
LncRNAs in the Dlk1-Dio3 Domain Are Essential for Mid-Embryonic Heart Development

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
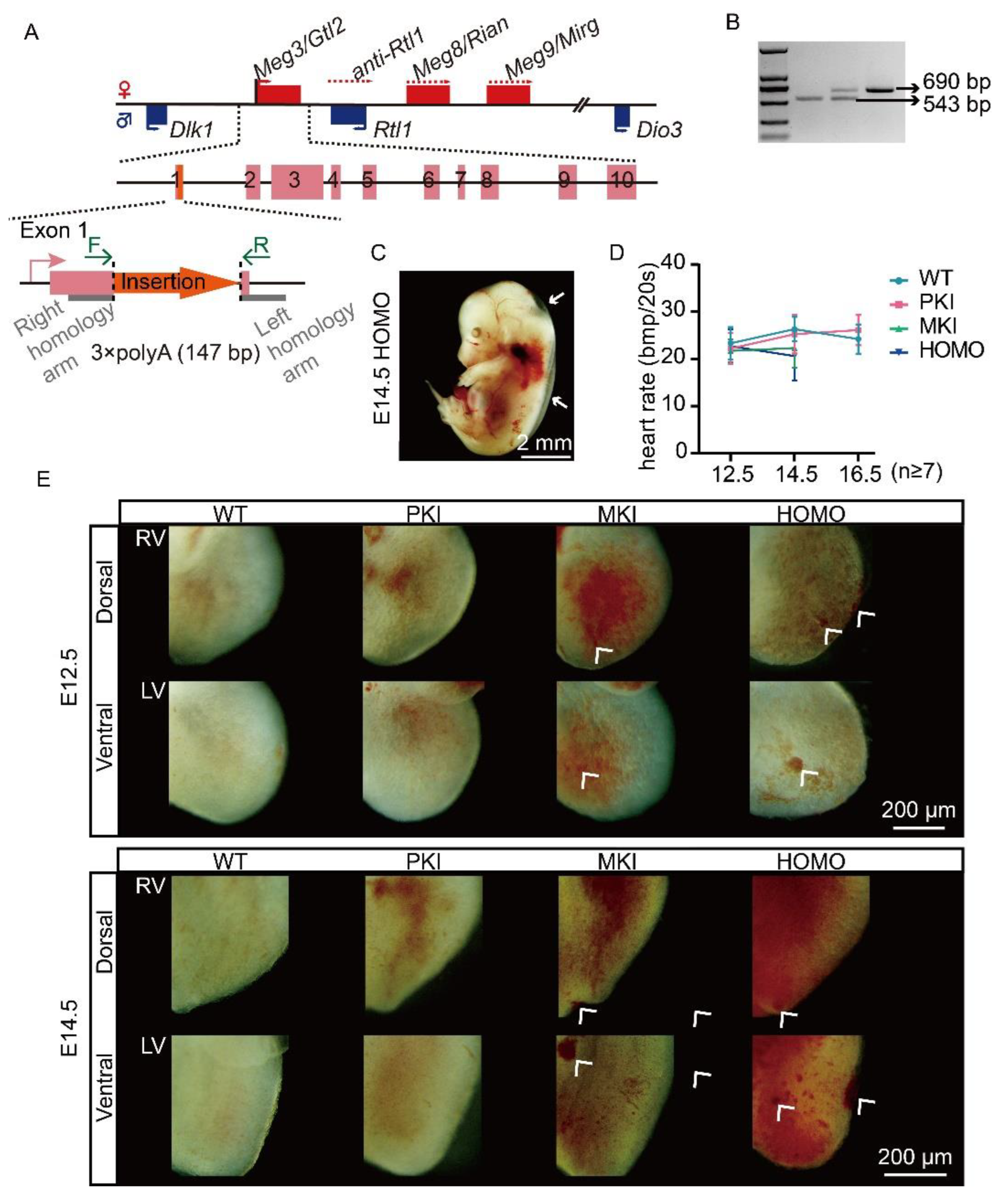
2.1. Construction of a Terminating Sequence Insertion Model
2.2. Dlk1-Dio3 Domain lncRNA Is Required for Normal Embryo Growth and Heart Development
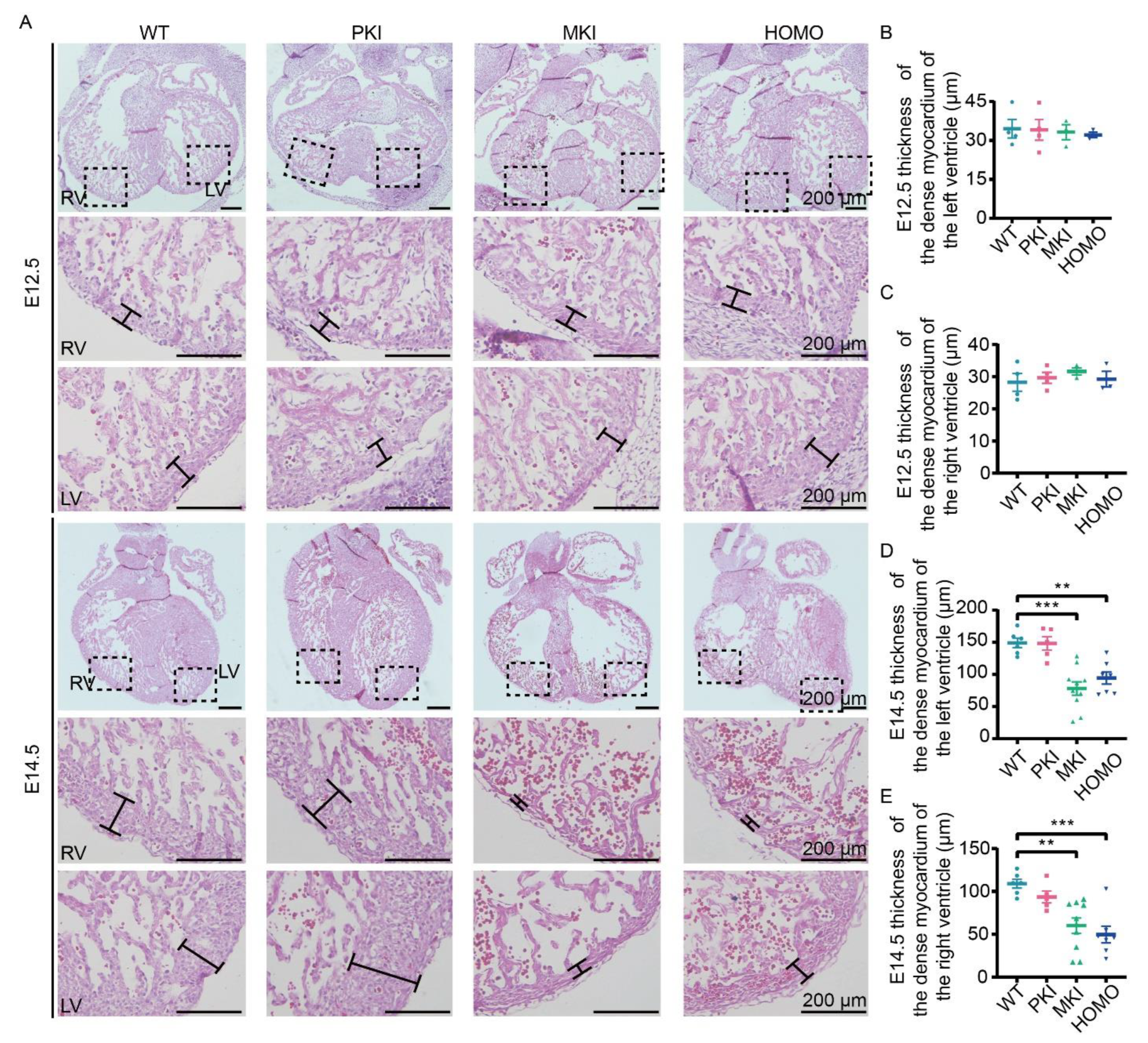
2.3. Dlk1-Dio3 Domain lncRNA Expression Deficiency Impairs Ventricular Wall Development
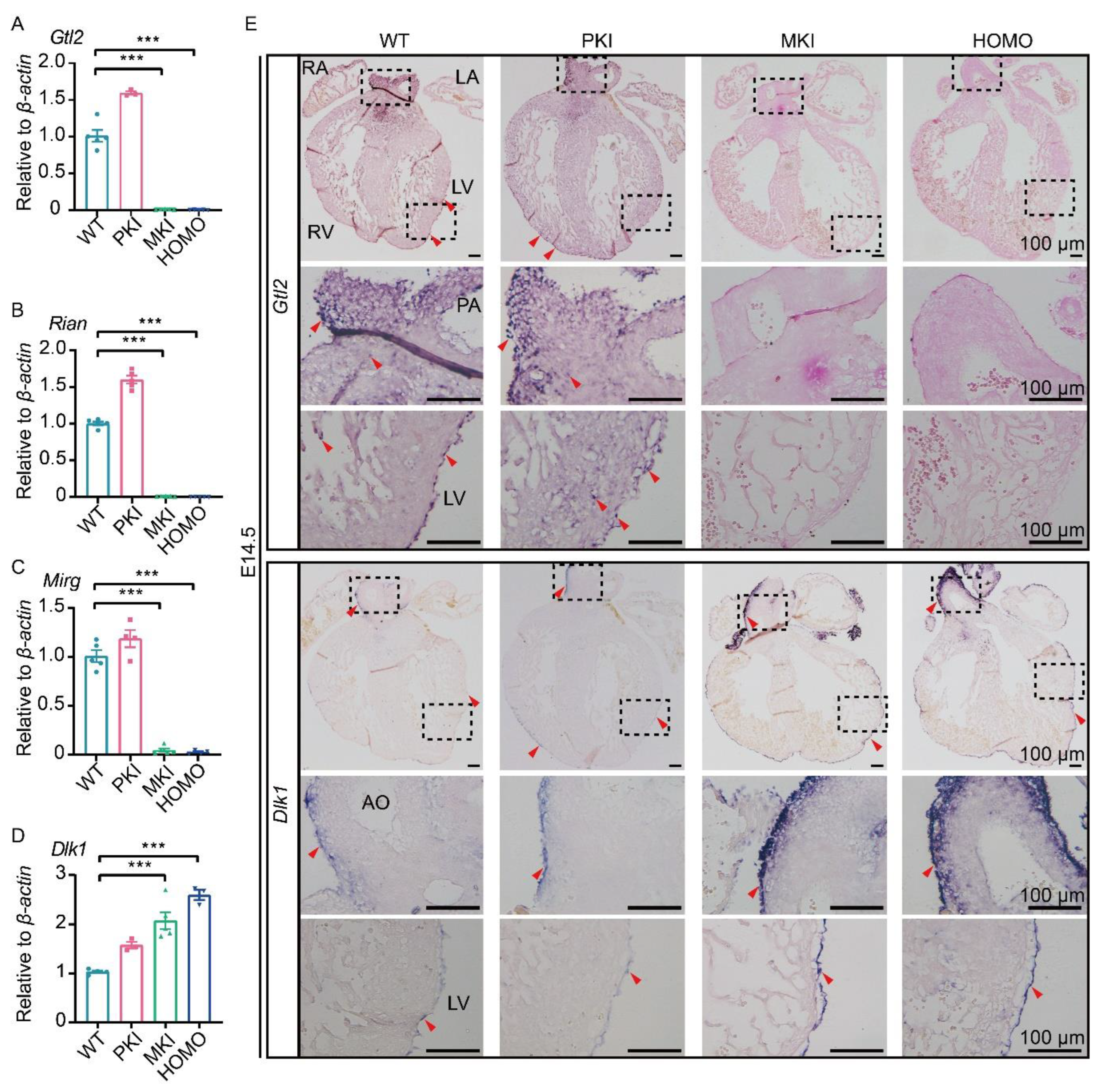
2.4. Expression of Genes within the Dlk1-Dio3 Domain in the Embryonic Heart
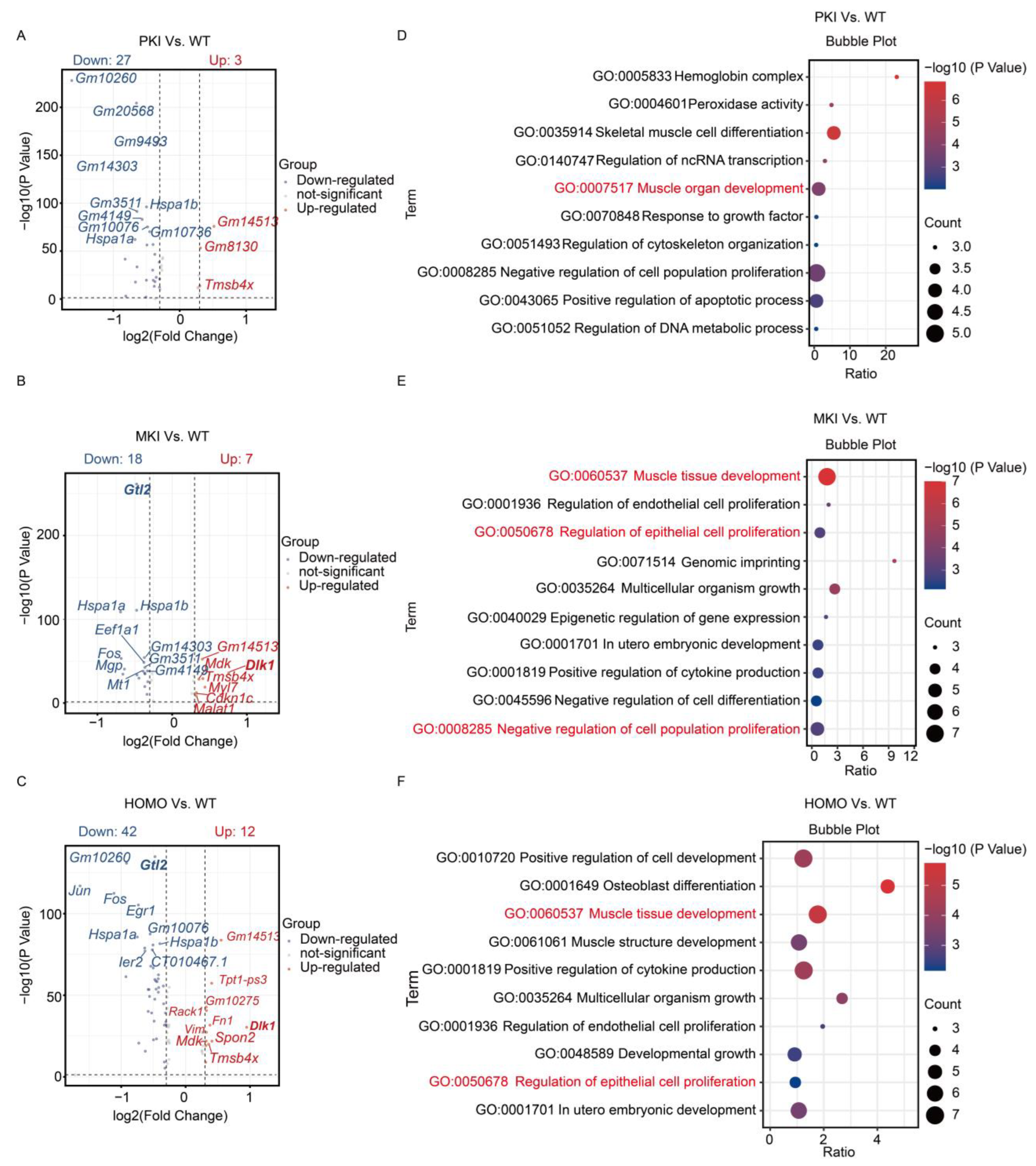
2.5. ScRNA-Seq Analysis and Validation of the E14.5 Heart
3. Discussion
4. Materials and Methods
4.1. Mouse Breeding, Timed Pregnancy Mating, and Genotyping
4.2. Histological Analysis and Quantification of Ventricular Wall Thickness
4.3. RNA ISH with Digoxigenin Labeling
4.4. RNA Isolation and qRT-PCR
4.5. scRNA-Seq Analysis
4.6. Isolation and Culture of Mice Epis
4.7. IF and IHC
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, M.C.; Kadow, Z.A.; Long, H.; Morikawa, Y.; Martin, T.J.; Birks, E.J.; Campbell, K.S.; Nerbonne, J.; Lavine, K.; Wadhwa, L.; et al. Integrated multi-omic characterization of congenital heart disease. Nature 2022, 608, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Shewale, B.; Dubois, N. Of form and function: Early cardiac morphogenesis across classical and emerging model systems. Semin. Cell Dev. Biol. 2021, 118, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bonet, F.; Añez, S.B.; Inácio, J.M.; Futschik, M.E.; Belo, J.A. CCBE1 Is Essential for Epicardial Function during Myocardium Development. Int. J. Mol. Sci. 2022, 23, 12642. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Deyett, A.; Ilmer, T.; Haendeler, S.; Torres Caballero, A.; Novatchkova, M.; Netzer, M.A.; Ceci Ginistrelli, L.; Mancheno Juncosa, E.; Bhattacharya, T.; et al. Multi-chamber cardioids unravel human heart development and cardiac defects. Cell 2023, 186, 5587–5605.e27. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Bezprozvannaya, S.; Hao, T.; Elnwasany, A.; Szweda, L.I.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Transcription factor NFYa controls cardiomyocyte metabolism and proliferation during mouse fetal heart development. Dev. Cell 2023, 58, 2867–2880.e7. [Google Scholar] [CrossRef]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef]
- Ding, S.; Zhang, X.; Qiu, H.; Wo, J.; Zhang, F.; Na, J. Non-cardiomyocytes in the heart in embryo development, health, and disease, a single-cell perspective. Front. Cell Dev. Biol. 2022, 10, 873264. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Bartelings, M.M.; Goumans, M.J.; DeRuiter, M.C.; Poelmann, R.E. The arterial and cardiac epicardium in development, disease and repair. Differ. Res. Biol. Divers. 2012, 84, 41–53. [Google Scholar] [CrossRef]
- Cao, J.; Poss, K.D. The epicardium as a hub for heart regeneration. Nat. Rev. Cardiol. 2018, 15, 631–647. [Google Scholar] [CrossRef]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Junghof, J.; Kogure, Y.; Yu, T.; Verdugo-Sivianes, E.M.; Narita, M.; Lucena-Cacace, A.; Yoshida, Y. CDH18 is a fetal epicardial biomarker regulating differentiation towards vascular smooth muscle cells. NPJ Regen. Med. 2022, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium During Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Masters, M.; Riley, P.R. The epicardium signals the way towards heart regeneration. Stem Cell Res. 2014, 13, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, F.C.; Charalambous, M. Genomic imprinting, growth and maternal-fetal interactions. J. Exp. Biol. 2018, 221, jeb164517. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Tello, J.; Yong, H.E.J.; Sandovici, I.; Dowsett, G.K.C.; Christoforou, E.R.; Salazar-Petres, E.; Boyland, R.; Napso, T.; Yeo, G.S.H.; Lam, B.Y.H.; et al. Fetal manipulation of maternal metabolism is a critical function of the imprinted Igf2 gene. Cell Metab. 2023, 35, 1195–1208.e6. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. TIG 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lin, C.; Woodfin, A.R.; Bartom, E.T.; Gao, X.; Smith, E.R.; Shilatifard, A. Regulation of the imprinted Dlk1-Dio3 locus by allele-specific enhancer activity. Genes Dev. 2016, 30, 92–101. [Google Scholar] [CrossRef]
- Kremer, V.; Stanicek, L.; van Ingen, E.; Bink, D.I.; Hilderink, S.; Tijsen, A.J.; Wittig, I.; Mägdefessel, L.; Nossent, A.Y.; Boon, R.A. The long non-coding RNA MEG8 induces an endothelial barrier through regulation of microRNA-370 and -494 processing. J. Cell Sci. 2022, 135, jcs259671. [Google Scholar] [CrossRef]
- Kremer, V.; Bink, D.I.; Stanicek, L.; van Ingen, E.; Gimbel, T.; Hilderink, S.; Günther, S.; Nossent, A.Y.; Boon, R.A. MEG8 regulates Tissue Factor Pathway Inhibitor 2 (TFPI2) expression in the endothelium. Sci. Rep. 2022, 12, 843. [Google Scholar] [CrossRef]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Shirai, M.; Obata, Y.; Ogawa, H.; Sotomaru, Y.; Kono, T. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Botticelli, E.M.; Kery, R.E.; Mao, Y.; Wang, X.; Yang, A.; Wang, X.; Zhou, J.; Zhang, X.; Soberman, R.J.; et al. Meg3-DMR, not the Meg3 gene, regulates imprinting of the Dlk1-Dio3 locus. Dev. Biol. 2019, 455, 10–18. [Google Scholar] [CrossRef]
- Zhang, X.; He, H.; Yu, H.; Teng, X.; Wang, Z.; Li, C.; Li, J.; Yang, H.; Shen, J.; Wu, T.; et al. Maternal RNA transcription in Dlk1-Dio3 domain is critical for proper development of the mouse placental vasculature. Commun. Biol. 2024, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Shamis, Y.; Cullen, D.E.; Liu, L.; Yang, G.; Ng, S.F.; Xiao, L.; Bell, F.T.; Ray, C.; Takikawa, S.; Moskowitz, I.P.; et al. Maternal and zygotic Zfp57 modulate NOTCH signaling in cardiac development. Proc. Natl. Acad. Sci. USA 2015, 112, E2020–E2029. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Bais, A.; He, H.; Rios, C.; Jiang, S.; Xu, J.; Chang, C.; Kostka, D.; Li, G. Single-cell transcriptomic analysis identifies murine heart molecular features at embryonic and neonatal stages. Nat. Commun. 2022, 13, 7960. [Google Scholar] [CrossRef]
- Yuan, P.; Cheedipudi, S.M.; Rouhi, L.; Fan, S.; Simon, L.; Zhao, Z.; Hong, K.; Gurha, P.; Marian, A.J. Single-Cell RNA Sequencing Uncovers Paracrine Functions of the Epicardial-Derived Cells in Arrhythmogenic Cardiomyopathy. Circulation 2021, 143, 2169–2187. [Google Scholar] [CrossRef]
- Goodyer, W.R.; Beyersdorf, B.M.; Paik, D.T.; Tian, L.; Li, G.; Buikema, J.W.; Chirikian, O.; Choi, S.; Venkatraman, S.; Adams, E.L.; et al. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ. Res. 2019, 125, 379–397. [Google Scholar] [CrossRef]
- Streef, T.J.; Groeneveld, E.J.; van Herwaarden, T.; Hjortnaes, J.; Goumans, M.J.; Smits, A.M. Single-cell analysis of human fetal epicardium reveals its cellular composition and identifies CRIP1 as a modulator of EMT. Stem Cell Rep. 2023, 18, 1421–1435. [Google Scholar] [CrossRef]
- Tian, X.; Li, Y.; He, L.; Zhang, H.; Huang, X.; Liu, Q.; Pu, W.; Zhang, L.; Li, Y.; Zhao, H.; et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat. Commun. 2017, 8, 87. [Google Scholar] [CrossRef]
- Rhee, S.; Paik, D.T.; Yang, J.Y.; Nagelberg, D.; Williams, I.; Tian, L.; Roth, R.; Chandy, M.; Ban, J.; Belbachir, N.; et al. Endocardial/endothelial angiocrines regulate cardiomyocyte development and maturation and induce features of ventricular non-compaction. Eur. Heart J. 2021, 42, 4264–4276. [Google Scholar] [CrossRef]
- Quijada, P.; Trembley, M.A.; Misra, A.; Myers, J.A.; Baker, C.D.; Pérez-Hernández, M.; Myers, J.R.; Dirkx, R.A., Jr.; Cohen, E.D.; Delmar, M.; et al. Coordination of endothelial cell positioning and fate specification by the epicardium. Nat. Commun. 2021, 12, 4155. [Google Scholar] [CrossRef]
- Viswanathan, G.; Kirshner, H.F.; Nazo, N.; Ali, S.; Ganapathi, A.; Cumming, I.; Zhuang, Y.; Choi, I.; Warman, A.; Jassal, C.; et al. Single-Cell Analysis Reveals Distinct Immune and Smooth Muscle Cell Populations that Contribute to Chronic Thromboembolic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2023, 207, 1358–1375. [Google Scholar] [CrossRef]
- Zhang, T.T.; Lei, Q.Q.; He, J.; Guan, X.; Zhang, X.; Huang, Y.; Zhou, Z.Y.; Fan, R.X.; Wang, T.; Li, C.X.; et al. Bestrophin3 Deficiency in Vascular Smooth Muscle Cells Activates MEKK2/3-MAPK Signaling to Trigger Spontaneous Aortic Dissection. Circulation 2023, 148, 589–606. [Google Scholar] [CrossRef]
- Hortells, L.; Meyer, E.C.; Thomas, Z.M.; Yutzey, K.E. Periostin-expressing Schwann cells and endoneurial cardiac fibroblasts contribute to sympathetic nerve fasciculation after birth. J. Mol. Cell. Cardiol. 2021, 154, 124–136. [Google Scholar] [CrossRef]
- Astanina, E.; Doronzo, G.; Corà, D.; Neri, F.; Oliviero, S.; Genova, T.; Mussano, F.; Middonti, E.; Vallariello, E.; Cencioni, C.; et al. The TFEB-TGIF1 axis regulates EMT in mouse epicardial cells. Nat. Commun. 2022, 13, 5191. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, C.; Rodriguez-Outeiriño, L.; Matias-Valiente, L.; Ramirez de Acuña, F.; Hernandez-Torres, F.; Lozano-Velasco, E.; Dominguez, J.N.; Franco, D.; Aranega, A.E. Regulation of Epicardial Cell Fate during Cardiac Development and Disease: An Overview. Int. J. Mol. Sci. 2022, 23, 3220. [Google Scholar] [CrossRef]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.O.; Hughes, J.R.; Hardison, R.C.; et al. Unlinking an lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef]
- Goyal, A.; Myacheva, K.; Groß, M.; Klingenberg, M.; Duran Arqué, B.; Diederichs, S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017, 45, e12. [Google Scholar] [CrossRef]
- Tierling, S.; Dalbert, S.; Schoppenhorst, S.; Tsai, C.E.; Oliger, S.; Ferguson-Smith, A.C.; Paulsen, M.; Walter, J. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 2006, 87, 225–235. [Google Scholar] [CrossRef]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Li, Z.; Gao, J.; Sun, D.; Jiao, Q.; Ma, J.; Cui, W.; Lou, Y.; Xu, F.; Li, S.; Li, H. LncRNA MEG3: Potential stock for precision treatment of cardiovascular diseases. Front. Pharmacol. 2022, 13, 1045501. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Yu, H.; Zeng, L.; Ma, H.; Cao, G. LncRNA Rian reduces cardiomyocyte pyroptosis and alleviates myocardial ischemia-reperfusion injury by regulating by the miR-17-5p/CCND1 axis. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2022, 45, 976–989. [Google Scholar] [CrossRef]
- Clark, A.L.; Naya, F.J. MicroRNAs in the Myocyte Enhancer Factor 2 (MEF2)-regulated Gtl2-Dio3 Noncoding RNA Locus Promote Cardiomyocyte Proliferation by Targeting the Transcriptional Coactivator Cited2. J. Biol. Chem. 2015, 290, 23162–23172. [Google Scholar] [CrossRef]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Zhu, J.G.; Liu, Y.Q.; Fan, Z.G.; Zhu, C.; Qian, L.M. Identification of the microRNA expression profile in the regenerative neonatal mouse heart by deep sequencing. Cell Biochem. Biophys. 2014, 70, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Hongjuan, H.; Ning, G.; Zhengbin, H.; Yanjiang, X.; Tiebo, Z.; Zhijun, H.; Qiong, W. miR-370 is stage-specifically expressed during mouse embryonic development and regulates Dnmt3a. FEBS Lett. 2013, 587, 775–781. [Google Scholar] [CrossRef]
- Wu, Y.H.; Zhao, H.; Zhou, L.P.; Zhao, C.X.; Wu, Y.F.; Zhen, L.X.; Li, J.; Ge, D.X.; Xu, L.; Lin, L.; et al. miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2. Int. J. Mol. Sci. 2015, 16, 25199–25213. [Google Scholar] [CrossRef]
- Sanli, I.; Lalevée, S.; Cammisa, M.; Perrin, A.; Rage, F.; Llères, D.; Riccio, A.; Bertrand, E.; Feil, R. Meg3 Non-coding RNA Expression Controls Imprinting by Preventing Transcriptional Upregulation in cis. Cell Rep. 2018, 23, 337–348. [Google Scholar] [CrossRef]
- Farhadova, S.; Ghousein, A.; Charon, F.; Surcis, C.; Gomez-Velazques, M.; Roidor, C.; Di Michele, F.; Borensztein, M.; De Sario, A.; Esnault, C.; et al. The long non-coding RNA Meg3 mediates imprinted gene expression during stem cell differentiation. Nucleic Acids Res. 2024, 52, 6183–6200. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, J.; Zhang, Y.; Han, N.; Di, X.; Xiao, T.; Cheng, S.; Gao, Y.; Liu, Y. DLK1 promotes lung cancer cell invasion through upregulation of MMP9 expression depending on Notch signaling. PLoS ONE 2014, 9, e91509. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Kuo, H.M.; Wu, P.C.; Cheng, S.H.; Chang, T.T.; Chang, Y.C.; Kung, M.L.; Wu, D.C.; Chuang, J.H.; Tai, M.H. Soluble delta-like 1 homolog (DLK1) stimulates angiogenesis through Notch1/Akt/eNOS signaling in endothelial cells. Angiogenesis 2018, 21, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.H.; Johnsen, R.H.; Eskildsen, T.; Baun, C.; Ellman, D.G.; Fang, S.; Bak, S.T.; Hvidsten, S.; Larsen, L.A.; Rosager, A.M.; et al. Pericardial delta like non-canonical NOTCH ligand 1 (Dlk1) augments fibrosis in the heart through epithelial to mesenchymal transition. Clin. Transl. Med. 2024, 14, e1565. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, L.; Goodyer, W.; Kort, E.J.; Buikema, J.W.; Xu, A.; Wu, J.C.; Jovinge, S.; Wu, S.M. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development 2019, 146, dev173476. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.C.; Kadow, Z.A.; Li, L.; Tran, T.T.; Wythe, J.D.; Martin, J.F. A cellular atlas of Pitx2-dependent cardiac development. Development 2019, 146, dev180398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, C.C.; Li, X.; Liu, Y.; Wang, Q.; Zhang, G.; Xiong, H.; He, A.; Ai, S. Novel enhancers conferring compensatory transcriptional regulation of Nkx2-5 in heart development. iScience 2023, 26, 106509. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- Ritacco, C.; Köse, M.C.; Courtois, J.; Canti, L.; Beguin, C.; Dubois, S.; Vandenhove, B.; Servais, S.; Caers, J.; Beguin, Y.; et al. Post-transplant cyclophosphamide prevents xenogeneic graft-versus-host disease while depleting proliferating regulatory T cells. iScience 2023, 26, 106085. [Google Scholar] [CrossRef] [PubMed]
- Rudat, C.; Kispert, A. Wt1 and epicardial fate mapping. Circ. Res. 2012, 111, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Bonet, F.; Pereira, P.N.G.; Bover, O.; Marques, S.; Inácio, J.M.; Belo, J.A. CCBE1 is required for coronary vessel development and proper coronary artery stem formation in the mouse heart. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Kiessling, I.; Heinicke, K.; Stallmach, T.; Ossent, P.; Vogel, O.; Aulmann, M.; Frietsch, T.; Schmid-Schönbein, H.; Kuschinsky, W.; et al. Transgenic mice overexpressing erythropoietin adapt to excessive erythrocytosis by regulating blood viscosity. Blood 2003, 102, 2278–2284. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, X.; He, H.; Yu, H.; Zhang, X.; Xing, J.; Shen, J.; Li, C.; Wang, M.; Shao, L.; Wang, Z.; et al. LncRNAs in the Dlk1-Dio3 Domain Are Essential for Mid-Embryonic Heart Development. Int. J. Mol. Sci. 2024, 25, 8184. https://doi.org/10.3390/ijms25158184
Teng X, He H, Yu H, Zhang X, Xing J, Shen J, Li C, Wang M, Shao L, Wang Z, et al. LncRNAs in the Dlk1-Dio3 Domain Are Essential for Mid-Embryonic Heart Development. International Journal of Molecular Sciences. 2024; 25(15):8184. https://doi.org/10.3390/ijms25158184
Chicago/Turabian StyleTeng, Xiangqi, Hongjuan He, Haoran Yu, Ximeijia Zhang, Jie Xing, Jiwei Shen, Chenghao Li, Mengyun Wang, Lan Shao, Ziwen Wang, and et al. 2024. "LncRNAs in the Dlk1-Dio3 Domain Are Essential for Mid-Embryonic Heart Development" International Journal of Molecular Sciences 25, no. 15: 8184. https://doi.org/10.3390/ijms25158184
APA StyleTeng, X., He, H., Yu, H., Zhang, X., Xing, J., Shen, J., Li, C., Wang, M., Shao, L., Wang, Z., Yang, H., Zhang, Y., & Wu, Q. (2024). LncRNAs in the Dlk1-Dio3 Domain Are Essential for Mid-Embryonic Heart Development. International Journal of Molecular Sciences, 25(15), 8184. https://doi.org/10.3390/ijms25158184