West, M.B.; Cook, P.F.; Hanigan, M.H. Gamma-glutamyl compounds: Substrate specificity of gamma-glutamyl transpeptidase enzymes. Anal. Biochem. 2011, 414, 208–214. [Google Scholar] [CrossRef]
- Hossian, A.K.M.N.; Zahra, F.T.; Poudel, S.; Abshire, C.F.; Polk, P.; Garai, J.; Zabaleta, J.; Mikelis, C.M.; Mattheolabakis, G. Advanced bioinformatic analysis and pathway prediction of NSCLC cells upon cisplatin resistance. Sci. Rep. 2021, 11, 6520. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Bivik, C.; Rosdahl, I.; Ollinger, K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis 2006, 28, 537–544. [Google Scholar] [CrossRef]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Tirodkar, T.S.; Lu, P.; Bai, A.; Scheffel, M.J.; Gencer, S.; Garrett-Mayer, E.; Bielawska, A.; Ogretmen, B.; Voelkel-Johnson, C. Expression of Ceramide Synthase 6 Transcriptionally Activates Acid Ceramidase in a c-Jun N-terminal Kinase (JNK)-dependent Manner. J. Biol. Chem. 2015, 290, 13157–13167. [Google Scholar] [CrossRef]
- Takehara, M.; Bandou, H.; Kobayashi, K.; Nagahama, M. Clostridium perfringens α-toxin specifically induces endothelial cell death by promoting ceramide-mediated apoptosis. Anaerobe 2020, 65, 102262. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef]
- Polymeropoulos, E.T.; Elliott, N.G.; Frappell, P.B. Hypoxic acclimation leads to metabolic compensation after reoxygenation in Atlantic salmon yolk-sac alevins. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2017, 213, 28–35. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Wang, H. The Metabolic Fates of Pyruvate in Normal and Neoplastic Cells. Cells 2021, 10, 762. [Google Scholar] [CrossRef]
- Jalloh, I.; Helmy, A.; Howe, D.J.; Shannon, R.J.; Grice, P.; Mason, A.; Gallagher, C.N.; Murphy, M.P.; Pickard, J.D.; Menon, D.K.; et al. A Comparison of Oxidative Lactate Metabolism in Traumatically Injured Brain and Control Brain. J. Neurotrauma 2018, 35, 2025–2035. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family—Role and regulation. IUBMB Life 2011, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.R.; McCallum, R.E. Identification of tumor necrosis factor as a transcriptional regulator of the phosphoenolpyruvate carboxykinase gene following endotoxin treatment of mice. Infect. Immun. 1992, 60, 4040–4050. [Google Scholar] [CrossRef]
- Hutton, J.C.; O’Brien, R.M. Glucose-6-phosphatase Catalytic Subunit Gene Family. J. Biol. Chem. 2009, 284, 29241–29245. [Google Scholar] [CrossRef]
- Mommsen, T.P.; Danulat, E.; Gavioli, M.E.; Foster, G.D.; Moon, T.W. Separation of enzymatically distinct populations of trout hepatocytes. Can. J. Zool. 1991, 69, 420–426. [Google Scholar] [CrossRef]
- Orho, M.; Bosshard, N.U.; Buist, N.R.; Gitzelmann, R.; Aynsley-Green, A.; Blümel, P.; Gannon, M.C.; Nuttall, F.Q.; Groop, L.C. Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0. J. Clin. Investig. 1998, 102, 507–515. [Google Scholar] [CrossRef]
- Imagawa, E.; Osaka, H.; Yamashita, A.; Shiina, M.; Takahashi, E.; Sugie, H.; Nakashima, M.; Tsurusaki, Y.; Saitsu, H.; Ogata, K.; et al. A hemizygous GYG2 mutation and Leigh syndrome: A possible link? Hum. Genet. 2013, 133, 225–234. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, S.; Lin, Y.; Xu, W.; Ye, D.; Xiong, Y.; Zhao, S.; Guan, K.L. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 2012, 15, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mommsen, T.P.; French, C.J.; Hochachka, P.W. Sites and patterns of protein and amino acid utilization during the spawning migration of salmon. Can. J. Zool. 1980, 58, 1785–1799. [Google Scholar] [CrossRef]
- Jiang, X.; Chang, H.; Zhou, Y. Expression, purification and preliminary crystallographic studies of human glutamate oxaloacetate transaminase 1 (GOT1). Protein Expr. Purif. 2015, 113, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Perfield, J.W.; Obin, M.S.; Greenberg, A.S. Adipose triglyceride lipase regulates basal lipolysis and lipid droplet size in adipocytes. J. Cell Biochem. 2008, 105, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Sollier, C.; Capel, E.; Aguilhon, C.; Smirnov, V.; Auclair, M.; Douillard, C.; Ladsous, M.; Defoort-Dhellemmes, S.; Gorwood, J.; Braud, L.; et al. LIPE-related lipodystrophic syndrome: Clinical features and disease modeling using adipose stem cells. Eur. J. Endocrinol. 2021, 184, 155–168. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2016, 13, 91–98. [Google Scholar] [CrossRef]
- Wei, X.; Schultz, K.; Bazilevsky, G.A.; Vogt, A.; Marmorstein, R. Molecular basis for acetyl-CoA production by ATP-citrate lyase. Nat. Struct. Mol. Biol. 2019, 27, 33–41. [Google Scholar] [CrossRef]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhu, S.G.; Xu, C.F. Biochemistry; Higher Education Press: Beijing, China, 2002; Chapter 28; pp. 230–256. (In Chinese) [Google Scholar]
- Liang, Y.-S.; Wu, R.-X.; Miao, B.-B.; Niu, S.-F.; Liang, Z.-B.; Zhai, Y. Integrated mRNA-Seq and miRNA-Seq analysis reveals the microRNAs-transcription factor-mRNA regulatory network of pearl gentian grouper under acute hypoxia. Aquaculture 2024, 578, 740120. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.C. Whole Genome Sequencing of Female Pompano (Trachinotus ovatus). Figshare. 2019. Available online: https://figshare.com/articles/dataset/Whole_genome_sequencing_of_female_pompano_Trachinotus_ovatus_/7570727/3 (accessed on 18 June 2022).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, 362–368. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Zhang, X.Q.; Huang, S.; Cao, Z.J.; Qin, Q.W.; Hu, W.T.; Sun, Y.; Zhou, Y.C. Selection of reference genes for quantitative real-time RT-PCR on gene expression in golden pompano (Trachinotus ovatus). Pol. J. Vet. Sci. 2017, 20, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Hy0 | Hy1 | Hy6 | Ro12 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hy0-1 | Hy0-2 | Hy0-3 | Hy1-1 | Hy1-2 | Hy1-3 | Hy6-1 | Hy6-2 | Hy6-3 | Ro12-1 | Ro12-2 | Ro12-3 | |
| Raw reads (×106) | 39.06 | 43.39 | 38.85 | 40.63 | 41.55 | 40.95 | 42.33 | 41.99 | 41.23 | 41.33 | 59.29 | 44.58 |
| Clean reads (×106) | 38.43 | 42.74 | 38.28 | 39.97 | 40.83 | 40.39 | 41.70 | 41.23 | 40.73 | 40.74 | 58.25 | 43.53 |
| Clean reads rate (%) | 98.39 | 98.50 | 98.53 | 98.38 | 98.27 | 98.63 | 98.51 | 98.19 | 98.79 | 98.57 | 98.25 | 97.64 |
| Raw bases (×109 bp) | 5.86 | 6.51 | 5.83 | 6.10 | 6.23 | 6.14 | 6.35 | 6.30 | 6.18 | 6.20 | 8.89 | 6.69 |
| Clean bases (×109 bp) | 5.73 | 6.38 | 5.72 | 5.97 | 6.09 | 6.03 | 6.23 | 6.15 | 6.08 | 6.09 | 8.70 | 6.49 |
| GC content (%) | 49.25 | 49.00 | 49.22 | 49.29 | 49.09 | 49.44 | 49.64 | 49.78 | 49.45 | 49.69 | 49.05 | 49.87 |
| Q20 (%) | 97.80 | 97.63 | 97.68 | 97.68 | 97.65 | 97.69 | 97.73 | 97.72 | 97.90 | 97.89 | 97.41 | 97.74 |
| Q30 (%) | 93.79 | 93.37 | 93.50 | 93.54 | 93.45 | 93.50 | 93.63 | 93.61 | 94.00 | 94.02 | 92.93 | 93.69 |
| High-quality reads (×106) | 38.41 | 42.72 | 38.27 | 39.94 | 40.81 | 40.37 | 41.67 | 41.21 | 40.71 | 40.72 | 58.22 | 43.51 |
| Total mapped (×106) | 35.83 | 40.02 | 35.80 | 37.46 | 38.15 | 38.06 | 39.18 | 36.97 | 38.35 | 37.54 | 54.07 | 40.79 |
| Total mapped rate (%) | 93.29 | 93.67 | 93.57 | 93.78 | 93.48 | 94.28 | 94.03 | 89.73 | 94.20 | 92.20 | 92.87 | 93.74 |
| Unique mapped (×106) | 34.09 | 37.80 | 33.91 | 35.53 | 36.41 | 35.93 | 37.04 | 35.36 | 36.57 | 35.80 | 51.30 | 38.51 |
| Unique mapped rate (%) | 88.75 | 88.50 | 88.62 | 88.95 | 89.22 | 89.01 | 88.90 | 85.81 | 89.84 | 87.92 | 88.11 | 88.51 |
| Genes/Metabolites | Description | Log2 (Fold Change) | |||
|---|---|---|---|---|---|
| Hy0 vs. Hy1 | Hy0 vs. Hy6 | Hy0 vs. Ro12 | Hy6 vs. Ro12 | ||
| Signal transduction | |||||
| FOS | proto-oncogene c-Fos | 9.16 ** | 7.05 ** | −0.18 | −7.22 ** |
| JUN | proto-oncogene c-Jun | 6.43 ** | 5.03 ** | 0.08 | −4.95 ** |
| Cell cycle arrest | |||||
| GADD45B | growth arrest and DNA damage-inducible protein GADD45 beta | 5.17 ** | 3.42 ** | 1.70 ** | −1.72 |
| CDKN1A | cyclin-dependent kinase inhibitor 1A | 1.18 ** | 1.52 ** | 0.25 | −1.28 * |
| Pro-apoptosis (intrinsic pathway) | |||||
| BAX | Bcl-2-associated X | 0.62 | 1.25 ** | 0.21 | −1.04 * |
| CYC-B | cytochrome c-b | 2.13 ** | 4.73 ** | 1.39 * | −3.35 ** |
| CASP3 | caspase-3 | −0.42 | 0.64 * | 0.97 ** | 0.34 |
| Pro-apoptosis (extrinsic pathway) | |||||
| TNFRSF10A | tumor necrosis factor receptor superfamily member 10A | 1.84 ** | 1.72 ** | 0.15 | −1.57 ** |
| Pro-apoptosis (glutathione metabolism) | |||||
| GPX7 | glutathione peroxidase 7 | −1.37 | −2.92 ** | 0.00 | 2.92 * |
| GGT5 | glutathione hydrolase 5 | 0.59 | 1.34 * | 1.91 ** | 0.57 |
| Glutathione | – | −2.40 # | −1.67 # | 0.58 | 2.25 # |
| L-Glutamic acid | – | −0.63 # | −0.90 # | −0.63 # | 0.27 |
| γ-Glutamylcysteine | – | 3.16 # | 1.38 | 0.06 | −1.32 |
| L-Cysteine | – | 0.31 | 1.17 | 2.93 # | 1.76 # |
| Pro-apoptosis (sphingosine metabolism) | |||||
| CERS6 | ceramide synthase 6 | 0.30 | −1.14 | 1.51 * | 2.65 ** |
| Sphingosine | – | 1.28 | 0.60 | 3.20 # | 2.60 # |
| Anti-apoptosis | |||||
| MCL1 | myeloid leukemia cell differentiation protein Mcl-1 | 1.48 ** | 1.77 ** | 0.07 | −1.70 ** |
| HSP70 | heat shock protein 70 | 8.28 ** | 9.83 ** | 2.00 | −7.83 ** |
| MDM2 | murine double minute 2 | 0.47 * | 1.32 ** | −0.41 | −1.72 ** |
| Glycolysis | |||||
| HK1 | hexokinase 1 | 1.42 ** | 2.28 ** | 0.95 | −1.33 * |
| PFKL | ATP-dependent 6-phosphofructokinase, liver type | −0.08 | 2.05 ** | 0.86 ** | −1.19 * |
| ALDOA | fructose-bisphosphate aldolase A | 1.52 ** | 3.22 ** | 1.40 ** | −1.82 ** |
| ALDOCB | fructose-bisphosphate aldolase C-B | 0.95 ** | 2.86 ** | 1.07 ** | −1.79 ** |
| GAPDH-2 | glyceraldehyde 3-phosphate dehydrogenase isoform 2 | 0.50 | 2.01 ** | 1.30 ** | −0.71 * |
| PGAM1 | phosphoglycerate mutase 1 | 1.27 * | 3.16 ** | 2.13 * | −1.03 |
| ENO1 | alpha-enolase | −0.15 | 1.97 ** | 1.55 ** | −0.42 |
| LDHA | lactate dehydrogenase A | 2.73 ** | 4.16 ** | 1.44 ** | −2.71 ** |
| Lactate transport | |||||
| SLC16A3 | solute carrier family 16 member 3 | 3.25 ** | 4.44 ** | 1.56 | −2.88 ** |
| Gluconeogensis | |||||
| G6PC | glucose-6-phosphatase | 2.02 ** | 1.29 ** | −0.26 | −1.56 ** |
| PCK1 | cytosolic phosphoenolpyruvate carboxykinase 1 | 4.95 ** | 4.70 ** | 1.39 * | −3.31 ** |
| Liver glycogen synthesis | |||||
| GYG2 | glycogenin-2 | 0.01 | 1.22 ** | −0.49 | −1.71 ** |
| GYS2 | glycogen synthase, liver | 0.18 | 0.86 ** | 0.39 | −0.47 |
| PHKA2 | phosphorylase b kinase regulatory subunit alpha, liver isoform | −0.61 | −1.85 ** | −0.95 * | 0.90 |
| PYGL | glycogen phosphorylase, liver form | −0.72 | −1.42 * | −0.59 | 0.83 |
| Amino acid metabolism | |||||
| GOT2 | L-aspartic acid aminotransferase 2 | 0.74 * | 1.62 ** | 0.29 | −1.33 ** |
| L-Aspartic acid | – | −0.37 # | −0.75 # | −0.56 # | 0.19 |
| Fat mobilization | |||||
| PNPLA2 | patatin-like phospholipase domain-containing protein 2 | 0.90 ** | 1.83 ** | −0.56 | −2.39 ** |
| LIPE | hormone-sensitive lipase-like | 0.98 * | 1.90 ** | −0.80 | −2.69 ** |
| Fatty acid biosynthesis | |||||
| ACSL4 | long-chain-fatty-acid-CoA ligase 4 | 2.19 * | 4.09 ** | −4.62 ** | −0.53 |
| ACLY | ATP-citrate lyase | 0.94 | 2.25 ** | 0.53 | −1.72 * |
| Fatty acid β-oxidation | |||||
| CPT1A | carnitine O-palmitoyltransferase 1, liver isoform | 0.35 | −0.05 | 1.17 ** | 1.22 * |
| CPT2 | carnitine O-palmitoyltransferase 2, mitochondrial | −0.80 * | −1.26 ** | 0.07 | 1.33 ** |
| TCA cycle | |||||
| Oxoglutaric acid | – | 0.48 | 0.30 | 1.01 # | 0.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.-H.; Wu, R.-X.; Ji, J.-N.; Zhang, J.; Niu, S.-F.; Tang, B.-G.; Miao, B.-B.; Liang, Z.-B. Integrated Transcriptomics and Metabolomics Reveal Changes in Cell Homeostasis and Energy Metabolism in Trachinotus ovatus in Response to Acute Hypoxic Stress. Int. J. Mol. Sci. 2024, 25, 1054. https://doi.org/10.3390/ijms25021054
Wang Q-H, Wu R-X, Ji J-N, Zhang J, Niu S-F, Tang B-G, Miao B-B, Liang Z-B. Integrated Transcriptomics and Metabolomics Reveal Changes in Cell Homeostasis and Energy Metabolism in Trachinotus ovatus in Response to Acute Hypoxic Stress. International Journal of Molecular Sciences. 2024; 25(2):1054. https://doi.org/10.3390/ijms25021054
Chicago/Turabian StyleWang, Qing-Hua, Ren-Xie Wu, Jiao-Na Ji, Jing Zhang, Su-Fang Niu, Bao-Gui Tang, Ben-Ben Miao, and Zhen-Bang Liang. 2024. "Integrated Transcriptomics and Metabolomics Reveal Changes in Cell Homeostasis and Energy Metabolism in Trachinotus ovatus in Response to Acute Hypoxic Stress" International Journal of Molecular Sciences 25, no. 2: 1054. https://doi.org/10.3390/ijms25021054
APA StyleWang, Q.-H., Wu, R.-X., Ji, J.-N., Zhang, J., Niu, S.-F., Tang, B.-G., Miao, B.-B., & Liang, Z.-B. (2024). Integrated Transcriptomics and Metabolomics Reveal Changes in Cell Homeostasis and Energy Metabolism in Trachinotus ovatus in Response to Acute Hypoxic Stress. International Journal of Molecular Sciences, 25(2), 1054. https://doi.org/10.3390/ijms25021054