
Osteoprotegerin Is Essential for the Development of Endothelial Dysfunction Induced by Angiotensin II in Mice
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
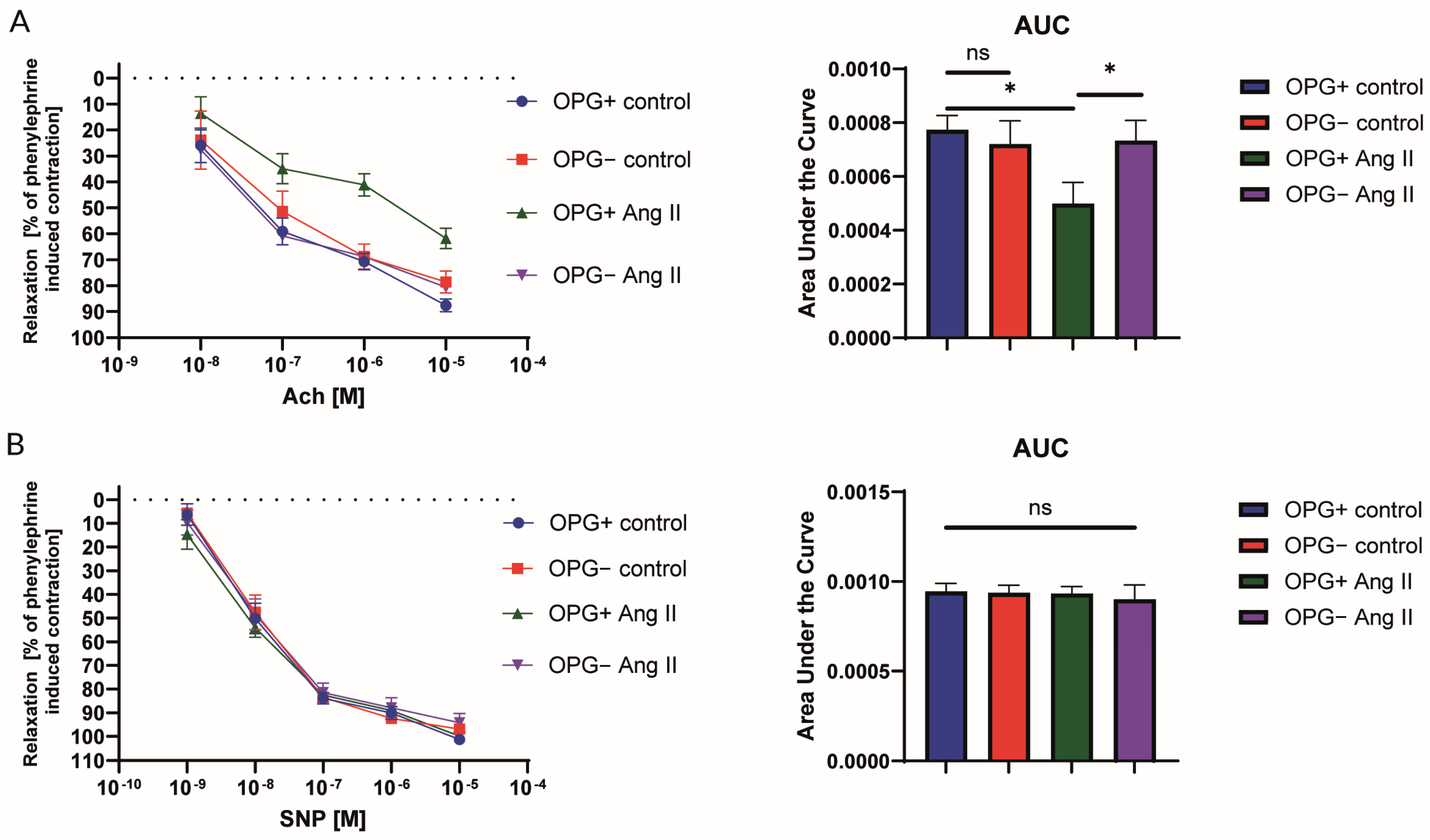
2.1. Nitric Oxide-Dependent Endothelial Function in the Isolated Murine Aorta
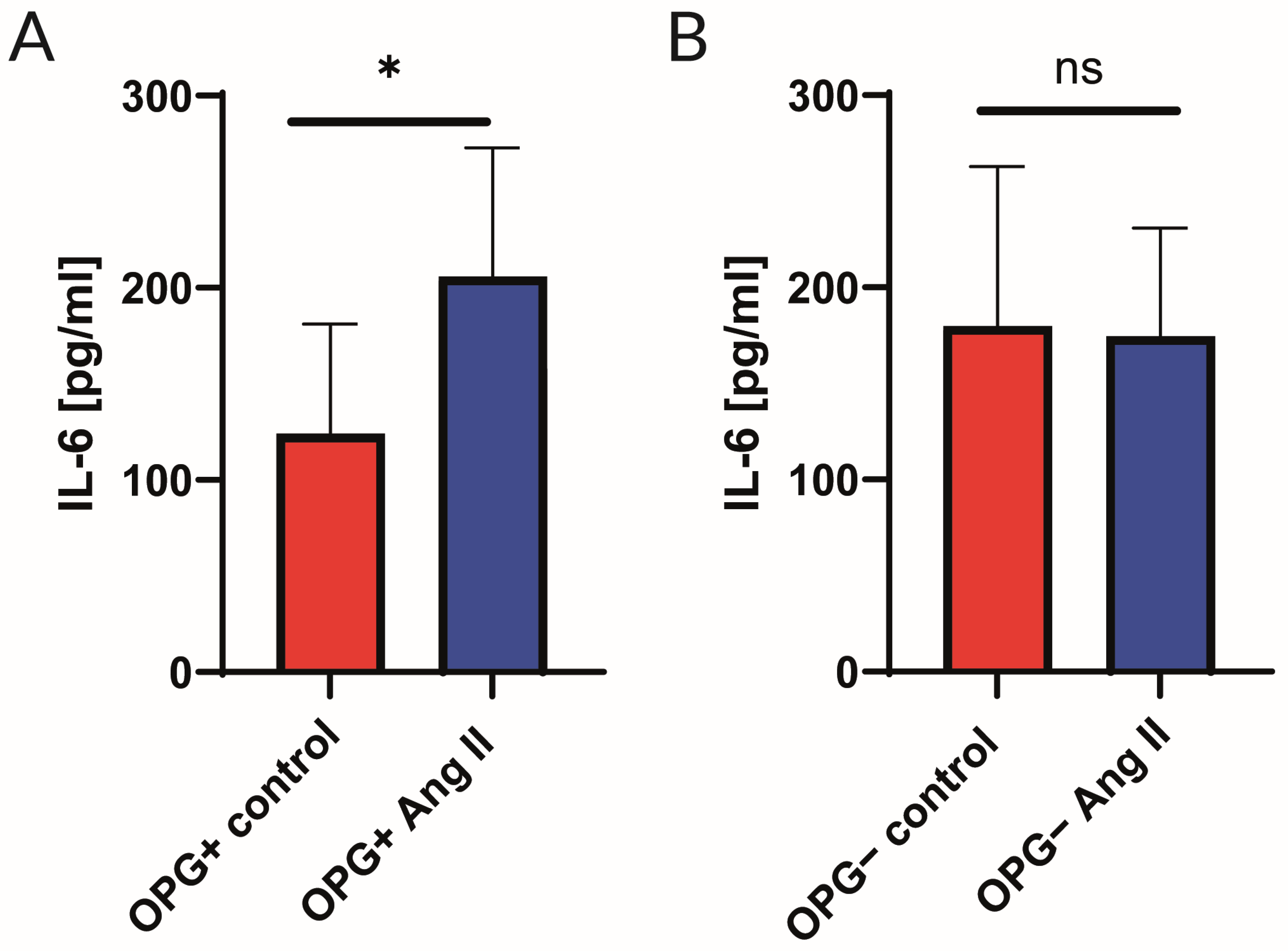
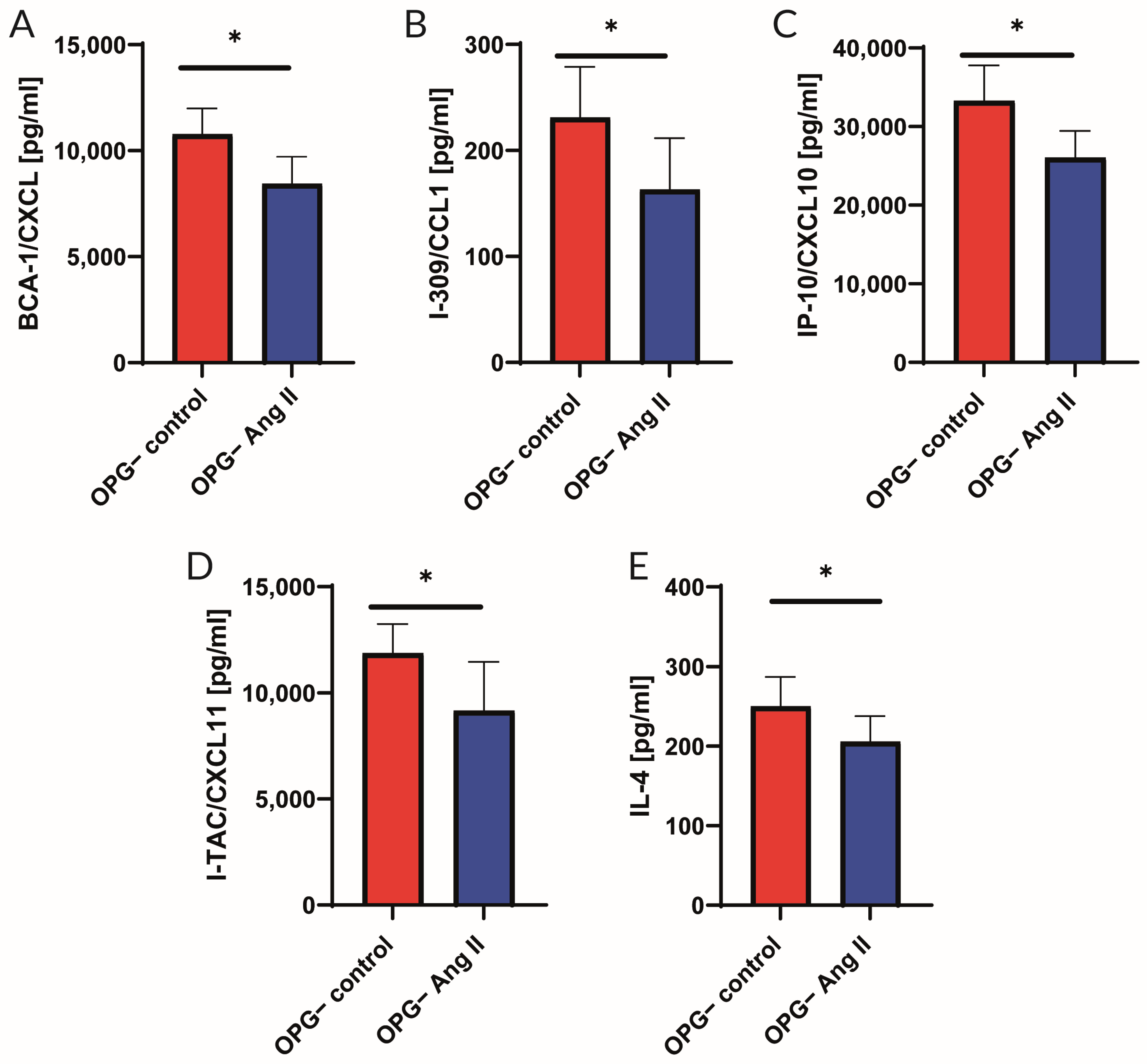
2.2. Analysis of Cytokine Panel
3. Discussion
3.1. Endothelial Function
3.2. Cytokine Profile
3.3. Mechanisms of Action of OPG in Vascular Wall
3.4. Conclusions, Potential Clinical Implications, and Future Perspectives
3.5. Limitation of the Study
4. Materials and Methods
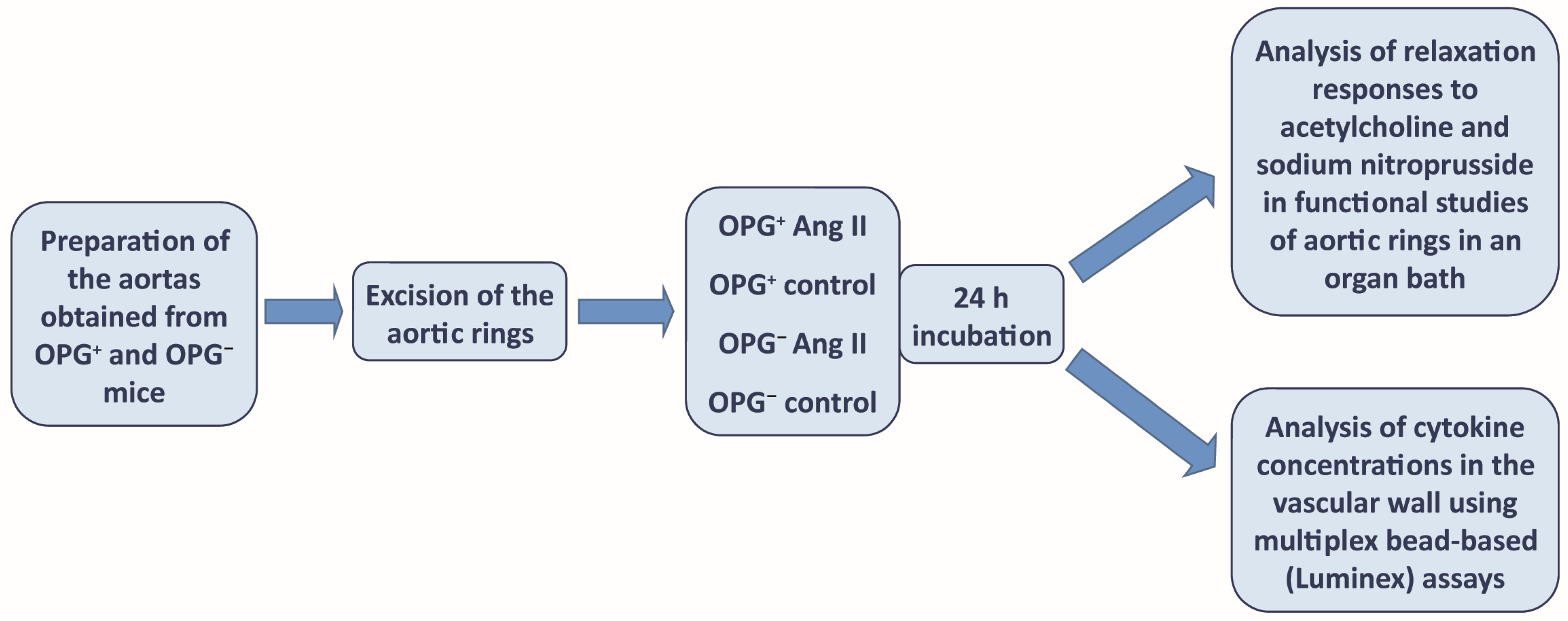
4.1. Evaluation of Nitric Oxide-Dependent Endothelial Function in the Isolated Murine Aorta
4.2. Fluorescent Bead-Based Luminex Cytokine Assay
4.3. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ueland, T.; Yndestad, A.; Dahl, C.P.; Gullestad, L.; Aukrust, P. TNF Revisited: Osteoprotegerin and TNF—Related Molecules in Heart Failure. Curr. Heart Fail. Rep. 2012, 9, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Korzon-Burakowska, A.; Burakowski, S. Oś osteoprotegeryna/RANKL/RANK—Rola w powikłaniach cukrzycy oraz w chorobie wieńcowej. Diabetol. Prakt. 2007, 8, 161–164. [Google Scholar]
- Bernardi, S.; Bossi, F.; Toffoli, B.; Fabris, B. Roles and Clinical Applications of OPG and TRAIL as Biomarkers in Cardiovascular Disease. Biomed. Res. Int. 2016, 2016, 1752854. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Cottin, Y.; Vergely, C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. Int. J. Mol. Sci. 2019, 20, 705. [Google Scholar] [CrossRef] [PubMed]
- Montagnana, M.; Lippi, G.; Danese, E.; Guidi, G.C. The role of osteoprotegerin in cardiovascular disease. Ann. Med. 2013, 45, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Celebi Torabfam, G.; Porsuk, M.H. The Role of the Receptor Activator of Nuclear Factor Kappa-B Ligand/Osteoprotegerin Ratio in Vascular Diseases: A Therapeutic Approach. Angiology 2024, 2024, 00033197231226275. [Google Scholar] [CrossRef] [PubMed]
- Corallini, F.; Rimondi, E.; Secchiero, P. TRAIL and osteoprotegerin: A role in endothelial physiopathology? Front. Biosci. 2008, 13, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Stępień, E. Osteoprotegerin as a possible novel predictor of cardiovascular dysfunction. Kardiochirurgia I Torakochirurgia Pol. 2012, 9, 82–85. [Google Scholar]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Mizuno, A.; Amizuka, N.; Irie, K.; Murakami, A.; Fujise, N.; Kanno, T.; Sato, Y.; Nakagawa, N.; Yasuda, H.; Mochizuki, S.; et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem. Biophys. Res. Comm. 1998, 247, 610–615. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Schoppet, M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular disease. JAMA 2004, 292, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M. Osteoprotegerin (OPG) as a biomarker for diabetic cardiovascular complications. SpringerPlus 2013, 2, 658. [Google Scholar] [CrossRef] [PubMed]
- Özkalaycı, F.; Gülmez, Ö.; Uğur-Altun, B.; Pandi-Perumal, S.R.; Altun, A. The Role of Osteoprotegerin as a Cardioprotective versus Reactive Inflammatory Marker: The Chicken or the Egg Paradox. Balkan Med. J. 2018, 35, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Shin, J.G.; Chung, C.H. Elevated Serum Osteoprotegerin Levels Are Associated with Vascular Endothelial Dysfunction in Type 2 Diabetes. Diabetes Care 2006, 9, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Corallini, F.; Beltrami, A.; Ceconi, C.; Bonasia, V.; Di Chiara, A.; Ferrari, R.; Zauli, G. An imbalanced OPG/TRAIL ratio is associated to severe acute myocardial infarction. Atherosclerosis 2010, 210, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M.; Munk, K.; Sloth, A.D.; Nielsen, S.T.; Flyvbjerg, A.; Bøtker, H.E. High osteoprotegerin leves predict MACCE in STEMI patients, but are not assotiated with myocardial salvage. Scand. Cardiovasc. J. 2014, 48, 209–215. [Google Scholar] [CrossRef]
- Pedersen, S.; Mogelvang, R.; Bjerre, M.; Frystyk, J.; Flyvbjerg, A.; Galatius, S.; Sørensen, T.B.; Iversen, A.; Hvelplund, A.; Jensen, J.S. Osteoprotegerin predicts long-term outcam in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Cardiology 2012, 123, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Erkol, A.; Oduncu, V.; Pala, S.; Kizilirmak, F.; Kilicgedik, A.; Yılmaz, F.; Güler, A.; Karabay, C.Y.; Kırma, C. Plasma osteoprotegerin level on admission is associated with no-reflow phenomenon after primary angioplasty and subsequent left ventricular remodeling in patients with acute ST-segment elevation myocardial infarction. Atherosclerosis 2012, 221, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Corallini, F.; Secchiero, P.; Beltrami, A.P.; Cesselli, D.; Puppato, E.; Ferrari, R.; Beltrami, C.A.; Zauli, G. TNF–alfa modulates the migratory response of mesenchymal stem cells to TRAIL. Cell. Mol. Life Sci. 2010, 67, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Suzuki, T.; Furniss, A.L.; Griswold, M.E.; Kullo, I.J.; Lindsey, M.L.; Winniford, M.D.; Butler, K.R.; Mosley, T.H.; Hall, M.E. Elevated serum osteoprotegerin is associated with increased left ventricular mass index and myocardial stiffness. J. Cardiovasc. Med. 2017, 18, 954–961. [Google Scholar] [CrossRef]
- Skrzypczyk, P.; Stelmaszczyk-Emmel, A.; Szyszka, M.; Ofiara, A.; Pańczyk-Tomaszewska, M. Circulating calcification inhibitors are associated with arterial damage in pediatric patients with primary hypertension. Pediatr. Nephrol. 2021, 36, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Toffoli, B.; Bossi, F.; Candido, R.; Stenner, E.; Carretta, R.; Barbone, F.; Fabris, B. Circulating osteoprotegerin is associated with chronic kidney disease in hypertensive patients. BMC Nephrol. 2017, 18, 219. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, A.N.; Nanda, N.; Suryanarayana, B.S.; Pal, G.K. Association of rs2073618 polymorphism and osteoprotegerin levels with hypertension and cardiovascular risks in patients with type 2 diabetes mellitus. Sci. Rep. 2023, 13, 17451. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Anderson, F.; Nelson, M.; Maloney, W.; Osdoby, P. Receptor Activator of NF-kB and Osteoprotegerin Expression by Human Microvascular Endothelial Cells, Regulation by Inflammatory Cytokines, and Role in Human Osteoclastogenesis. J. Biol. Chem. 2001, 276, 20659–20672. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Corallini, F.; Bossi, F.; Fischetti, F.; Durigutto, P.; Celeghini, C.; Tedesco, F.; Secchiero, P. Osteoprotegerin increases leukocyte adhesion to endothelial cells both in vitro and in vivo. Blood 2007, 110, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Malyankar, U.M.; Scatena, M.; Suchland, K.L.; Yun, T.J.; Clark, E.A.; Giachelli, C.M. Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem. 2000, 275, 20959–20962. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, B.; Pickering, R.J.; Tsorotes, D.; Wang, B.; Bernardi, S.; Kantharidis, P.; Fabris, B.; Zauli, G.; Secchiero, P.; Thomas, M.C. Osteoprotegerin promotes vascular fibrosis via a TGF-β1 autocrine loop. Atherosclerosis 2011, 218, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, M.; Myles, D.; Zhu, X.; Du, J.; Cao, X.; Chen, Y.E. PDGF induces osteoprotegerin expression in vascular smooth muscle cells by multiple signal pathways. FEBS Lett. 2002, 521, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.S.; McCann, M.; Karan, M.; Norman, P.; Ketheesan, N.; Golledge, J. Association of osteoprotegerin with human abdominal aortic aneurysm progression. Circulation 2005, 111, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.S.; Cullen, B.; Campbell, J.H.; Golledge, J. Interaction between angiotensin II, osteoprotegerin, and peroxisome proliferator-activated receptor-γ in abdominal aortic aneurysm. J. Vasc. Res. 2009, 46, 209–217. [Google Scholar] [CrossRef]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- van der Vorst, E.P.C.; Daissormont, I.; Aslani, M.; Seijkens, T.; Wijnands, E.; Lutgens, E.; Duchene, J.; Santovito, D.; Döring, Y.; Halvorsen, B.; et al. Interruption of the CXCL13/CXCR5 Chemokine Axis Enhances Plasma IgM Levels and Attenuates Atherosclerosis Development. Thromb. Haemost. 2020, 120, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhu, R.; Yu, T.; Fan, H.; Hu, Y.; Mohanta, S.K.; Hu, D. Chronic Inflammation: A Common Promoter in Tertiary Lymphoid Organ Neogenesis. Front. Immunol. 2019, 10, 2938. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, H.; Chen, M.; Lu, Y.; Zheng, L.; Ma, L. CCL24/CCR3 axis plays a central role in angiotensin II-induced heart failure by stimulating M2 macrophage polarization and fibroblast activation. Cell Biol. Toxicol. 2023, 39, 1413–1431. [Google Scholar] [CrossRef] [PubMed]
- Boukhris, R.; Becker, K.L. Calcification of the aorta and osteoporosis. A roentgenographic study. JAMA 1972, 219, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Kado, D.M.; Browner, W.S.; Blackwell, T.; Gore, R.; Cummings, R.S. Rate of bone loss is associated with mortality in older women: A prospective study. J. Bone Miner. Res. 2000, 15, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Tsuda, E.; Washida, N.; Kobayashi, F.; Goto, M.; Harada, A.; Ikeda, K.; Higashio, K.; Yamada, Y. Immunological characterization of circulating osteoprotegerin/osteoclasto-genesis inhibitory factor: Increased serum concentrations in postmenopausal women with osteoporosis. J. Bone Miner. Res. 1999, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; June, H.H.; Buckley, J.R.; Williamson, M.K. Osteoprotegerin inhibits artery calcification induced by warfarin and by vitamin D. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Browner, W.S.; Lui, L.Y.; Cummings, S.R. Associations of serum osteoprotegerin levels with diabetes, stroke, bone density, fractures, and mortality in elderly women. J. Clin. Endocrinol. Metab. 2001, 86, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; Ikari, Y.; Shioi, A.; Mori, K.; Miki, T.; Hara, K.; Nishizawa, Y. Serum osteoprotegerin levels are associated with the presence and severity of coronary artery disease. Circulation 2002, 106, 1192–1194. [Google Scholar] [CrossRef]
- Knudsen, S.T.; Foss, C.H.; Poulsen, P.L.; Andersen, N.H.; Mogensen, C.E.; Rasmussen, L.M. Increased plasma concentrations of osteoprotegerin in type 2 diabetic patients with microvascular complications. Eur. J. Endocrinol. 2003, 149, 39–42. [Google Scholar] [CrossRef]
- Dutka, M.; Bobiński, R.; Wojakowski, W.; Francuz, T.; Pająk, C.; Zimmer, K. Osteoprotegerin and RANKL-RANK-OPG-TRAIL signalling axis in heart failure and other cardiovascular diseases. Heart Fail. Rev. 2022, 27, 1395–1411. [Google Scholar] [CrossRef]
- Wright, H.L.; McCarthy, H.S.; Middleton, J.; Marshall, M.J. RANK, RANKL and osteoprotegerin in bone biology and disease. Curr. Rev. Musculoskelet. Med. 2009, 2, 56–64. [Google Scholar] [CrossRef]
- Clancy, P.; Koblar, S.A.; Golledge, J. Angiotensin receptor 1 blockade reduces secretion of inflammation associated cytokines from cultured human carotid atheroma and vascular cells in association with reduced extracellular signal regulated kinase expression and activation. Atherosclerosis 2014, 236, 108–115. [Google Scholar] [CrossRef]
- Chen, S.; Grover, M.; Sibai, T.; Black, J.; Rianon, N.; Rajagopal, A.; Munivez, E.; Bertin, T.; Dawson, B.; Chen, Y.; et al. Losartan increases bone mass and accelerates chondrocyte hypertrophy in developing skeleton. Mol. Genet. Metab. 2015, 115, 53–60. [Google Scholar] [CrossRef]
- Min, J.K.; Kim, Y.M.; Kim, Y.M.; Kim, E.C.; Gho, Y.S.; Kang, I.J.; Lee, S.Y.; Kong, Y.Y.; Kwon, Y.G. Vascular endothelial growth factor up-regulates expression of receptor activator of NF-kappa B (RANK) in endothelial cells. Concomitant increase of angiogenic responses to RANK ligand. J. Biol. Chem. 2003, 278, 39548–39557. [Google Scholar] [CrossRef]
- Potente, M.; Carmeliet, P. The Link between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Di Pietro, R.; Mariggiò, M.A.; Guarnieri, S.; Sancilio, S.; Giardinelli, A.; Di Silvestre, S.; Consoli, A.; Zauli, G.; Pandolfi, A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) regulates endothelial nitric oxide synthase (eNOS) activity and its localization within the human vein endothelial cells (HUVEC) in culture. J. Cell. Biochem. 2006, 97, 782–794. [Google Scholar] [CrossRef]
- Bernardi, S.; Fabris, B.; Thomas, M.; Toffoli, B.; Tikellis, C.; Candido, R.; Catena, C.; Mulatero, P.; Barbone, F.; Radillo, O.; et al. Osteoprotegerin increases in metabolic syndrome and promotes adipose tissue proinflammatory changes. Mol. Cell. Endocrinol. 2014, 394, 13–20. [Google Scholar] [CrossRef]
- Mangan, S.H.; Van Campenhout, A.; Rush, C.; Golledge, J. Osteoprotegerin upregulates endothelial cell adhesion molecule response to tumor necrosis factor-alpha associated with induction of angiopoietin-2. Cardiovasc. Res. 2007, 76, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Scatena, M.; Kirk, E.A.; Rattazzi, M.; Varon, R.M.; Averill, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. The regulatory role of nitric oxide in proinflammatory cytokine expression during the induction and resolution of inflammation. J. Leukoc. Biol. 2010, 88, 1157–1162. [Google Scholar] [CrossRef]
- Quercioli, A.; Mach, F.; Bertolotto, M.; Lenglet, S.; Vuilleumier, N.; Galan, K.; Pagano, S.; Braunersreuther, V.; Pelli, G.; Pistoia, V.; et al. Receptor activator of NF-κB ligand (RANKL) increases the release of neutrophil products associated with coronary vulnerability. Thromb. Haemost. 2012, 107, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Moreno, C.; Jiménez-Ferrer, E.; Castro-Martínez, G.; Méndez-Martínez, M.; Santana, M.A.; Arrellín-Rosas, G.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Hernández-Téllez, B.; Ramírez-Pliego, O.; et al. Characterization of a murine model of endothelial dysfunction induced by chronic intraperitoneal administration of angiotensin II. Sci. Rep. 2021, 11, 21193. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutka, M.; Garczorz, W.; Kosowska, A.; Buczek, E.; Godek, P.; Wojakowski, W.; Francuz, T. Osteoprotegerin Is Essential for the Development of Endothelial Dysfunction Induced by Angiotensin II in Mice. Int. J. Mol. Sci. 2024, 25, 6434. https://doi.org/10.3390/ijms25126434
Dutka M, Garczorz W, Kosowska A, Buczek E, Godek P, Wojakowski W, Francuz T. Osteoprotegerin Is Essential for the Development of Endothelial Dysfunction Induced by Angiotensin II in Mice. International Journal of Molecular Sciences. 2024; 25(12):6434. https://doi.org/10.3390/ijms25126434
Chicago/Turabian StyleDutka, Mieczysław, Wojciech Garczorz, Agnieszka Kosowska, Elzbieta Buczek, Piotr Godek, Wojciech Wojakowski, and Tomasz Francuz. 2024. "Osteoprotegerin Is Essential for the Development of Endothelial Dysfunction Induced by Angiotensin II in Mice" International Journal of Molecular Sciences 25, no. 12: 6434. https://doi.org/10.3390/ijms25126434
APA StyleDutka, M., Garczorz, W., Kosowska, A., Buczek, E., Godek, P., Wojakowski, W., & Francuz, T. (2024). Osteoprotegerin Is Essential for the Development of Endothelial Dysfunction Induced by Angiotensin II in Mice. International Journal of Molecular Sciences, 25(12), 6434. https://doi.org/10.3390/ijms25126434