Quantitative Characterization of the Impact of Protein–Protein Interactions on Ligand–Protein Binding: A Multi-Chain Dynamics Perturbation Analysis Method


Abstract
1. Introduction
2. Results and Discussion
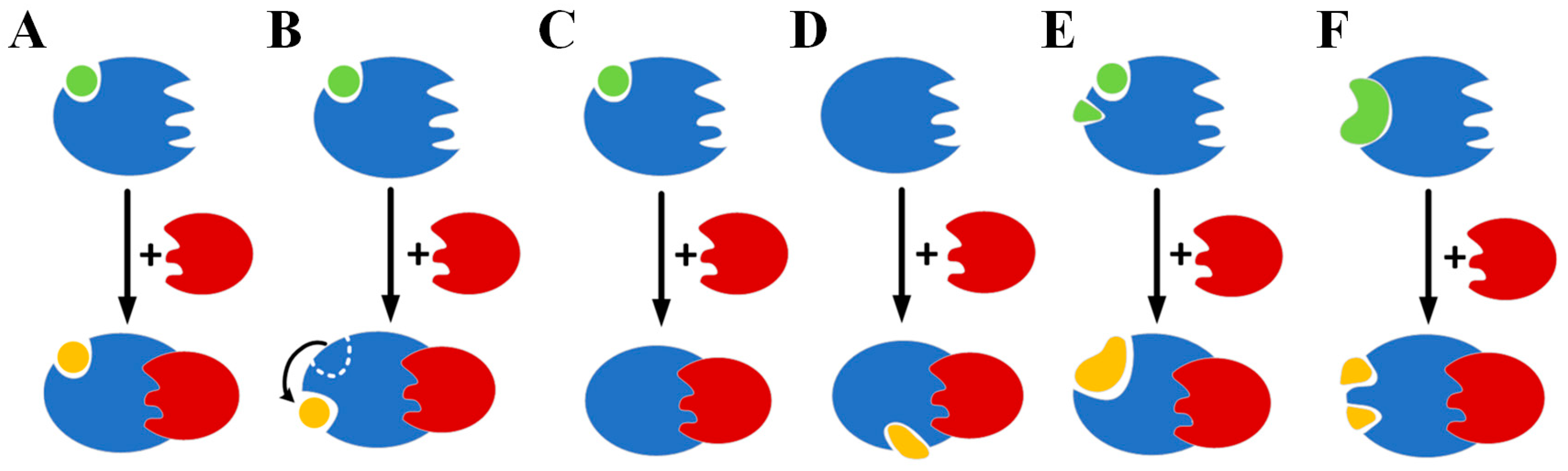
2.1. Protein–Protein Interactions Have Diverse Effects on the Binding of Ligands to Proteins
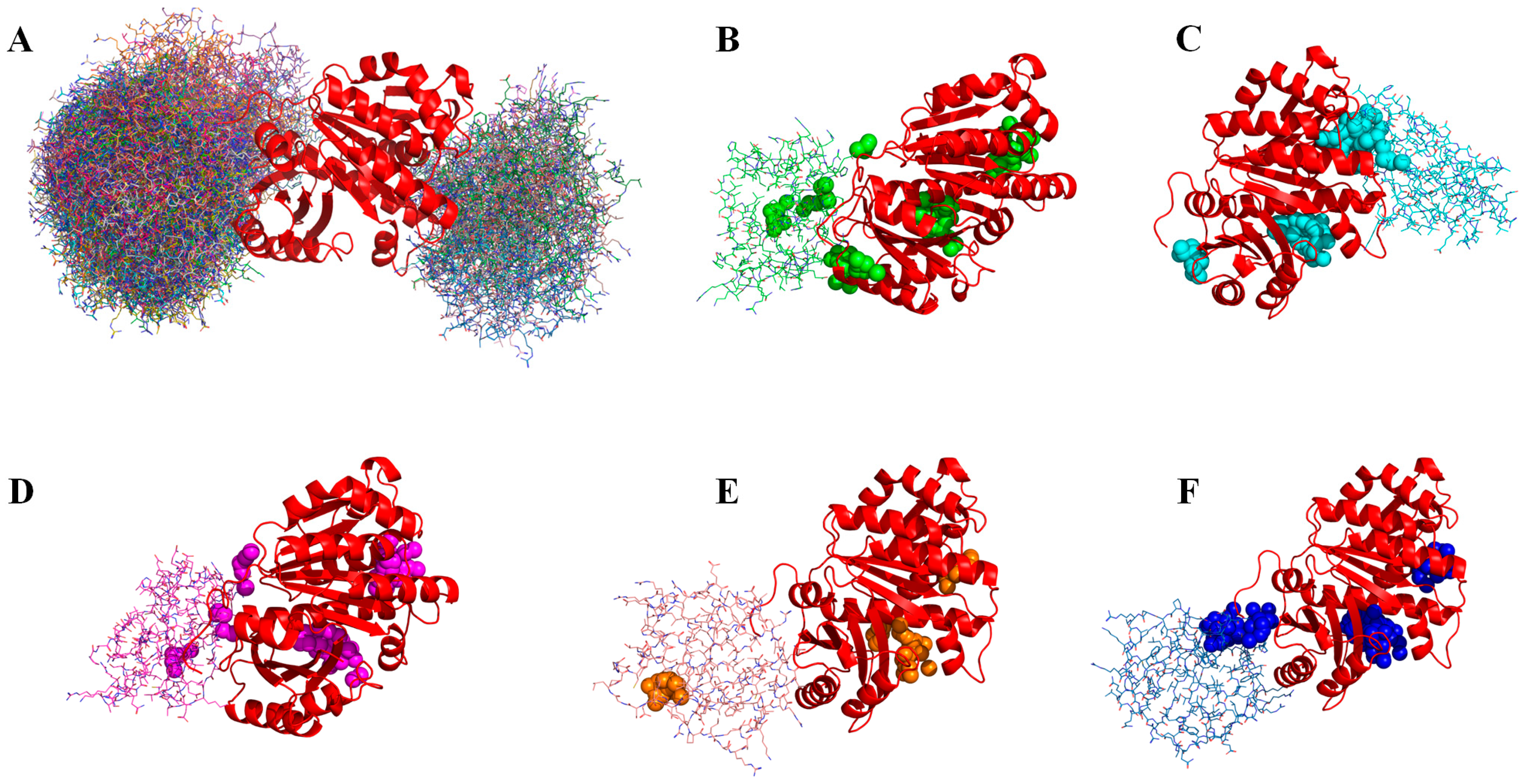
2.2. Evaluating the Prediction of mcDPA for Small-Molecule Binding Pockets of Protein–Protein Complexes from the Benchmark
2.3. Comparison with Binding Pockets of FDA-Approved Small-Molecule Drugs Targeting Protein–Protein Complexes
2.4. Modeling the Effect of Protein–Protein Interactions on the Prediction of Ligand-Binding Regions
2.5. The Effect of Protein–Protein Docking Orientation in the Predicted Binding Regions
3. Materials and Methods
3.1. The Test Dataset
3.2. Implementing mcDPA
3.3. Structural Characterization of the Effects of PPIs on LPIs
3.4. Computational Characterization of the Effect of PPI on Ligand–Receptor Binding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nero, T.L.; Morton, C.J.; Holien, J.K.; Wielens, J.; Parker, M.W. Oncogenic protein interfaces: Small molecules, big challenges. Nat. Rev. Cancer 2014, 14, 248–262. [Google Scholar] [CrossRef]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef]
- Dequeker, C.; Laine, E.; Carbone, A. Decrypting protein surfaces by combining evolution, geometry, and molecular docking. Proteins 2019, 87, 952–965. [Google Scholar] [CrossRef]
- Wang, T.; Yang, N.; Liang, C.; Xu, H.; An, Y.; Xiao, S.; Zheng, M.; Liu, L.; Wang, G.; Nie, L. Detecting Protein-Protein Interaction Based on Protein Fragment Complementation Assay. Curr. Protein Pept. Sci. 2020, 21, 598–610. [Google Scholar] [CrossRef]
- Skolnick, J.; Zhou, H. Implications of the Essential Role of Small Molecule Ligand Binding Pockets in Protein-Protein Interactions. J. Phys. Chem. B 2022, 126, 6853–6867. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-S.; Yang, G.-J.; Wang, W.; Leung, C.-H.; Ma, D.-L. The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 2020, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef]
- Tonddast-Navaei, S.; Skolnick, J. Are protein-protein interfaces special regions on a protein’s surface? J. Chem. Phys. 2015, 143, 243149. [Google Scholar] [CrossRef]
- Namboodiri, H.V.; Bukhtiyarova, M.; Ramcharan, J.; Karpusas, M.; Lee, Y.; Springman, E.B. Analysis of imatinib and sorafenib binding to p38alpha compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry 2010, 49, 3611–3618. [Google Scholar] [CrossRef]
- Simard, J.R.; Getlik, M.; Grütter, C.; Pawar, V.; Wulfert, S.; Rabiller, M.; Rauh, D. Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J. Am. Chem. Soc. 2009, 131, 13286–13296. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. A comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput. Biol. 2013, 9, e1003302. [Google Scholar] [CrossRef]
- Bessman, N.J.; Bagchi, A.; Ferguson, K.M.; Lemmon, M.A. Complex relationship between ligand binding and dimerization in the epidermal growth factor receptor. Cell Rep. 2014, 9, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Skolnick, J. The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc. Natl. Acad. Sci. USA 2012, 109, 3784–3789. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Morris, R.J.; Laskowski, R.A.; Thornton, J.M. Shape variation in protein binding pockets and their ligands. J. Mol. Biol. 2007, 368, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Morris, R.J.; Laskowski, R.A.; Favia, A.D.; Thornton, J.M. On the diversity of physicochemical environments experienced by identical ligands in binding pockets of unrelated proteins. Proteins 2010, 78, 1120–1136. [Google Scholar] [CrossRef]
- Kokh, D.B.; Czodrowski, P.; Rippmann, F.; Wade, R.C. Perturbation Approaches for Exploring Protein Binding Site Flexibility to Predict Transient Binding Pockets. J. Chem. Theory Comput. 2016, 12, 4100–4113. [Google Scholar] [CrossRef]
- Gu, L.; Li, B.; Ming, D. A multilayer dynamic perturbation analysis method for predicting ligand–protein interactions. BMC Bioinform. 2022, 23, 456. [Google Scholar] [CrossRef]
- Oliva, M.A.; Trambaiolo, D.; Löwe, J. Structural insights into the conformational variability of FtsZ. J. Mol. Biol. 2007, 373, 1229–1242. [Google Scholar] [CrossRef]
- Cordell, S.C.; Robinson, E.J.; Lowe, J. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc. Natl. Acad. Sci. USA 2003, 100, 7889–7894. [Google Scholar] [CrossRef]
- Hirshberg, M.; Stockley, R.W.; Dodson, G.; Webb, M.R. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 1997, 4, 147. [Google Scholar] [CrossRef]
- Würtele, M.; Wolf, E.; Pederson, K.J.; Buchwald, G.; Ahmadian, M.R.; Barbieri, J.T.; Wittinghofer, A. How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat. Struct. Biol. 2001, 8, 23–26. [Google Scholar] [PubMed]
- Bubb, M.R.; Govindasamy, L.; Yarmola, E.G.; Vorobiev, S.M.; Almo, S.C.; Somasundaram, T.; Chapman, M.S.; Agbandje-McKenna, M.; McKenna, R. Polylysine induces an antiparallel actin dimer that nucleates filament assembly: Crystal structure at 3.5-Å resolution. J. Biol. Chem. 2002, 277, 20999–21006. [Google Scholar] [CrossRef]
- Otterbein, L.R.; Cosio, C.; Graceffa, P.; Dominguez, R. Crystal structures of the vitamin D-binding protein and its complex with actin: Structural basis of the actin-scavenger system. Proc. Natl. Acad. Sci. USA 2002, 99, 8003–8008. [Google Scholar] [CrossRef] [PubMed]
- Lennon, B.W.; Williams, C.H., Jr.; Ludwig, M.L. Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 1999, 8, 2366–2379. [Google Scholar] [CrossRef]
- Lennon, B.W.; Williams, C.H., Jr.; Ludwig, M.L. Twists in catalysis: Alternating conformations of Escherichia coli thioredoxin reductase. Science 2000, 289, 1190–1194. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Casasnovas, J.M.; Larvie, M.; Stehle, T. Crystal structure of two CD46 domains reveals an extended measles virus-binding surface. Embo J. 1999, 18, 2911–2922. [Google Scholar] [CrossRef]
- Cupelli, K.; Müller, S.; Persson, B.D.; Jost, M.; Arnberg, N.; Stehle, T. Structure of adenovirus type 21 knob in complex with CD46 reveals key differences in receptor contacts among species B adenoviruses. J. Virol. 2010, 84, 3189–3200. [Google Scholar] [CrossRef]
- Vreven, T.; Moal, I.H.; Vangone, A.; Pierce, B.G.; Kastritis, P.L.; Torchala, M.; Chaleil, R.; Jiménez-García, B.; Bates, P.A.; Fernandez-Recio, J. Updates to the integrated protein–protein interaction benchmarks: Docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol. 2015, 427, 3031–3041. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Marchand, J.-R.; Pirard, B.; Ertl, P.; Sirockin, F. CAVIAR: A method for automatic cavity detection, description and decomposition into subcavities. J. Comput.-Aided Mol. Des. 2021, 35, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein–ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Ming, D.; Cohn, J.D.; Wall, M.E. Fast dynamics perturbation analysis for prediction of protein functional sites. BMC Struct. Biol. 2008, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Ming, D.; Wall, M.E. Allostery in a coarse-grained model of protein dynamics. Phys. Rev. Lett. 2005, 95, 198103. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cat. | A-L1 | L1 | B-A-L2 | L2 | ARMSD | L1 Prediction | L2 Prediction | ||
|---|---|---|---|---|---|---|---|---|---|
| Precision | Recall | Precision | Recall | ||||||
| OG | 1QG4(A) | GDP | 1A2K(D,A) | GDP | 0.76 | 0.00 | 0.00 | 0.67 | 0.36 |
| OG | 1AZT(A) | GSP | 1AZS(C,B) | GSP | 0.52 | 0.27 | 0.60 | 0.00 | 0.00 |
| OG | 1MH1(A) | GNP | 1E96(A,B) | GTP | 1.18 | 0.43 | 0.79 | 0.26 | 0.45 |
| OG | 1TND(A) | GSP | 1FQJ(A,B) | GDP | 0.45 @ | 0.00 | 0.00 | 0.00 | 0.00 |
| OG | 1GIA(A) | GSP | 1GP2(A,B) | GDP | 1.56 | 0.27 | 0.12 | 0.13 | 0.54 |
| OG | 1A4R(A) | GDP | 1GRN(A,B) | GDP | 1.50 | 0.60 | 0.79 | 0.13 | 0.45 |
| OG | 1MH1(A) | GNP | 1HE1(C,A) | GDP | 1.02 | 0.43 | 0.79 | 0.31 | 0.94 |
| OG | 821P(A) | GNP | 1HE8(B,A) | GNP | 0.97 | 0.57 | 0.70 | 0.00 | 0.00 |
| OG | 1MH1(A) | GNP | 1I4D(D,A) | GDP | 1.11 | 0.43 | 0.79 | 0.67 | 0.38 |
| OG | 1QG4(A) | GDP | 1IBR(A,B) | GNP | 1.47 | 0.00 | 0.00 | 0.00 | 0.00 |
| OG | 1O3Y(A) | GTP | 1J2J(A,B) | GTP | 0.37 | 0.82 | 0.82 | 0.67 | 1.00 |
| OG | 1RRP(A) | GNP | 1K5D(A,B) | GNP | 1.32 | 0.00 | 0.00 | 0.00 | 0.00 |
| OG | 5P21(A) | GNP | 1LFD(B,A) | GNP | 0.99 | 0.52 | 0.88 | 1.00 | 0.33 |
| OG | 1HUR(A) | GDP | 1R8S(A,E) | GDP | 3.63 | 0.00 | 0.00 | 0.33 | 1.00 |
| OG | 6Q21(A) | GCP | 1WQ1(R,G) | GDP | 1.41 | 1.00 | 0.48 | 0.00 | 0.00 |
| OG | 2BME(A) | GNP | 1Z0K(A,B) | GTP | 0.26 | 0.21 | 0.75 | 0.33 | 0.18 |
| OG | 1MH1(A) | GNP | 2FJU(A,B) | GSP | 0.75 | 0.43 | 0.79 | 0.27 | 0.36 |
| OG | 1Z06(A) | GNP | 2G77(B,A) | GDP | 1.72 | 0.86 | 0.94 | 0.29 | 0.21 |
| OG | 1GFI(A) | GDP | 2GTP(A,D) | GDP | 1.52 | 0.11 | 0.12 | 0.00 | 0.00 |
| OG | 1MH1(A) | GNP | 2H7V(A,C) | GDP | 1.54 | 0.43 | 0.79 | 0.41 | 0.79 |
| OG | 1G16(A) | GDP | 3CPH(A,G) | GDP | 0.53 | 0.70 | 0.69 | 0.00 | 0.00 |
| OX | 1IJJ(A) | ATP | 1ATN(A,D) | ATP | 2.18 | 0.80 | 0.31 | 0.00 | 0.00 |
| OX | 3DNI(A) | BMA | 1ATN(D,A) | BMA | 1.63 | 0.00 | 0.00 | 0.00 | 0.00 |
| OX | 1QRQ(A) | NDP | 1EXB(A,E) | NDP | 0.49 | 0.27 | 0.54 | 0.44 | 0.13 |
| OX | 1IJJ(A) | ATP | 1H1V(A,G) | ATP | 1.73 | 0.80 | 0.31 | 0.00 | 0.00 |
| OX | 1KUY(A) | COT | 1IB1(E,A) | COT | 0.95 | 0.68 | 0.89 | 0.83 | 0.57 |
| OX | 1IJJ(A) | ATP | 1KXP(A,D) | ATP | 1.79 | 0.80 | 0.31 | 0.00 | 0.00 |
| OX | 1IAM(A) | NAG | 1MQ8(A,B) | NAG | 1.27 | 0.00 | 0.00 | 0.00 | 0.00 |
| OX | 3MIN(A) | HCA | 1N2C(A,B) | HCA | 0.62 | 0.75 | 0.97 | 0.71 | 0.41 |
| OX | 2VAW(A) | GDP | 1OFU(A,X) | GDP | 0.86 | 0.76 | 0.96 | 0.78 | 0.86 |
| OX | 2FXU(A) | ATP | 1Y64(A,B) | ATP | 1.61 | 0.75 | 0.4 | 0.00 | 0.00 |
| OX | 1IJJ(A) | ATP | 2BTF(A,P) | ATP | 1.48 | 0.80 | 0.31 | 0.00 | 0.00 |
| OX | 1NG1(A) | GDP | 2J7P(A,D) | GNP | 2.37 | 0.35 | 0.69 | 0.50 | 0.58 |
| OX | 2IYL(D) | GDP | 2J7P(D,A) | GNP | 2.42 | 0.00 | 0.00 | 0.50 | 0.52 |
| OX | 3BIX(A) | NAG | 3BIW(A,E) | NAG | 1.92 | 0.14 | 0.75 | 0.00 | 0.00 |
| OX | 1IJJ(A) | ATP | 3DAW(A,B) | ATP | 1.64 | 0.80 | 0.31 | 0.00 | 0.00 |
| OX | 3ODQ(A) | HEM | 3SZK(D,F) | HEM | 1.03 | 0.77 | 1.0 | 0.83 | 0.91 |
| ES | 1E1N(A) | FAD | 1E6E(A,B) | FAD | 0.62 | 0.80 | 0.49 | 0.00 | 0.00 |
| ES | 1CL0(A) | FAD | 1F6M(A,C) | FAD | 4.66 | 0.33 | 0.09 | 0.75 | 0.47 |
| ES | 1B39(A) | ATP | 1FQ1(B,A) | ATP | 3.24 | 0.37 | 0.72 | 0.18 | 0.94 |
| ES | 1XK9(A) | P34 | 1ZM4(B,A) | TAD | 10.27 § | 0.43 | 0.95 | 1.00 | 0.67 |
| ES | 1U90(A) | GDP | 2A9K(A,B) | GDP | 1.08 | 0.00 | 0.00 | 0.01 | 0.07 |
| ES | 1J54(A) | TMP | 2IDO(A,B) | TMP | 1.19 | 0.59 | 0.96 | 0.59 | 0.74 |
| ES | 1CCP(A) | HEM | 2PCC(A,B) | HEM | 0.25 | 0.88 | 0.93 | 1.00 | 0.72 |
| ES | 1YCC(A) | HEM | 2PCC(B,A) | HEM | 0.31 | 0.85 | 0.78 | 1.00 | 0.72 |
| ES | 1GIQ(A) | NAD | 4H03(A,B) | NAD | 2.15 | 0.92 | 0.89 | 0.61 | 0.82 |
| ES | 1IJJ(A) | ATP | 4H03(B,A) | ATP | 0.64 | 0.80 | 0.31 | 0.00 | 0.00 |
| ER | 1JMJ(A) | NAG | 1JMO(A,H) | NDG | 5.29 | 0.00 | 0.00 | 0.00 | 0.00 |
| ER | 2CN0(H) | F25 | 1JMO(H,A) | NAG | 24.32 ‡ | 0.82 | 0.95 | 0.00 | 0.00 |
| ER | 3C13(A) | EMO | 1JWH(A,C) | ANP | 6.07 § | 0.43 | 1.00 | 0.69 | 0.87 |
| ER | 1V8Z(A) | PLP | 1WDW(B,A) | PLP | 0.33 | 0.00 | 0.00 | 0.00 | 0.00 |
| ER | 1E3T(A) | NAP | 2OOR(C,A) | TXP | 7.39 | 0.13 | 0.12 | 0.00 | 0.00 |
| ER | 1L7E(A) | NAI | 2OOR(A,C) | NAD | 1.18 | 0.28 | 0.95 | 0.26 | 1.00 |
| ER | 2YVF(A) | FAD | 2YVJ(A,B) | FAD | 1.31 | 0.83 | 0.25 | 1.00 | 0.19 |
| AA | 1HRC(A) | HEM | 1WEJ(F,H) | HEM | 3.42 | 0.84 | 0.95 | 1.00 | 0.69 |
| AA | 1YWH(A) | NAG | 2FD6(U,A) | NAG | 3.90 | 0.00 | 0.00 | 0.00 | 0.00 |
| AA | 3TGT(A) | NAG | 3SE8(G,H) | NAG | 1.09 | 0.00 | 0.00 | 0.12 | 0.11 |
| AA | 3TGT(A) | NAG | 3U7Y(G,H) | NAG | 0.71 | 0.00 | 0.00 | 0.00 | 0.00 |
| AA | 4GT7(A) | NAG | 5HYS(G,I) | NAG | 1.33 | 0.14 | 0.64 | 0.00 | 0.00 |
| OR | 1JX6(A) | AI2 | 1ZHH(A,B) | NHE | 19.90 ‡ | 0.47 | 1.00 | 0.50 | 0.15 |
| OR | 1R42(A) | NAG | 2AJF(A,E) | NAG | 1.53 | 0.00 | 0.00 | 0.29 | 0.29 |
| OR | 1YWH(A) | NAG | 2I9B(E,A) | NAG | 5.26 | 0.00 | 0.00 | 0.00 | 0.00 |
| OR | 1CKL(A) | NAG | 3L89(M,A) | NAG | 4.85 | 0.00 | 0.00 | 0.00 | 0.00 |
| Activity | Drug | Target PPIs | Target PDB Code | Prediction | |
|---|---|---|---|---|---|
| Precision | Recall | ||||
| Inhibitor | Navitoclax | BCL2/BAX | 4LVT | 0.35 | 0.81 |
| Inhibitor | Venetoclax | BCL2/BAX | 6O0K | 0.46 | 1.00 |
| Inhibitor | ABT-737 | BCLXUBAK | 2YXJ | 0.41 | 0.89 |
| Inhibitor | Maraviroc | CCR5/gp120 | 4MBS | 1.00 | 0.48 |
| Inhibitor | Tirofiban | FGG/ITGA2B/ITGB3 | 2VDM | 0.34 | 0.80 |
| Inhibitor | BIO8898 | CD40−CD40L | 3LKJ | 1.00 | 0.36 |
| Inhibitor | Tacrolimus | FKBP12/CNA/CNB | 1BKF | 0.81 | 1.00 |
| Inhibitor | Sirolimus | FKBP12/MTOR | 1FAP | 0.88 | 0.98 |
| Inhibitor | Pevonedistat | NEDD8/APPBPI/UBA3 | 3GZN | 0.11 | 0.42 |
| Inhibitor | AMG-232 | P53/MDM2 | 4OAS | 1.00 | 0.95 |
| Inhibitor | CGM097 | P53/MDM2 | 4ZYF | 0.70 | 0.96 |
| Inhibitor | Nutin-2 | P53/MDM2 | 1RV1 | 0.83 | 0.97 |
| Inhibitor | RO-5045337 | P53/MDM2 | 4IPF | 0.76 | 1.00 |
| Inhibitor | SAR-405838 | P53/MDM2 | 5TRF | 0.92 | 0.88 |
| Inhibitor | honokiol | RXR/TIF2 | 4OC7 | 0.00 | 0.00 |
| Stabilizer | Epibestatin | 14-3-3/PMA2 | 3M50 | 0.41 | 0.36 |
| Stabilizer | Pyrrolidone1 | 14-3-3/PMA2 | 3M51 | 0.59 | 0.43 |
| Stabilizer | BMS-202 | PD-1L/PD-1L | 5J89 | 0.90 | 0.73 |
| Stabilizer | BMS-8 | PD-1L/PD-1L | 5J8O | 0.97 | 0.75 |
| Stabilizer | Compound 3 | 14-3-3/ChREBP | 6YGJ | 1.00 | 0.52 |
| Stabilizer | Fusicoccin | 14-3-3/H+-ATPase | 2O98 | 0.49 | 0.53 |
| Stabilizer | Lenalidomide | CK1α/CRL4 | 5FQD | 0.00 | 0.00 |
| Stabilizer | CC0651 | Cdc34/Ubiquitin 1α | 4MDK | 0.86 | 0.49 |
| Stabilizer | GW6471 | PPARα/SMRT | 1KKQ | 0.67 | 0.80 |
| Stabilizer | 2x RO-2443 | MDM4/MDM4 | 3U15 | 1.00 | 0.49 |
| Stabilizer | 2x RO-2443 | MDM2/MDM2 | 3VBG | 1.00 | 0.49 |
| Stabilizer | 2x Tafamidis | TTR/TTR | 3TCT | 0.00 | 0.00 |
| Stabilizer | 4xTrifluoperazine | S100A4/S100A4 | 3KO0(A,B) | 0.33 | 0.11 |
| Stabilizer | (R,R)-2a | iGluR2/iGluR2 | 3BBR | 0.00 | 0.00 |
| Stabilizer | Coumarin | lambda-6A/lambda-6A | 6MG5 | 0.05 | 0.24 |
| Stabilizer | 2x NS309 | CaM/CaMBD2-a | 4J9Z | 1.00 | 0.64 |
| Stabilizer | Inositol tetraphosphate | HDAC3/SMRT | 4A69 | 0.85 | 0.92 |
| Stabilizer | FK506 | FKBP12/calcineurin | 1TCO | 0.67 | 0.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Li, H.; Su, T.; Ming, D. Quantitative Characterization of the Impact of Protein–Protein Interactions on Ligand–Protein Binding: A Multi-Chain Dynamics Perturbation Analysis Method. Int. J. Mol. Sci. 2024, 25, 9172. https://doi.org/10.3390/ijms25179172
Li L, Li H, Su T, Ming D. Quantitative Characterization of the Impact of Protein–Protein Interactions on Ligand–Protein Binding: A Multi-Chain Dynamics Perturbation Analysis Method. International Journal of Molecular Sciences. 2024; 25(17):9172. https://doi.org/10.3390/ijms25179172
Chicago/Turabian StyleLi, Lu, Hao Li, Ting Su, and Dengming Ming. 2024. "Quantitative Characterization of the Impact of Protein–Protein Interactions on Ligand–Protein Binding: A Multi-Chain Dynamics Perturbation Analysis Method" International Journal of Molecular Sciences 25, no. 17: 9172. https://doi.org/10.3390/ijms25179172
APA StyleLi, L., Li, H., Su, T., & Ming, D. (2024). Quantitative Characterization of the Impact of Protein–Protein Interactions on Ligand–Protein Binding: A Multi-Chain Dynamics Perturbation Analysis Method. International Journal of Molecular Sciences, 25(17), 9172. https://doi.org/10.3390/ijms25179172