
Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids

 ,
,  , ,
, ,  , ,
, ,  , and
, and 
Abstract
1. Introduction
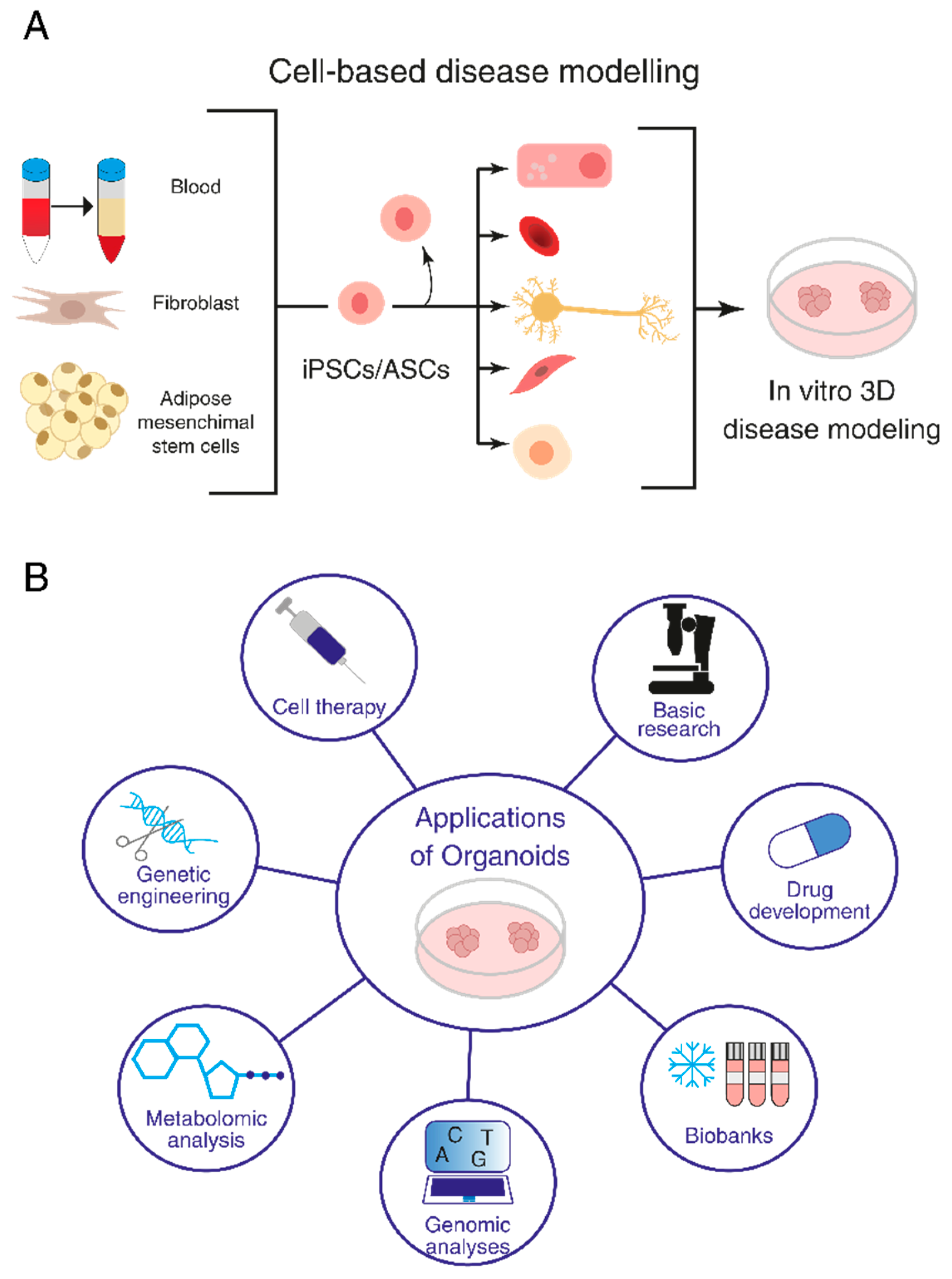
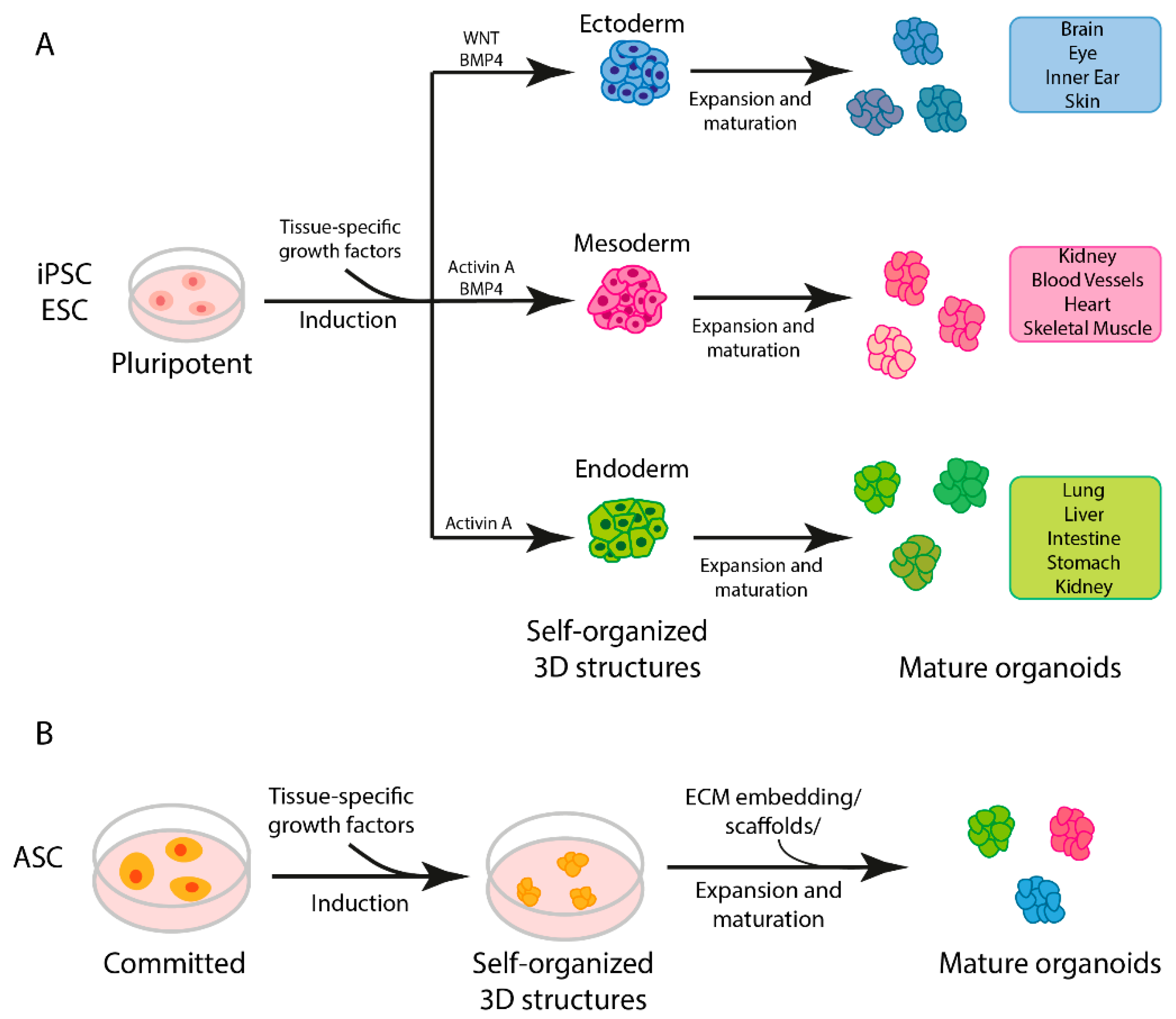
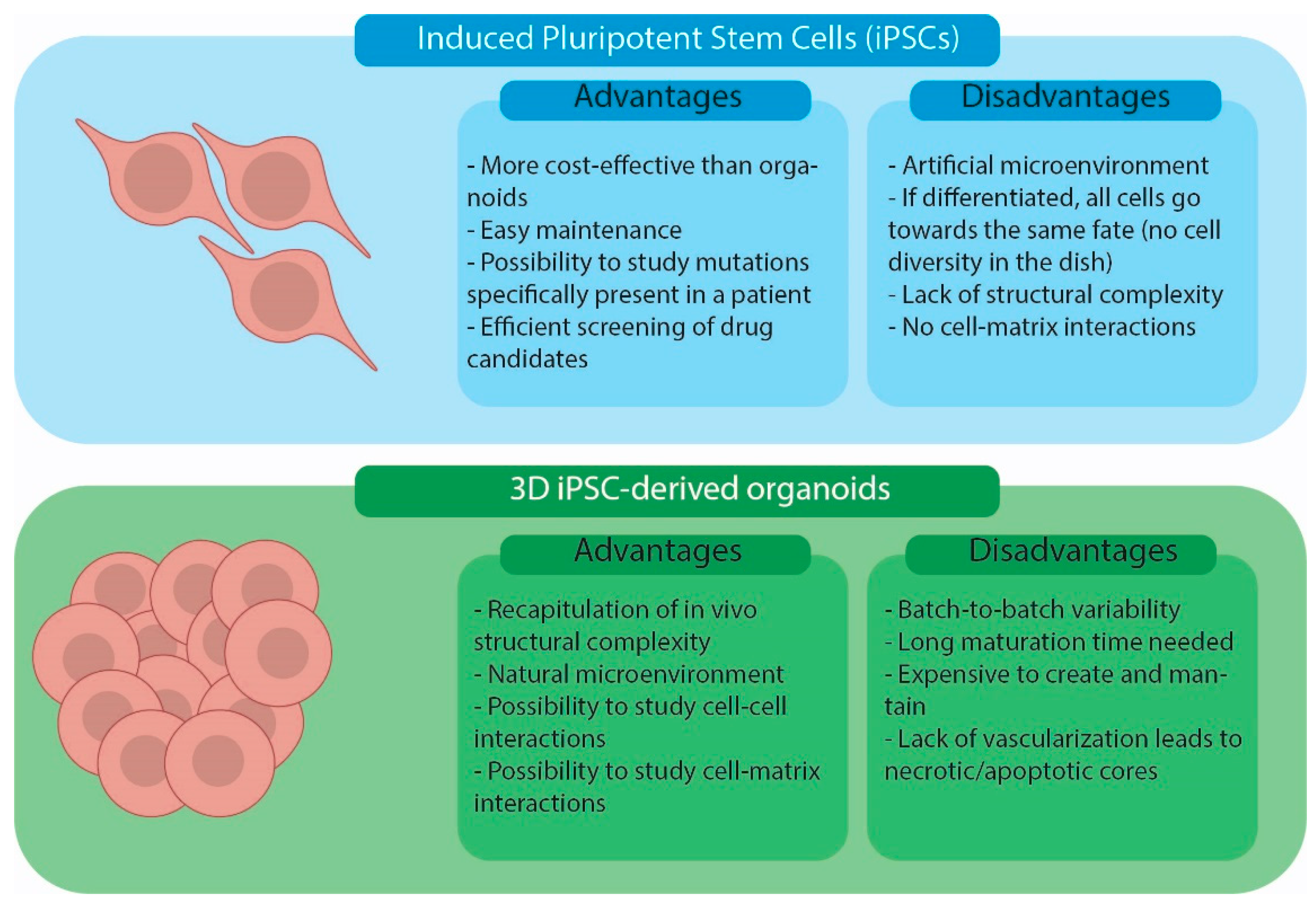
1.1. General Strategies Used for Organoid Production
1.2. Biomedical Applications
1.3. Cutting-Edge Technologies for Genetic Modifications of iPSCs and Organoids
1.4. Quality Controls
2. Organoids and iPSC-Based Models for Rare Neurodegenerative, Neuromuscular and Skeletal Diseases
2.1. Modeling Rare Neurodegenerative Diseases and Tauopathies with iPSC-Derived Organoids
2.1.1. Autosomal Recessive Primary Microcephaly (MCPH3)
2.1.2. Miller–Dieker Syndrome (MDS)
2.1.3. Leukoencephalopathy with Vanishing White Matter (VWM)
2.1.4. Huntington Disease (HD)
2.1.5. Creutzfeldt–Jakob Disease (CJD)
2.1.6. Retinitis Pigmentosa (RP)
2.1.7. Charcot–Marie–Tooth (CMT) Disease
2.1.8. Frontotemporal Dementia (FTD)
2.2. hiPSCs, Organoids and Novel Platforms for Neuromuscular Disorder Modeling
2.3. iPSCs and Organoids for Modeling Bone Disorders
2.3.1. Bone and Cartilage Organoid Models
2.3.2. Bone and Cartilage Disorders Modeling
2.4. Organoids Models for Brain Tumors
3. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Novelli, G.; Spitalieri, P.; Murdocca, M.; Centanini, E.; Sangiuolo, F. Organoid Factory: The Recent Role of the Human Induced Pluripotent Stem Cells (hiPSCs) in Precision Medicine. Front. Cell Dev. Biol. 2022, 10, 1059579. [Google Scholar] [CrossRef]
- Ma, M.; Moulton, M.J.; Lu, S.; Bellen, H.J. “Fly-Ing” from Rare to Common Neurodegenerative Disease Mechanisms. Trends Genet. TIG 2022, 38, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Adamson, K.I.; Sheridan, E.; Grierson, A.J. Use of Zebrafish Models to Investigate Rare Human Disease. J. Med. Genet. 2018, 55, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bai, X. Stem Cell-Based Disease Modeling and Cell Therapy. Cells 2020, 9, 2193. [Google Scholar] [CrossRef] [PubMed]
- Argentati, C.; Tortorella, I.; Bazzucchi, M.; Morena, F.; Martino, S. Harnessing the Potential of Stem Cells for Disease Modeling: Progress and Promises. J. Pers. Med. 2020, 10, 8. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Corrò, C.; Novellasdemunt, L.; Li, V.S.W. A Brief History of Organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Silva-Pedrosa, R.; Salgado, A.J.; Ferreira, P.E. Revolutionizing Disease Modeling: The Emergence of Organoids in Cellular Systems. Cells 2023, 12, 930. [Google Scholar] [CrossRef]
- Andrews, M.G.; Kriegstein, A.R. Challenges of Organoid Research. Annu. Rev. Neurosci. 2022, 45, 23–39. [Google Scholar] [CrossRef]
- Calà, G.; Sina, B.; De Coppi, P.; Giobbe, G.G.; Gerli, M.F.M. Primary Human Organoids Models: Current Progress and Key Milestones. Front. Bioeng. Biotechnol. 2023, 11, 1058970. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef]
- Teriyapirom, I.; Batista-Rocha, A.S.; Koo, B.-K. Genetic Engineering in Organoids. J. Mol. Med. Berl. Ger. 2021, 99, 555–568. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.-K. Modeling Mouse and Human Development Using Organoid Cultures. Dev. Camb. Engl. 2015, 142, 3113–3125. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Xiang, Y.; Xiao, J.; Zheng, W.; Ebrahimkhani, M.R.; Yang, C.; Wu, A.R.; Liu, P.; Huang, Y.; Sugimura, R. Organoid-Based Single-Cell Spatiotemporal Gene Expression Landscape of Human Embryonic Development and Hematopoiesis. Signal Transduct. Target. Ther. 2023, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, E.L.; Heredero Berzal, A.; Boon, C.J.F.; Quinn, P.M.J.; Ten Asbroek, A.L.M.A.; Bergen, A.A. The Role of Small Molecules and Their Effect on the Molecular Mechanisms of Early Retinal Organoid Development. Int. J. Mol. Sci. 2021, 22, 7081. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.-H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling Human Development and Disease in Pluripotent Stem-Cell-Derived Gastric Organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed Differentiation of Human Pluripotent Stem Cells into Intestinal Tissue In Vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Rothenbücher, T.S.P.; Gürbüz, H.; Pereira, M.P.; Heiskanen, A.; Emneus, J.; Martinez-Serrano, A. Next Generation Human Brain Models: Engineered Flat Brain Organoids Featuring Gyrification. Biofabrication 2021, 13, 011001. [Google Scholar] [CrossRef] [PubMed]
- Rispoli, P.; Scandiuzzi Piovesan, T.; Decorti, G.; Stocco, G.; Lucafò, M. iPSCs as a Groundbreaking Tool for the Study of Adverse Drug Reactions: A New Avenue for Personalized Therapy. WIREs Mech. Dis. 2023, e1630. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.-G. 2D- and 3D-Based Intestinal Stem Cell Cultures for Personalized Medicine. Cells 2018, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Pérez-González, C.; Ceada, G.; Greco, F.; Matejčić, M.; Gómez-González, M.; Castro, N.; Menendez, A.; Kale, S.; Krndija, D.; Clark, A.G.; et al. Mechanical Compartmentalization of the Intestinal Organoid Enables Crypt Folding and Collective Cell Migration. Nat. Cell Biol. 2021, 23, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, S.; Irwin, C.; Singh, K.K. Human Pluripotent Stem Cell (hPSC) and Organoid Models of Autism: Opportunities and Limitations. Transl. Psychiatry 2023, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhao, M.; Chen, H.; Zhou, X.; Lenahan, C.; Ou, Y.; He, Y. The Application of Brain Organoid Technology in Stroke Research: Challenges and Prospects. Front. Cell. Neurosci. 2021, 15, 646921. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.-N.; Xu, T.-Y.; Miao, Z.-W.; Su, D.-F.; Miao, C.-Y. Organoid Technology for Brain and Therapeutics Research. CNS Neurosci. Ther. 2017, 23, 771–778. [Google Scholar] [CrossRef]
- Yang, H. Tau and Stathmin Proteins in Breast Cancer: A Potential Therapeutic Target. Clin. Exp. Pharmacol. Physiol. 2022, 49, 445–452. [Google Scholar] [CrossRef]
- Xiang, K.; Zhuang, H. Liver Organoid Potential Application for Hepatitis E Virus Infection. Adv. Exp. Med. Biol. 2023, 1417, 133–139. [Google Scholar] [CrossRef]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium Infection in Human Small Intestinal and Lung Organoids. Nat. Microbiol. 2018, 3, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Artegiani, B.; Clevers, H. Use and Application of 3D-Organoid Technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of Small-Molecule Inhibitors of Zika Virus Infection and Induced Neural Cell Death via a Drug Repurposing Screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Zilbauer, M. Biobanking of Human Gut Organoids for Translational Research. Exp. Mol. Med. 2021, 53, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and Establishment of Self-Renewing Human and Mouse Adult Liver and Pancreas 3D Organoids and Their Genetic Manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Munro, M.J.; Tan, S.T.; Gray, C. Applications for Colon Organoid Models in Cancer Research. Organoids 2023, 2, 37–49. [Google Scholar] [CrossRef]
- Flood, P.; Hanrahan, N.; Nally, K.; Melgar, S. Human Intestinal Organoids: Modeling Gastrointestinal Physiology and Immunopathology—Current Applications and Limitations. Eur. J. Immunol. 2023, e2250248. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Roselli, R.; Biagioli, M.; Bordoni, M.; Ricci, P.; Zampella, A.; Distrutti, E.; Donini, A.; Fiorucci, S. Modeling Inflammatory Bowel Disease by Intestinal Organoids. Recent Adv. Inflamm. Allergy Drug Discov. 2023, 17, 39–53. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, W.; Wang, R.; Hu, Y.; Yi, K.; Sun, X.; Wang, G.; Xu, X. Tumor Organoid Model of Colorectal Cancer (Review). Oncol. Lett. 2023, 26, 328. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef]
- Del Dosso, A.; Urenda, J.-P.; Nguyen, T.; Quadrato, G. Upgrading the Physiological Relevance of Human Brain Organoids. Neuron 2020, 107, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ye, Z.; Jang, Y.-Y. Convergence of Human Pluripotent Stem Cell, Organoid, and Genome Editing Technologies. Exp. Biol. Med. 2021, 246, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Heide, M.; Huttner, W.B. Genetic Modification of Brain Organoids. Front. Cell. Neurosci. 2019, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.-L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-Dependent Selection Yields AAV Variants for Widespread Gene Transfer to the Adult Brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449.e4. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of Functionally Integrated Human Forebrain Spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef]
- Kawasaki, K.; Fujii, M.; Sugimoto, S.; Ishikawa, K.; Matano, M.; Ohta, Y.; Toshimitsu, K.; Takahashi, S.; Hosoe, N.; Sekine, S.; et al. Chromosome Engineering of Human Colon-Derived Organoids to Develop a Model of Traditional Serrated Adenoma. Gastroenterology 2020, 158, 638–651.e8. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling Colorectal Cancer Using CRISPR-Cas9-Mediated Engineering of Human Intestinal Organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Whittle, J.R.; Vaillant, F.; Chen, H.-R.; Dawson, C.; Liu, K.; Geurts, M.H.; Herold, M.J.; Clevers, H.; Lindeman, G.J.; et al. Modeling Breast Cancer Using CRISPR-Cas9-Mediated Engineering of Human Breast Organoids. J. Natl. Cancer Inst. 2020, 112, 540–544. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.-J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Nieto-Estévez, V.; Kyrychenko, S.; Schneider, J.W.; Hsieh, J. Retinoblastoma Protein Controls Growth, Survival and Neuronal Migration in Human Cerebral Organoids. Dev. Camb. Engl. 2017, 144, 1025–1034. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human Brain Organoids on a Chip Reveal the Physics of Folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Xiaoshuai, L.; Qiushi, W.; Rui, W. Advantages of CRISPR-Cas9 Combined Organoid Model in the Study of Congenital Nervous System Malformations. Front. Bioeng. Biotechnol. 2022, 10, 932936. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective Metabolic Programming Impairs Early Neuronal Morphogenesis in Neural Cultures and an Organoid Model of Leigh Syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef]
- Bendriem, R.M.; Singh, S.; Aleem, A.A.; Antonetti, D.A.; Ross, M.E. Tight Junction Protein Occludin Regulates Progenitor Self-Renewal and Survival in Developing Cortex. eLife 2019, 8, e49376. [Google Scholar] [CrossRef]
- Bhatia, S.; Pooja; Yadav, S.K. CRISPR-Cas for Genome Editing: Classification, Mechanism, Designing and Applications. Int. J. Biol. Macromol. 2023, 238, 124054. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Zhang, T. Computational Approaches for Effective CRISPR Guide RNA Design and Evaluation. Comput. Struct. Biotechnol. J. 2020, 18, 35–44. [Google Scholar] [CrossRef]
- Kang, S.-Y.; Kimura, M.; Shrestha, S.; Lewis, P.; Lee, S.; Cai, Y.; Joshi, P.; Acharya, P.; Liu, J.; Yang, Y.; et al. A Pillar and Perfusion Plate Platform for Robust Human Organoid Culture and Analysis. Adv. Healthc. Mater. 2023, e2302502. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Lichtman, J.W. Clarifying Tissue Clearing. Cell 2015, 162, 246–257. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Mousavi Shaegh, S.A.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-Integrated Organs-on-Chips Platform for Automated and Continual in Situ Monitoring of Organoid Behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef]
- Takebe, T.; Zhang, R.-R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a Vascularized and Functional Human Liver from an iPSC-Derived Organ Bud Transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhang, J.; Gao, K.; Zhou, L.; Jiang, Y.; Wang, J.; Wu, Y. Human-Induced Pluripotent Stem Cell-Derived Cerebral Organoid of Leukoencephalopathy with Vanishing White Matter. CNS Neurosci. Ther. 2023, 29, 1049–1066. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Costamagna, G.; Tamanini, S.; Corti, S. Back to the Origins: Human Brain Organoids to Investigate Neurodegeneration. Brain Res. 2020, 1727, 146561. [Google Scholar] [CrossRef]
- Groveman, B.R.; Foliaki, S.T.; Orru, C.D.; Zanusso, G.; Carroll, J.A.; Race, B.; Haigh, C.L. Sporadic Creutzfeldt-Jakob Disease Prion Infection of Human Cerebral Organoids. Acta Neuropathol. Commun. 2019, 7, 90. [Google Scholar] [CrossRef]
- Gao, M.-L.; Lei, X.-L.; Han, F.; He, K.-W.; Jin, S.-Q.; Zhang, Y.-Y.; Jin, Z.-B. Patient-Specific Retinal Organoids Recapitulate Disease Features of Late-Onset Retinitis Pigmentosa. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Su, T.; Liang, L.; Zhang, L.; Wang, J.; Chen, L.; Su, C.; Cao, J.; Yu, Q.; Deng, S.; Chan, H.F.; et al. Retinal Organoids and Microfluidic Chip-Based Approaches to Explore the Retinitis Pigmentosa with USH2A Mutations. Front. Bioeng. Biotechnol. 2022, 10. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Jin, Z.-B. Retinal Organoids as Models for Development and Diseases. Cell Regen. 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-L.; Gao, M.-L.; Lei, X.-L.; Lv, J.-N.; Zhao, H.; He, K.-W.; Xia, X.-X.; Li, L.-Y.; Chen, Y.-C.; Li, Y.-P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Deng, W.-L.; Jin, Z.-B. Modeling Retinitis Pigmentosa through Patient-Derived Retinal Organoids. STAR Protoc. 2021, 2, 100438. [Google Scholar] [CrossRef]
- Kandoi, S.; Martinez, C.; Chen, K.X.; Mehine, M.; Mansfield, B.C.; Duncan, J.L.; Lamba, D.A. Disease Modeling and Pharmacological Rescue of Autosomal Dominant Retinitis Pigmentosa Associated with RHO Copy Number Variation. medRxiv 2023. [Google Scholar] [CrossRef]
- Rodrigues, A.; Slembrouck-Brec, A.; Nanteau, C.; Terray, A.; Tymoshenko, Y.; Zagar, Y.; Reichman, S.; Xi, Z.; Sahel, J.-A.; Fouquet, S.; et al. Modeling PRPF31 Retinitis Pigmentosa Using Retinal Pigment Epithelium and Organoids Combined with Gene Augmentation Rescue. NPJ Regen. Med. 2022, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, J.; Vendredy, L.; Adriaenssens, E.; Da Silva Authier, T.; Asselbergh, B.; Kaji, M.; Weckhuysen, S.; Van Den Bosch, L.; Baets, J.; Timmerman, V. Downregulation of PMP22 Ameliorates Myelin Defects in iPSC-Derived Human Organoid Cultures of CMT1A. Brain 2023, 146, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Van Den Bosch, L.; De Vos, W.H.; et al. Induced Pluripotent Stem Cell-Derived Motor Neurons of CMT Type 2 Patients Reveal Progressive Mitochondrial Dysfunction. Brain 2021, 144, 2471–2485. [Google Scholar] [CrossRef]
- Bowles, K.R.; Silva, M.C.; Whitney, K.; Bertucci, T.; Berlind, J.E.; Lai, J.D.; Garza, J.C.; Boles, N.C.; Mahali, S.; Strang, K.H.; et al. ELAVL4, Splicing, and Glutamatergic Dysfunction Precede Neuron Loss in MAPT Mutation Cerebral Organoids. Cell 2021, 184, 4547–4563.e17. [Google Scholar] [CrossRef]
- Glasauer, S.M.K.; Goderie, S.K.; Rauch, J.N.; Guzman, E.; Audouard, M.; Bertucci, T.; Joy, S.; Rommelfanger, E.; Luna, G.; Keane-Rivera, E.; et al. Human Tau Mutations in Cerebral Organoids Induce a Progressive Dyshomeostasis of Cholesterol. Stem Cell Rep. 2022, 17, 2127–2140. [Google Scholar] [CrossRef]
- Lines, G.; Casey, J.M.; Preza, E.; Wray, S. Modelling Frontotemporal Dementia Using Patient-Derived Induced Pluripotent Stem Cells. Mol. Cell. Neurosci. 2020, 109, 103553. [Google Scholar] [CrossRef]
- de Majo, M.; Koontz, M.; Marsan, E.; Salinas, N.; Ramsey, A.; Kuo, Y.-M.; Seo, K.; Li, H.; Dräger, N.; Leng, K.; et al. Granulin Loss of Function in Human Mature Brain Organoids Implicates Astrocytes in TDP-43 Pathology. Stem Cell Rep. 2023, 18, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.-C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells Form Neuromuscular Junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef]
- Massih, B.; Veh, A.; Schenke, M.; Mungwa, S.; Seeger, B.; Selvaraj, B.T.; Chandran, S.; Reinhardt, P.; Sterneckert, J.; Hermann, A.; et al. A 3D Cell Culture System for Bioengineering Human Neuromuscular Junctions to Model ALS. Front. Cell Dev. Biol. 2023, 11, 996952. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Faustino Martins, J.-M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.-L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 26, 172–186.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, B.; Zhan, R.-Z.; Rao, L.; Bursac, N. Exercise Mimetics and JAK Inhibition Attenuate IFN-γ-Induced Wasting in Engineered Human Skeletal Muscle. Sci. Adv. 2021, 7, eabd9502. [Google Scholar] [CrossRef]
- Salazar-Noratto, G.E.; Nations, C.C.; Stevens, H.Y.; Xu, M.; Gaynard, S.; Dooley, C.; de Nijs, N.; McDonagh, K.; Shen, S.; Willimon, S.C.; et al. Patient-Specific iPSC-Derived Models Link Aberrant Endoplasmic Reticulum Stress Sensing and Response to Juvenile Osteochondritis Dissecans Etiology. Stem Cells Transl. Med. 2023, 12, 293–306. [Google Scholar] [CrossRef]
- Cui, X.; Cui, Y.; Shi, L.; Luan, J.; Zhou, X.; Han, J. A Preliminary Study on the Mechanism of Skeletal Abnormalities in Turner Syndrome Using Inducing Pluripotent Stem Cells (iPS)-Based Disease Models. Intractable Rare Dis. Res. 2019, 8, 113–119. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Ng, E.S.; Cameron, T.L.; Kung, L.H.W.; Sampurno, L.; Rowley, L.; Lilianty, J.; Patria, Y.N.; Stenta, T.; Hanssen, E.; et al. Modeling Human Skeletal Development Using Human Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2211510120. [Google Scholar] [CrossRef]
- Lilianty, J.; Bateman, J.F.; Lamandé, S.R. Generation of a Heterozygous COL2A1 (p.G1113C) Hypochondrogenesis Mutation iPSC Line, MCRIi019-A-7, Using CRISPR/Cas9 Gene Editing. Stem Cell Res. 2021, 56, 102515. [Google Scholar] [CrossRef]
- Howden, S.; Hosseini Far, H.; Motazedian, A.; Elefanty, A.G.; Stanley, E.G.; Lamandé, S.R.; Bateman, J.F. The Use of Simultaneous Reprogramming and Gene Correction to Generate an Osteogenesis Imperfecta Patient COL1A1 c. 3936 G>T iPSC Line and an Isogenic Control iPSC Line. Stem Cell Res. 2019, 38, 101453. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Chessler, S.D.; Golub, S.B.; Byers, P.H.; Chan, D.; Cole, W.G.; Sillence, D.O.; Bateman, J.F. Endoplasmic Reticulum-Mediated Quality Control of Type I Collagen Production by Cells from Osteogenesis Imperfecta Patients with Mutations in the pro Alpha 1 (I) Chain Carboxyl-Terminal Propeptide Which Impair Subunit Assembly. J. Biol. Chem. 1995, 270, 8642–8649. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-Y.; Ko, J.M.; Park, M.-H.; Koo, S.K. Generation of a Patient-Specific Induced Pluripotent Stem Cell Line, KSCBi006-A, for Osteogenesis Imperfecta Type I with the COL1A1, c.3162delT Mutation. Stem Cell Res. 2019, 41, 101622. [Google Scholar] [CrossRef]
- Saito, A.; Ooki, A.; Nakamura, T.; Onodera, S.; Hayashi, K.; Hasegawa, D.; Okudaira, T.; Watanabe, K.; Kato, H.; Onda, T.; et al. Targeted Reversion of Induced Pluripotent Stem Cells from Patients with Human Cleidocranial Dysplasia Improves Bone Regeneration in a Rat Calvarial Bone Defect Model. Stem Cell Res. Ther. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.; Mathew, R.K.; Polson, E.S.; Williams, J.; Wurdak, H. Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov. Adv. Life Sci. R D 2018, 23, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling Medulloblastoma In Vivo and with Human Cerebellar Organoids. Nat. Commun. 2020, 11, 583. [Google Scholar] [CrossRef]
- Huang, M.; Xu, S.; Li, Y.; Shang, L.; Zhan, X.; Qin, C.; Su, J.; Zhao, Z.; He, Y.; Qin, L.; et al. Novel Human Meningioma Organoids Recapitulate the Aggressiveness of the Initiating Cell Subpopulations Identified by ScRNA-Seq. Adv. Sci. 2023, 10, e2205525. [Google Scholar] [CrossRef]
- Chan, H.S.C.; Ng, H.K.; Chan, A.K.-Y.; Cheng, S.H.; Chow, C.; Wong, N.; Wong, G.K.C. Establishment and Characterization of Meningioma Patient-Derived Organoid. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2021, 94, 192–199. [Google Scholar] [CrossRef]
- Yamazaki, S.; Ohka, F.; Hirano, M.; Shiraki, Y.; Motomura, K.; Tanahashi, K.; Tsujiuchi, T.; Motomura, A.; Aoki, K.; Shinjo, K.; et al. Newly Established Patient-Derived Organoid Model of Intracranial Meningioma. Neuro-Oncol. 2021, 23, 1936–1948. [Google Scholar] [CrossRef]
- Li, Y.-P.; Wang, Y.-T.; Wang, W.; Zhang, X.; Shen, R.-J.; Jin, K.; Jin, L.-W.; Jin, Z.-B. Second Hit Impels Oncogenesis of Retinoblastoma in Patient-Induced Pluripotent Stem Cell-Derived Retinal Organoids: Direct Evidence for Knudson’s Theory. PNAS Nexus 2022, 1, pgac162. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Y.; Zhang, Y.-Y.; Li, Y.-P.; Hua, Z.-Q.; Zhang, C.-J.; Wu, K.-C.; Yu, F.; Zhang, Y.; Su, J.; et al. Human Embryonic Stem Cell-Derived Organoid Retinoblastoma Reveals a Cancerous Origin. Proc. Natl. Acad. Sci. USA 2020, 117, 33628–33638. [Google Scholar] [CrossRef] [PubMed]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Retinoblastoma from Human Stem Cell-Derived Retinal Organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-M.; Ma, C.; Jin, K.; Jin, Z.-B. Retinal Organoid and Gene Editing for Basic and Translational Research. Vision Res. 2023, 210, 108273. [Google Scholar] [CrossRef] [PubMed]
- Rozanska, A.; Cerna-Chavez, R.; Queen, R.; Collin, J.; Zerti, D.; Dorgau, B.; Beh, C.S.; Davey, T.; Coxhead, J.; Hussain, R.; et al. pRB-Depleted Pluripotent Stem Cell Retinal Organoids Recapitulate Cell State Transitions of Retinoblastoma Development and Suggest an Important Role for pRB in Retinal Cell Differentiation. Stem Cells Transl. Med. 2022, 11, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.A.; Depla, J.A.; Sridhar, A.; Wolthers, K.; Pajkrt, D.; Vieira de Sá, R. A Beginner’s Guide on the Use of Brain Organoids for Neuroscientists: A Systematic Review. Stem Cell Res. Ther. 2023, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Wynshaw-Boris, A. Lissencephaly and LIS1: Insights into the Molecular Mechanisms of Neuronal Migration and Development. Clin. Genet. 2007, 72, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.M.C.; van der Lei, H.D.W.; Vermeulen, G.; Gerver, J.A.M.; Lourenço, C.M.; Naidu, S.; Mierzewska, H.; Gemke, R.J.B.J.; de Vet, H.C.W.; Uitdehaag, B.M.J.; et al. Natural History of Vanishing White Matter. Ann. Neurol. 2018, 84, 274–288. [Google Scholar] [CrossRef]
- Trimouille, A.; Marguet, F.; Sauvestre, F.; Lasseaux, E.; Pelluard, F.; Martin-Négrier, M.-L.; Plaisant, C.; Rooryck, C.; Lacombe, D.; Arveiler, B.; et al. Foetal Onset of EIF2B Related Disorder in Two Siblings: Cerebellar Hypoplasia with Absent Bergmann Glia and Severe Hypomyelination. Acta Neuropathol. Commun. 2020, 8, 48. [Google Scholar] [CrossRef]
- Conforti, P.; Besusso, D.; Bocchi, V.D.; Faedo, A.; Cesana, E.; Rossetti, G.; Ranzani, V.; Svendsen, C.N.; Thompson, L.M.; Toselli, M.; et al. Faulty Neuronal Determination and Cell Polarization Are Reverted by Modulating HD Early Phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E762–E771. [Google Scholar] [CrossRef]
- The HD iPSC Consortium. Developmental Alterations in Huntington’s Disease Neural Cells and Pharmacological Rescue in Cells and Mice. Nat. Neurosci. 2017, 20, 648–660. [Google Scholar] [CrossRef]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096.e6. [Google Scholar] [CrossRef]
- Groveman, B.R.; Race, B.; Foliaki, S.T.; Williams, K.; Hughson, A.G.; Baune, C.; Zanusso, G.; Haigh, C.L. Sporadic Creutzfeldt–Jakob Disease Infected Human Cerebral Organoids Retain the Original Human Brain Subtype Features Following Transmission to Humanized Transgenic Mice. Acta Neuropathol. Commun. 2023, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Takashima, H. The Current State of Charcot–Marie–Tooth Disease Treatment. Genes 2023, 14, 1391. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in Vivo Model of Functional and Vascularized Human Brain Organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Uzquiano, A.; Kedaigle, A.J.; Pigoni, M.; Paulsen, B.; Adiconis, X.; Kim, K.; Faits, T.; Nagaraja, S.; Antón-Bolaños, N.; Gerhardinger, C.; et al. Proper Acquisition of Cell Class Identity in Organoids Allows Definition of Fate Specification Programs of the Human Cerebral Cortex. Cell 2022, 185, 3770–3788.e27. [Google Scholar] [CrossRef]
- Velasco, S. Modeling Brain Disorders Using Transplanted Organoids: Beyond the Short Circuit. Cell Stem Cell 2022, 29, 1617–1618. [Google Scholar] [CrossRef]
- Yoon, S.-J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of Human Cortical Organoid Generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD Brain Organoid Slice Cultures Display Distinct Early Astrocyte and Targetable Neuronal Pathology. Nat. Neurosci. 2021, 24, 1542–1554. [Google Scholar] [CrossRef]
- Corsi, A.; Bombieri, C.; Valenti, M.T.; Romanelli, M.G. Tau Isoforms: Gaining Insight into MAPT Alternative Splicing. Int. J. Mol. Sci. 2022, 23, 15383. [Google Scholar] [CrossRef]
- Bertucci, T.; Bowles, K.R.; Lotz, S.; Qi, L.; Stevens, K.; Goderie, S.K.; Borden, S.; Oja, L.M.; Lane, K.; Lotz, R.; et al. Improved Protocol for Reproducible Human Cortical Organoids Reveals Early Alterations in Metabolism with MAPT Mutations. BioRxiv 2023. [Google Scholar] [CrossRef]
- Nakamura, M.; Shiozawa, S.; Tsuboi, D.; Amano, M.; Watanabe, H.; Maeda, S.; Kimura, T.; Yoshimatsu, S.; Kisa, F.; Karch, C.M.; et al. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Rep. 2019, 13, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kritskiy, O.; Watson, L.A.; Barker, S.J.; Dey, D.; Raja, W.K.; Lin, Y.-T.; Ko, T.; Cho, S.; Penney, J.; et al. Inhibition of P25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 9917–9924. [Google Scholar] [CrossRef] [PubMed]
- Iberite, F.; Gruppioni, E.; Ricotti, L. Skeletal Muscle Differentiation of Human iPSCs Meets Bioengineering Strategies: Perspectives and Challenges. NPJ Regen. Med. 2022, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Jalal, S.; Dastidar, S.; Tedesco, F.S. Advanced Models of Human Skeletal Muscle Differentiation, Development and Disease: Three-Dimensional Cultures, Organoids and Beyond. Curr. Opin. Cell Biol. 2021, 73, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Joshi, J.M.; Sundaravadivelu, P.K.; Raina, K.; Lenka, N.; Kaveeshwar, V.; Thummer, R.P. An Overview on Promising Somatic Cell Sources Utilized for the Efficient Generation of Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 1954–1974. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Gerli, M.F.M.; Ragazzi, M.; Dastidar, S.; Benedetti, S.; Loperfido, M.; VandenDriessche, T.; Chuah, M.K.; Tedesco, F.S. Efficient Derivation and Inducible Differentiation of Expandable Skeletal Myogenic Cells from Human ES and Patient-Specific iPS Cells. Nat. Protoc. 2015, 10, 941–958. [Google Scholar] [CrossRef]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering Human Pluripotent Stem Cells into a Functional Skeletal Muscle Tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef]
- Fernández-Costa, J.M.; Tejedera-Vilafranca, A.; Fernández-Garibay, X.; Ramón-Azcón, J. Muscle-on-a-Chip Devices: A New Era for in Vitro Modelling of Muscular Dystrophies. Dis. Model. Mech. 2023, 16, dmm050107. [Google Scholar] [CrossRef]
- Pinton, L.; Khedr, M.; Lionello, V.M.; Sarcar, S.; Maffioletti, S.M.; Dastidar, S.; Negroni, E.; Choi, S.; Khokhar, N.; Bigot, A.; et al. 3D Human Induced Pluripotent Stem Cell-Derived Bioengineered Skeletal Muscles for Tissue, Disease and Therapy Modeling. Nat. Protoc. 2023, 18, 1337–1376. [Google Scholar] [CrossRef]
- Zschüntzsch, J.; Meyer, S.; Shahriyari, M.; Kummer, K.; Schmidt, M.; Kummer, S.; Tiburcy, M. The Evolution of Complex Muscle Cell In Vitro Models to Study Pathomechanisms and Drug Development of Neuromuscular Disease. Cells 2022, 11, 1233. [Google Scholar] [CrossRef] [PubMed]
- Caron, L.; Kher, D.; Lee, K.L.; McKernan, R.; Dumevska, B.; Hidalgo, A.; Li, J.; Yang, H.; Main, H.; Ferri, G.; et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl. Med. 2016, 5, 1145–1161. [Google Scholar] [CrossRef]
- Laberthonnière, C.; Novoa-Del-Toro, E.-M.; Delourme, M.; Chevalier, R.; Broucqsault, N.; Mazaleyrat, K.; Streichenberger, N.; Manel, V.; Bernard, R.; Salort Campana, E.; et al. Facioscapulohumeral Dystrophy Weakened Sarcomeric Contractility Is Mimicked in Induced Pluripotent Stem Cells-Derived Innervated Muscle Fibres. J. Cachexia Sarcopenia Muscle 2022, 13, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Caputo, L.; Granados, A.; Lenzi, J.; Rosa, A.; Ait-Si-Ali, S.; Puri, P.L.; Albini, S. Acute Conversion of Patient-Derived Duchenne Muscular Dystrophy iPSC into Myotubes Reveals Constitutive and Inducible over-Activation of TGFβ-Dependent pro-Fibrotic Signaling. Skelet. Muscle 2020, 10, 13. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lim, H.T.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.P.; Kim, Y.J.; Kim, H.; et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived Using a Human iPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Mournetas, V.; Massouridès, E.; Dupont, J.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis Modelled by Human Pluripotent Stem Cells: A Multi-omic Study of Duchenne Myopathy Early Onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Asano, T.; Nakata, T.; Hotta, A.; Sakurai, H. A Muscle Fatigue-like Contractile Decline Was Recapitulated Using Skeletal Myotubes from Duchenne Muscular Dystrophy Patient-Derived iPSCs. Cell Rep. Med. 2021, 2, 100298. [Google Scholar] [CrossRef] [PubMed]
- Bruge, C.; Geoffroy, M.; Benabides, M.; Pellier, E.; Gicquel, E.; Dhiab, J.; Hoch, L.; Richard, I.; Nissan, X. Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies. Biomedicines 2022, 10, 1428. [Google Scholar] [CrossRef]
- Mateos-Aierdi, A.J.; Dehesa-Etxebeste, M.; Goicoechea, M.; Aiastui, A.; Richaud-Patin, Y.; Jiménez-Delgado, S.; Raya, A.; Naldaiz-Gastesi, N.; López de Munain, A. Patient-Specific iPSC-Derived Cellular Models of LGMDR1. Stem Cell Res. 2021, 53, 102333. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and Reproducible Myogenic Differentiation from Human iPS Cells: Prospects for Modeling Miyoshi Myopathy in Vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Kawada, R.; Jonouchi, T.; Kagita, A.; Sato, M.; Hotta, A.; Sakurai, H. Establishment of Quantitative and Consistent In Vitro Skeletal Muscle Pathological Models of Myotonic Dystrophy Type 1 Using Patient-Derived iPSCs. Sci. Rep. 2023, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Gonzalez, R.; Perlingeiro, R.C.R. Recapitulating Muscle Disease Phenotypes with Myotonic Dystrophy 1 Induced Pluripotent Stem Cells: A Tool for Disease Modeling and Drug Discovery. Dis. Model. Mech. 2018, 11, dmm034728. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front. Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, E.; Herrero-Hernandez, P.; Wan, R.; Broeders, M.; In ’t Groen, S.L.M.; van Gestel, T.J.M.; van IJcken, W.F.J.; Cheung, T.H.; van der Ploeg, A.T.; Schaaf, G.J.; et al. Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Rep. 2018, 10, 1975–1990. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Awaya, T.; Jonouchi, T.; Kimura, R.; Kimura, S.; Era, T.; Heike, T.; Sakurai, H. A Skeletal Muscle Model of Infantile-Onset Pompe Disease with Patient-Specific iPS Cells. Sci. Rep. 2017, 7, 13473. [Google Scholar] [CrossRef] [PubMed]
- Ortuño-Costela, M.D.C.; Cerrada, V.; Moreno-Izquierdo, A.; García-Consuegra, I.; Laberthonnière, C.; Delourme, M.; Garesse, R.; Arenas, J.; Fuster García, C.; García García, G.; et al. Generation of the First Human In Vitro Model for McArdle Disease Based on iPSC Technology. Int. J. Mol. Sci. 2022, 23, 13964. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, T.; Osafune, K.; Sakurai, H.; Asaka, I.; Tanaka, A.; Yamaguchi, S.; Yamada, K.; Hitomi, H.; Arai, S.; Kurose, Y.; et al. Functional Analysis of iPSC-Derived Myocytes from a Patient with Carnitine Palmitoyltransferase II Deficiency. Biochem. Biophys. Res. Commun. 2014, 448, 175–181. [Google Scholar] [CrossRef]
- Gartz, M.; Haberman, M.; Sutton, J.; Slick, R.A.; Luttrell, S.M.; Mack, D.L.; Lawlor, M.W. ACTA1 H40Y Mutant iPSC-Derived Skeletal Myocytes Display Mitochondrial Defects in an In Vitro Model of Nemaline Myopathy. Exp. Cell Res. 2023, 424, 113507. [Google Scholar] [CrossRef]
- Shahriyari, M.; Islam, M.R.; Sakib, S.M.; Rinn, M.; Rika, A.; Krüger, D.; Kaurani, L.; Gisa, V.; Winterhoff, M.; Anandakumar, H.; et al. Engineered Skeletal Muscle Recapitulates Human Muscle Development, Regeneration and Dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 3106–3121. [Google Scholar] [CrossRef]
- Afshar Bakooshli, M.; Lippmann, E.S.; Mulcahy, B.; Iyer, N.; Nguyen, C.T.; Tung, K.; Stewart, B.A.; van den Dorpel, H.; Fuehrmann, T.; Shoichet, M.; et al. A 3D Culture Model of Innervated Human Skeletal Muscle Enables Studies of the Adult Neuromuscular Junction. eLife 2019, 8, e44530. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D Model of Amyotrophic Lateral Sclerosis (ALS) from Human iPS-Derived Muscle Cells and Optogenetic Motor Neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, I.; Seol, Y.-J.; Ko, I.K.; Yoo, J.J.; Atala, A.; Lee, S.J. Neural Cell Integration into 3D Bioprinted Skeletal Muscle Constructs Accelerates Restoration of Muscle Function. Nat. Commun. 2020, 11, 1025. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, R.; Spijkers, X.M.; Pasteuning-Vuhman, S.; Vulto, P.; Pasterkamp, R.J. Neuromuscular Junction-on-a-Chip: ALS Disease Modeling and Read-out Development in Microfluidic Devices. J. Neurochem. 2021, 157, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Torun, T.; Maffioletti, S.M.; Serio, A.; Tedesco, F.S. Bioengineering Human Skeletal Muscle Models: Recent Advances, Current Challenges and Future Perspectives. Exp. Cell Res. 2022, 416, 113133. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Li, X.; Zheng, F.; Liu, H.; Wang, C.; Wang, X.; Liao, Y.; Liu, J.; Meng, K.; Yu, J.; et al. Advances in In Vitro Models of Neuromuscular Junction: Focusing on Organ-on-a-Chip, Organoids, and Biohybrid Robotics. Adv. Mater. Deerfield Beach Fla. 2023, 35, e2211059. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K. Chapter 1—Vertebrate Skeletal Tissues. In Bones and Cartilage, 2nd ed.; Hall, B.K., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 3–16. ISBN 978-0-12-416678-3. [Google Scholar]
- Dalle Carbonare, L.; Innamorati, G.; Valenti, M.T. Transcription Factor Runx2 and Its Application to Bone Tissue Engineering. Stem Cell Rev. Rep. 2012, 8, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Lu, A.; Liang, C. Organoids as Innovative Models for Bone and Joint Diseases. Cells 2023, 12, 1590. [Google Scholar] [CrossRef]
- Bartosh, T.J.; Ylöstalo, J.H.; Mohammadipoor, A.; Bazhanov, N.; Coble, K.; Claypool, K.; Lee, R.H.; Choi, H.; Prockop, D.J. Aggregation of Human Mesenchymal Stromal Cells (MSCs) into 3D Spheroids Enhances Their Antiinflammatory Properties. Proc. Natl. Acad. Sci. USA 2010, 107, 13724–13729. [Google Scholar] [CrossRef]
- Kaushik, G.; Ponnusamy, M.P.; Batra, S.K. Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models. Stem Cells 2018, 36, 1329–1340. [Google Scholar] [CrossRef]
- Merimi, M.; El-Majzoub, R.; Lagneaux, L.; Moussa Agha, D.; Bouhtit, F.; Meuleman, N.; Fahmi, H.; Lewalle, P.; Fayyad-Kazan, M.; Najar, M. The Therapeutic Potential of Mesenchymal Stromal Cells for Regenerative Medicine: Current Knowledge and Future Understandings. Front. Cell Dev. Biol. 2021, 9, 661532. [Google Scholar] [CrossRef]
- Akiva, A.; Melke, J.; Ansari, S.; Liv, N.; van der Meijden, R.; van Erp, M.; Zhao, F.; Stout, M.; Nijhuis, W.H.; de Heus, C.; et al. An Organoid for Woven Bone. Adv. Funct. Mater. 2021, 31, 2010524. [Google Scholar] [CrossRef]
- Kassem, M.; Ankersen, L.; Eriksen, E.F.; Clark, B.F.; Rattan, S.I. Demonstration of Cellular Aging and Senescence in Serially Passaged Long-Term Cultures of Human Trabecular Osteoblasts. Osteoporos. Int. 1997, 7, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Giffin, J.L.; Gaitor, D.; Franz-Odendaal, T.A. The Forgotten Skeletogenic Condensations: A Comparison of Early Skeletal Development Amongst Vertebrates. J. Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Freitas Mendes, L.; Chen, X.; Lesage, R.; Van Hoven, I.; Leysen, E.; Kerckhofs, G.; Bosmans, K.; Chai, Y.C.; Yamashita, A.; et al. Human Pluripotent Stem Cell-Derived Cartilaginous Organoids Promote Scaffold-Free Healing of Critical Size Long Bone Defects. Stem Cell Res. Ther. 2021, 12, 513. [Google Scholar] [CrossRef] [PubMed]
- Frenz, S.; Goek, I.; Buser, M.; Salewskij, K.; Fairley, S.; Conca, R.; Drexler, N.; Jonsson, G.; Thomas, M.; Mizoguchi, Y.; et al. Generation of Human Induced Pluripotent Stem Cell-Derived Bone Marrow Organoids. Blood 2022, 140, 1682–1683. [Google Scholar] [CrossRef]
- O’Connor, S.K.; Katz, D.B.; Oswald, S.J.; Groneck, L.; Guilak, F. Formation of Osteochondral Organoids from Murine Induced Pluripotent Stem Cells. Tissue Eng. Part A 2021, 27, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Freisinger, P.; Ala-Kokko, L.; LeGuellec, D.; Franc, S.; Bouvier, R.; Ritvaniemi, P.; Prockop, D.J.; Bonaventure, J. Mutation in the COL2A1 Gene in a Patient with Hypochondrogenesis. Expression of Mutated COL2A1 Gene Is Accompanied by Expression of Genes for Type I Procollagen in Chondrocytes. J. Biol. Chem. 1994, 269, 13663–13669. [Google Scholar] [CrossRef]
- Liang, G.; Lian, C.; Huang, D.; Gao, W.; Liang, A.; Peng, Y.; Ye, W.; Wu, Z.; Su, P.; Huang, D. Endoplasmic Reticulum Stress-Unfolding Protein Response-Apoptosis Cascade Causes Chondrodysplasia in a Col2a1 p.Gly1170Ser Mutated Mouse Model. PLoS ONE 2014, 9, e86894. [Google Scholar] [CrossRef]
- Lin, S.; Li, K.; Qi, L. Cancer Stem Cells in Brain Tumors: From Origin to Clinical Implications. MedComm 2023, 4, e341. [Google Scholar] [CrossRef]
- Miller, K.; Ostrom, Q.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.; Waite, K.; Jemal, A.; Siegel, R.; et al. Brain and Other Central Nervous System Tumor Statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Andreatta, F.; Beccaceci, G.; Fortuna, N.; Celotti, M.; De Felice, D.; Lorenzoni, M.; Foletto, V.; Genovesi, S.; Rubert, J.; Alaimo, A. The Organoid Era Permits the Development of New Applications to Study Glioblastoma. Cancers 2020, 12, 3303. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, H.; Patel, B.C. Retinoblastoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Seigel, G.M.; Vergara, M.N.; Furey, K.; Shah, D. A Retinal Organoid Model of Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2023, 64, 1317. [Google Scholar]
- Srimongkol, A.; Laosillapacharoen, N.; Saengwimol, D.; Chaitankar, V.; Rojanaporn, D.; Thanomchard, T.; Borwornpinyo, S.; Hongeng, S.; Kaewkhaw, R. Sunitinib Efficacy with Minimal Toxicity in Patient-Derived Retinoblastoma Organoids. J. Exp. Clin. Cancer Res. 2023, 42, 39. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Cell Model | Reference |
|---|---|---|---|
| NEURODEGENERATIVE/DISORDERS | |||
| Autosomal recessive primary microcephaly (MCPH3) OMIM #604804 | CDK5RAP2 | iPSC-derived cerebral organoids | [19] |
| Miller–Dieker Syndrome (MDS; severe form of lissencephaly) OMIM #247200 | deletion of 17p13.3 involving LIS1 and YWHAE genes | patient-derived iPSC cortical organoids; patient-specific forebrain organoids + CRISPR/Cas9 genome editing | [45,52] |
| Leukoencephalopathy with vanishing white matter 1-5 (VWM1-5) OMIM #603896; #620312; #620313; #620314; #620315 | EIF2B1-5 | iPSC-derived brain organoids with and without eIF2B gene mutations | [66] |
| Huntington’s disease (HD) OMIM #143100 | HTT | iPSC-derived neurons and brain organoids, both carrying HTT mutations | [67] |
| Sporadic Creutzfeld–Jacob prion disease (CJD) OMIM# 123400 | PRNP | cerebral organoids infected with CJD prions | [68] |
| Retinitis pigmentosa (RP) OMIM#268000 | several genes | Patient-derived iPSC retinal organoids; iPSC-derived retinal pigment epithelial cells + CRISPR/Cas9 editing | [69,70,71,72,73,74,75] |
| Charcot–Marie–Toot 1A (CMT1A) OMIM #118220 | PMP22 | Patient-derived iPSC PNS organoids | [76] |
| Axonal Charcot–Marie–Tooth (CMT type 2A1; 2E; 2F; and 2L) OMIM #118210; #607684; #606595; #608373 | several genes | iPSC-derived neurons from CMT2 patients + CRISPR/Cas9 editing | [77] |
| Frontotemporal dementia (FTD) with or without Parkinsonism OMIM#600274 | MAPT | iPSC-derived cerebral organoids from patients carrying MAPT mutations; patient-derived iPSC lines with MAPT mutations + CRISPR/Cas9 editing | [78,79,80] |
| NEUROMUSCULAR DISORDERS | |||
| Amyotrophic lateral sclerosis overlapping with frontotemporal dementia (ALS/FTD) OMIM# 612069 | TARDBP (TDP43) | patient-derived iPSC cortical organoids iPSC-derived co-cultured neurons and astrocytes | [81] |
| Amyotrophic lateral sclerosis (ALS) OMIM #105400 | SOD1 | patient-derived iPSC sensorimotor organoids; ALS iPSCs with SOD1 mutations and primary myoblasts co-culture | [82,83] |
| Duchenne muscular dystrophy (DMD) OMIM #310200 | DMD | patient-derived iPSC skeletal muscle organoids + CRISPR/Cas9 editing; 3D co-culture | [84] |
| Limb–girdle muscular dystrophy type R3 (LGMDR3) OMIM# 608099 | SGCA | 3D co-culture | [84] |
| LAMIN A/C (LMNA)-related muscular dystrophies OMIM# 613205 | LMNA | 3D co-culture | [84] |
| Myasthenia gravis (MG) OMIM #254200 | --- | human iPSC-derived neuromuscular organoids containing neuromuscular junctions | [85] |
| Idiopathic inflammatory myopathies | --- | 3D myobundle | [86] |
| SKELETAL DISORDERS | |||
| Juvenile osteochondritis disseccans (JOCD) | --- | patient-derived iPSC lines, 3D chondrogenic modeling | [87] |
| Turner syndrome | X chromosome | patient-derived iPSC lines | [88] |
| Hypochondrogenesis (achondrogenesis type II) OMIM #200610 | COL2A1 | cartilage produced from patient-derived iPSCs | [89,90] |
| Osteogenesis imperfecta (OI) OMIM#120150 | COL1A1 | patient-derived iPSC lines | [91,92,93] |
| Cleidocranial dysplasia (CCD) OMIM # 119600 | RUNX2 | patient-derived iPSC lines + CRISPR/Cas9 editing | [94] |
| BRAIN TUMORS | |||
| Glioblastoma (GB) | HRAS TP53 | patient-derived glioblastoma organoids; human-ESC-derived glioma + CRISPR/Cas9 editing; glioblastoma spheroid invasion of a brain organoid (GLICO) | [51,95,96] |
| Medulloblastoma (MB) | Otx2/c-MYC | genetically engineered iPSC cerebellar organoids | [97] |
| Meningioma | --- | patient-derived meningioma organoids | [98,99,100] |
| Retinoblastoma | RB1 | human-ESC-derived and patient-derived retinal organoids + CRISPR/Cas9 editing | [101,102,103,104,105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bombieri, C.; Corsi, A.; Trabetti, E.; Ruggiero, A.; Marchetto, G.; Vattemi, G.; Valenti, M.T.; Zipeto, D.; Romanelli, M.G. Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids. Int. J. Mol. Sci. 2024, 25, 1014. https://doi.org/10.3390/ijms25021014
Bombieri C, Corsi A, Trabetti E, Ruggiero A, Marchetto G, Vattemi G, Valenti MT, Zipeto D, Romanelli MG. Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids. International Journal of Molecular Sciences. 2024; 25(2):1014. https://doi.org/10.3390/ijms25021014
Chicago/Turabian StyleBombieri, Cristina, Andrea Corsi, Elisabetta Trabetti, Alessandra Ruggiero, Giulia Marchetto, Gaetano Vattemi, Maria Teresa Valenti, Donato Zipeto, and Maria Grazia Romanelli. 2024. "Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids" International Journal of Molecular Sciences 25, no. 2: 1014. https://doi.org/10.3390/ijms25021014
APA StyleBombieri, C., Corsi, A., Trabetti, E., Ruggiero, A., Marchetto, G., Vattemi, G., Valenti, M. T., Zipeto, D., & Romanelli, M. G. (2024). Advanced Cellular Models for Rare Disease Study: Exploring Neural, Muscle and Skeletal Organoids. International Journal of Molecular Sciences, 25(2), 1014. https://doi.org/10.3390/ijms25021014