


Progressive Evolutionary Dynamics of Gene-Specific ω Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization


Abstract
1. Introduction
2. Results
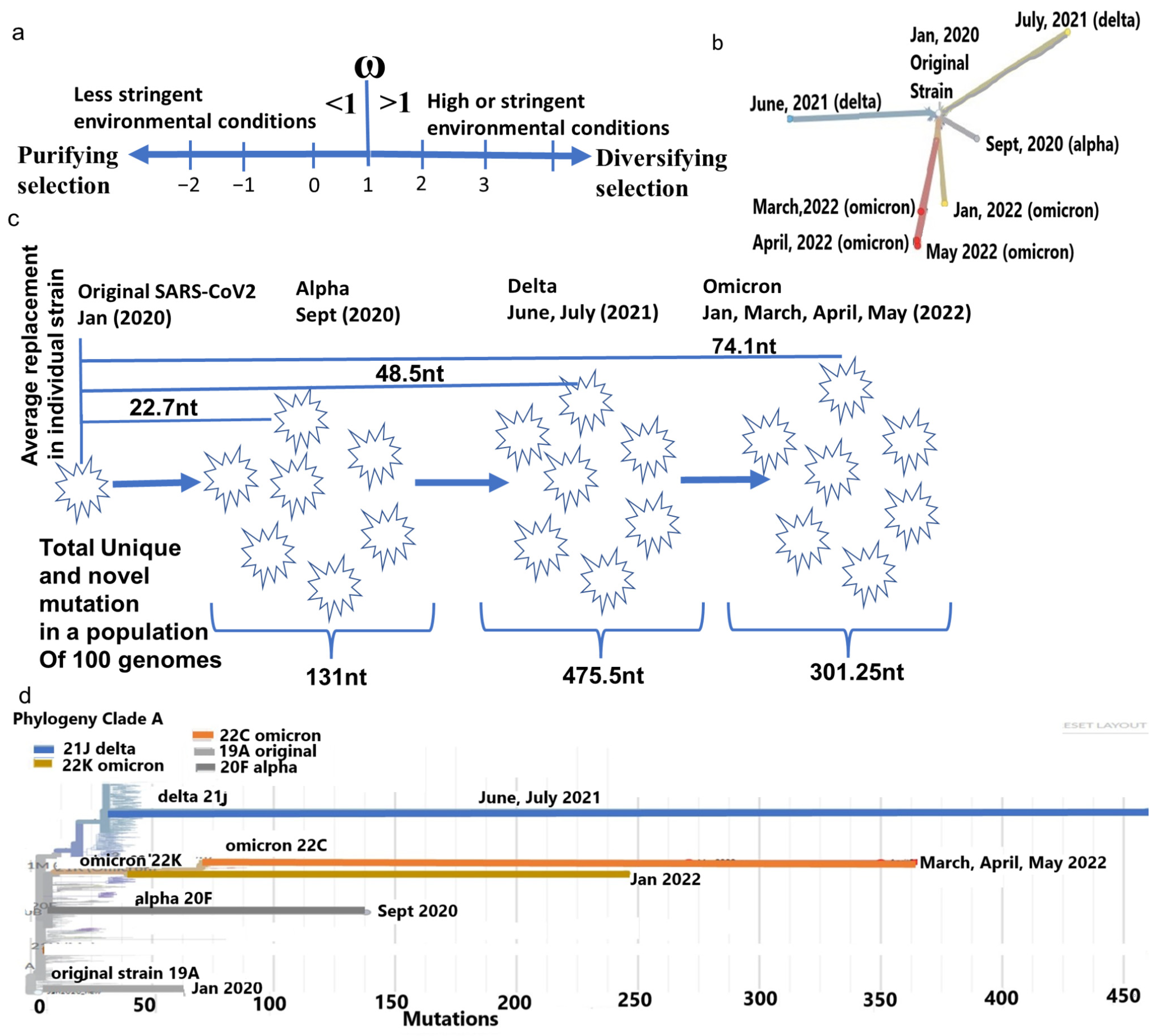
2.1. Cladistic Phylogenetic Differences of Major Strains of SARS-CoV-2
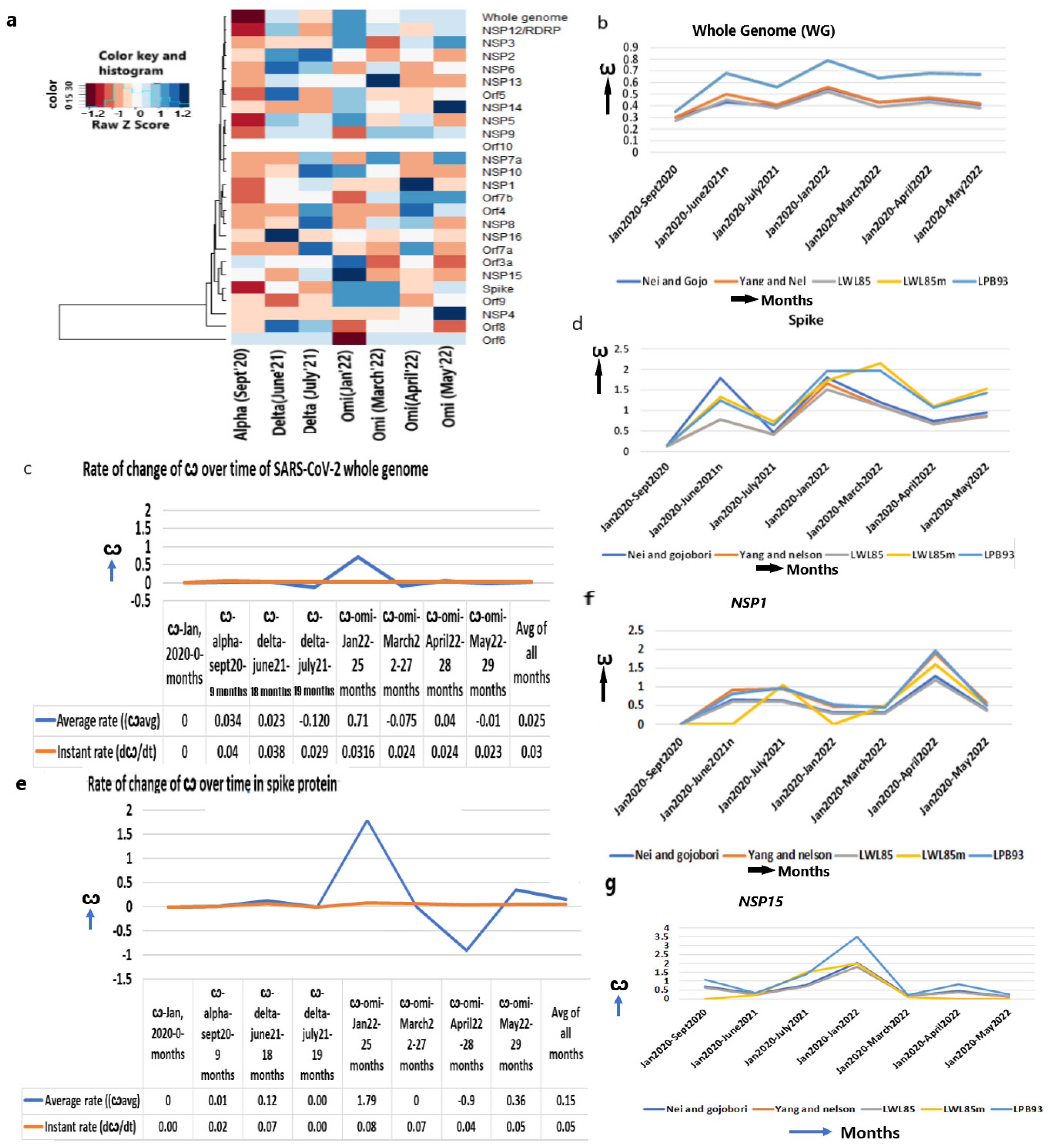
2.2. The ω of the Whole SARS-CoV-2 Genome Is Increased since Its Emergence with an Approach to Diversifying Selection
2.3. The Increase in the ω of Spike (S) Gene Increases Infection
2.4. The ω of the Other Viral Entry Associated Genes Are Continuously Increasing
2.5. The ω of Replication and Multiplication Machinery Assisted Genes Are Markedly Increased
2.6. The ω of Virus Assembly and Release Proteins Are Increased
2.7. Genes with Unchanged or Decreased ω Implicate Well Adaptation in Human Host with Efficient Functions
2.8. The Increased Virulence of Delta Could Be Attributed to Increased ω in Immunomodulatory Genes
2.9. The ω of Several Genes Are Highest in Initial Omicron but Decreased in Subsequent Months
3. Discussion
4. Materials and Methods
4.1. Genomic Sequences
4.2. Identification of Mutations
4.3. Estimation of ω
4.4. Rate of Change in ω in Various Strains
4.5. Phylogenetic Analysis
4.6. Statistical Analysis, Graphs, and Charts
4.7. Visualization of Data and Rate of ω
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977, 267, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Yasunaga, T.; Nishida, T. Nucleotide sequence divergence and functional constraint in mRNA evolution. Proc. Natl. Acad. Sci. USA 1980, 77, 7328–7332. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef]
- Maiti, A.K. Evolutionary shift from purifying selection towards divergent selection of SARS-CoV-2 favors its invasion into multiple human organs. Virus Res. 2022, 313, 198712. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Bernard, D.W.; Davis, J.J.; Shukla, M.; Nguyen, M.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; et al. Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area. mBio 2020, 11, e02707-20. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef] [PubMed]
- Andeweg, S.P.; Vennema, H.; Veldhuijzen, I.; Smorenburg, N.; Schmitz, D.; Zwagemaker, F.; Van Gageldonk-Lafeber, A.B.; Hahné, S.J.M.; Reusken, C.; Knol, M.J.; et al. Elevated risk of infection with SARS-CoV-2 Beta, Gamma, and Delta variant compared to Alpha variant in vaccinated individuals. Sci. Transl. Med. 2022, 15, eabn4338. [Google Scholar] [CrossRef] [PubMed]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell 2021, 184, 2201–2211.e7. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Tenforde, M.W.; Chappell, J.D.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Clinical severity of, and effectiveness of mRNA vaccines against, COVID-19 from omicron, delta, and alpha SARS-CoV-2 variants in the United States: Prospective observational study. BMJ 2022, 376, e069761. [Google Scholar] [CrossRef]
- Alefishat, E.; Jelinek, H.F.; Mousa, M.; Tay, G.K.; Alsafar, H.S. Immune response to SARS-CoV-2 variants: A focus on severity, susceptibility, and preexisting immunity. J. Infect. Public Health 2022, 15, 277–288. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Papakyriakou, A.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF. Viruses 2022, 14, 707. [Google Scholar] [CrossRef]
- Zheng, S.-Y.; Zhang, Y.P.; Liu, Y.X.; Zhao, W.; Peng, X.L.; Zheng, Y.P.; Fu, Y.H.; Yu, J.M.; He, J.S. Tracking of Mutational Signature of SARS-CoV-2 Omicron on Distinct Continents and Little Difference was Found. Viruses 2023, 15, 321. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Earnest, R.; Uddin, R.; Matluk, N.; Renzette, N.; Turbett, S.E.; Siddle, K.J.; Loreth, C.; Adams, G.; Tomkins-Tinch, C.H.; Petrone, M.E.; et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. Cell. Rep. Med. 2022, 3, 100583. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hause, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: Viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Mou, K.; Mukhtar, F.; Khan, M.T.; Darwish, D.B.; Peng, S.; Muhammad, S.; Al-Sehemi, A.G.; Wei, D.-Q. Emerging Mutations in Nsp1 of SARS-CoV-2 and Their Effect on the Structural Stability. Pathogens 2021, 10, 1285. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Akter, S.; Rashid, A.A.; Khair, S.; Alam, A.R.U. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathog. 2022, 170, 105699. [Google Scholar] [CrossRef]
- Abbas, Q.; Kusakin, A.; Sharrouf, K.; Jyakhwo, S.; Komissarov, A.S. Follow-up investigation and detailed mutational characterization of the SARS-CoV-2 Omicron variant lineages (BA.1, BA.2, BA.3 and BA.1.1). bioRxiv 2022. [Google Scholar] [CrossRef]
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.K. The African-American population with a low allele frequency of SNP rs1990760 (T allele) in IFIH1 predicts less IFN-beta expression and potential vulnerability to COVID-19 infection. Immunogenetics 2020, 72, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, L.; Zhao, Y.; Feng, D.; Cheng, J.; Zhang, G. Coronavirus Endoribonuclease Ensures Efficient Viral Replication and Prevents Protein Kinase R Activation. J. Virol. 2020, 95, e02103-20. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Negi, S.S.; Bhargava, A.; Kolla, V.P.; Arora, R.D. A Preliminary Genomic Analysis of the Omicron Variants of SARS-CoV-2 in Central India During the third wave of the COVID-19 Pandemic. Arch. Med. Res. 2022, 53, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Reshamwala, S.M.S.; Likhite, V.; Degani, M.S.; Deb, S.S.; Noronha, S.B. Mutations in SARS-CoV-2 nsp7 and nsp8 proteins and their predicted impact on replication/transcription complex structure. J. Med. Virol. 2021, 93, 4616–4619. [Google Scholar] [CrossRef] [PubMed]
- Brandal, L.T.; MacDonald, E.; Veneti, L.; Ravlo, T.; Lange, H.; Naseer, U.; Feruglio, S.; Bragstad, K.; Hungnes, O.; Ødeskaug, L.E.; et al. Outbreak caused by the SARS-CoV-2 Omicron variant in Norway, November to December 2021. Euro Surveill. 2021, 26, 2101147. [Google Scholar] [CrossRef] [PubMed]
- te Velthuis, A.J.; van den Worm, S.H.; Snijder, E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012, 40, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Prüβ, B.M. Variants of SARS-CoV-2: Mutations, transmissibility, virulence, drug resistance, and antibody/vaccine sensitivity. Front. Biosci. (Landmark Ed.) 2022, 27, 65. [Google Scholar] [CrossRef]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef]
- Bansal, K.; Kumar, S. Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. Virus Res. 2022, 315, 198765. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Kawachi, K.; Terada, Y.; Omori, H.; Matsuura, Y.; Kamitani, W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 2017, 510, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.-K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Fung, S.Y.; Siu, K.-L.; Lin, H.; Chan, C.-P.; Yeung, M.L.; Jin, D.-Y. SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1. Cell. Biosci. 2022, 12, 36. [Google Scholar] [CrossRef]
- Kaur, M.; Sharma, A.; Kumar, S.; Singh, G.; Barnwal, R.P. SARS-CoV-2: Insights into its structural intricacies and functional aspects for drug and vaccine development. Int. J. Biol. Macromol. 2021, 179, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.; Abdalla, M.; Wei, D.Q.; Khan, M.T.; Lodhi, M.S.; Darwish, D.B.; Sharaf, M.; Tu, X. Emerging mutations in envelope protein of SARS-CoV-2 and their effect on thermodynamic properties. Inform. Med. Unlocked 2021, 25, 100675. [Google Scholar] [CrossRef]
- Zaffagni, M.; Harris, J.M.; Patop, I.L.; Pamudurti, N.R.; Nguyen, S.; Kadener, S. SARS-CoV-2 Nsp14 mediates the effects of viral infection on the host cell transcriptome. Elife 2022, 11, e71945. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.A. Mutations in the genome of severe acute respiratory syndrome coronavirus 2: Implications for COVID-19 severity and progression. J. Int. Med. Res. 2022, 50, 3000605221086433. [Google Scholar] [CrossRef]
- Fogeron, M.; Montserret, R.; Zehnder, J.; Nguyen, M.-H.; Dujardin, M.; Brigandat, L.; Cole, L.; Ninot-Pedrosa, M.; Lecoq, L.; Meier, B.H.; et al. SARS-CoV-2 ORF7b: Is a bat virus protein homologue a major cause of COVID-19 symptoms? bioRxiv 2021. [Google Scholar] [CrossRef]
- Hassan, S.S.; Attrish, D.; Ghosh, S.; Choudhury, P.P.; Uversky, V.N.; Aljabali, A.A.; Lundstrom, K.; Uhal, B.D.; Rezaei, N.; Seyran, M.; et al. Notable sequence homology of the ORF10 protein introspects the architecture of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 181, 801–809. [Google Scholar] [CrossRef]
- Pancer, K.; Milewska, A.; Owczarek, K.; Dabrowska, A.; Kowalski, M.; Łabaj, P.P.; Branicki, W.; Sanak, M.; Pyrc, K. The SARS-CoV-2 ORF10 is not essential in vitro or in vivo in humans. PLoS Pathog. 2020, 16, e1008959. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Fu, B.; Yin, S.; Li, Z.; Liu, H.; Zhang, H.; Xing, N.; Wang, Y.; Xue, W.; Xiong, Y. ORF8 contributes to cytokine storm during SARS-CoV-2 infection by activating IL-17 pathway. iScience 2021, 24, 102293. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2021, 2, 99–112.e7. [Google Scholar] [CrossRef]
- Wang, H.; Rizvi, S.R.A.; Dong, D.; Lou, J.; Wang, Q.; Sopipong, W.; Su, Y.; Najar, F.; Agarwal, P.K.; Kozielski, F.; et al. Emerging variants of SARS-CoV-2 NSP10 highlight strong functional conservation of its binding to two nonstructural proteins, NSP14 and NSP16. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yang, X.-J. Omicron variant of SARS-CoV-2 gains new mutations in New Zealand and Hong Kong. bioRxiv 2022. [Google Scholar] [CrossRef]
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011, 7, e1002059. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell. Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.A.K.; Medina, P.M.B. Temporal changes in the accessory protein mutations of SARS-CoV-2 variants and their predicted structural and functional effects. J. Med. Virol. 2022, 94, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell. Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, A.; Abdullaev, A.; Abdurakhimov, A.; Abdurakhimov, A.; Mirakbarova, Z.; Mirakbarova, Z.; Ibragimova, S.; Ibragimova, S.; Tsoy, V.; Tsoy, V.; et al. Genome sequence diversity of SARS-CoV-2 obtained from clinical samples in Uzbekistan. PLoS ONE 2022, 17, e0270314. [Google Scholar] [CrossRef] [PubMed]
- Nealon, J.; Cowling, B.J. Omicron severity: Milder but not mild. Lancet 2022, 399, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Yeh, M.T.; Zinger, T.; Smith, M.; Wright, C.; Ling, G.; Nielsen, R.; Macadam, A.; Andino, R. The Evolutionary Pathway to Virulence of an RNA Virus. Cell 2017, 169, 35–46.e19. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef]
- Shafer, S.L. Intrinsic Severity of the SARS-CoV-2 Omicron Variant. N. Engl. J. Med. 2022, 386, 1867. [Google Scholar] [PubMed]
- Dhawan, M.; Sharma, A.; Priyanka, N.; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vaccin. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Liu, Y.; Ziarnik, M.; Seo, S.; Cao, Y.; Zhang, X.F.; Im, W. Binding of Human ACE2 and RBD of Omicron Enhanced by Unique Interaction Patterns among SARS-CoV-2 Variants of Concern. J. Comput. Chem. 2023, 44, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell. Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Dargham, S.R.; Chemaitelly, H.; Al Khal, A.; Tang, P.; Hasan, M.R.; Coyle, P.V.; Thomas, A.G.; Borham, A.M.; Concepcion, E.G.; et al. Severity of Illness in Persons Infected with the SARS-CoV-2 Delta Variant vs. Beta Variant in Qatar. JAMA Intern. Med. 2022, 182, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Dcosta, B.M.; Samanovic-Golden, M.; Herati, R.S.; Cornelius, A.; Mulligan, M.J.; Landau, N.R. Neutralization of viruses with European, South African, and United States SARS-CoV-2 variant spike proteins by convalescent sera and BNT162b2 mRNA vaccine-elicited antibodies. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell. Host. Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Lubinski, B.; Jaimes, J.A.; Whittaker, G.R. Intrinsic furin-mediated cleavability of the spike S1/S2 site from SARS-CoV-2 variant B.1.1.529 (Omicron). bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lorente-González, M.; Suarez-Orti, M.; Landete, P. Evolution and clinical trend of SARS-CoV-2 variants. Open Respir. Arch. 2022, 4, 100169. [Google Scholar] [CrossRef] [PubMed]
- Sheikhi, F.; Yousefian, N.; Tehranipoor, P.; Kowsari, Z. Estimation of the basic reproduction number of Alpha and Delta variants of COVID-19 pandemic in Iran. PLoS ONE 2022, 17, e0265489. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rocklöv, J. The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus. J. Travel. Med. 2021, 28, taab124. [Google Scholar] [CrossRef] [PubMed]
- Tandel, D.; Sah, V.; Singh, N.K.; Potharaju, P.S.; Gupta, D.; Shrivastava, S.; Sowpati, D.T.; Harshan, K.H. SARS-CoV-2 Variant Delta Potently Suppresses Innate Immune Response and Evades Interferon-Activated Antiviral Responses in Human Colon Epithelial Cells. Microbiol. Spectr. 2022, 10, e0160422. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e0207721. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Pamilo, P.; Bianchi, N.O. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evol. 1993, 10, 271–281. [Google Scholar] [PubMed]
- Maiti, A.K.; (Mydnavar, Southfield, MI, USA). The ω of SARS-CoV-2 does change markedly with a less number of genome study (<100 genome). 2024; material not intended for publication. [Google Scholar]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [PubMed]
- Li, W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Shayestehpour, M.; Mirzaei, H. The impact of spike mutated variants of SARS-CoV2 [Alpha, Beta, Gamma, Delta, and Lambda] on the efficacy of subunit recombinant vaccines. Braz. J. Infect. Dis. 2021, 25, 101606. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Azumaya, C.M.; Moritz, M.; Pourmal, S.; Diallo, A.; Merz, G.E.; Jang, G.; Bouhaddou, M.; Fossati, A.; Brilot, A.F.; et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Sahni, C.; Chowdhury, P.B.R.; Devadas, D.; Ashish, A.; Singh, N.K.; Yadav, A.; Kaur, M.; Mishra, S.; Vishwakarma, S.; Singh, R.; et al. SARS-CoV-2 Mutations Responsible for Immune Evasion Leading to Breakthrough Infection. Cureus 2022, 14, e29544. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello, F.; Soares, V.C.; Dias, S.S.; et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K. Identification of novel mutations in the methyltransferase complex (Nsp10-Nsp16) of SARS-CoV-2. Biochem. Biophys. Rep. 2020, 24, 100833. [Google Scholar] [CrossRef]
- Ilmjärv, S.; Abdul, F.; Acosta-Gutiérrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 13705. [Google Scholar] [CrossRef]
- Eskier, D.; Karakülah, G.; Suner, A.; Oktay, Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ 2020, 8, e9587. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; He, L.; Tian, Y.; Qin, Y.; Sun, H.; Ding, W.; Gui, L.; Wu, P. Molecular epidemiology analysis of early variants of SARS-CoV-2 reveals the potential impact of mutations P504L and Y541C (NSP13) in the clinical COVID-19 outcomes. Infect. Genet. Evol. 2021, 92, 104831. [Google Scholar] [CrossRef] [PubMed]
- Eskier, D.; Suner, A.; Oktay, Y.; Karakülah, G. Mutations of SARS-CoV-2 nsp14 exhibit strong association with increased genome-wide mutation load. PeerJ 2020, 8, e10181. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Papadimitriou, K.; Ioannidis, A.; Papandreou, N.C.; Milona, R.S.; Iconomidou, V.A.; Chatzipanagiotou, S. SARS-CoV-2 Amino Acid Mutations Detection in Greek Patients Infected in the First Wave of the Pandemic. Microorganisms 2022, 10, 1430. [Google Scholar] [CrossRef] [PubMed]
- Tasakis, R.N.; Samaras, G.; Jamison, A.; Lee, M.; Paulus, A.; Whitehouse, G.; Verkoczy, L.; Papavasiliou, F.N.; Diaz, M. SARS-CoV-2 variant evolution in the United States: High accumulation of viral mutations over time likely through serial Founder Events and mutational bursts. PLoS ONE 2021, 16, e0255169. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N.Z.; Sundar, D.; Rahman, S.A.; Hira, S.; Hasnain, S.E. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 2021, 13, 439. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 228. [Google Scholar]
- Mishra, S. Computational Structural and Functional Analyses of ORF10 in Novel Coronavirus SARS-CoV-2 Variants to Understand Evolutionary Dynamics. Evol. Bioinform. Online 2022, 18, 11769343221108218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ω-Alpha- Sept20 | ω-Delta- June21 | ω-Delta- July21 | ω-Omi- Jan22 | ω-Omi- March22 | ω-Omi- April22 | ω-Omi- May22 |
|---|---|---|---|---|---|---|---|
| Whole Genome (WG) | 0.35 | 0.68 | 0.56 | 0.79 | 0.64 | 0.68 | 0.67 |
| Spike | 0.15 | 1.24 | 0.64 | 1.97 | 1.97 | 1.07 | 1.43 |
| NSP1 | 0 | 0.83 | 0.97 | 0.51 | 0.49 | 1.96 | 0.5 |
| NSP2 | 0.47 | 0.6 | 0.62 | 0.5 | 0.44 | 0.51 | 0.45 |
| NSP3 | 0.48 | 0.56 | 0.53 | 0.75 | 0.41 | 0.67 | 0.75 |
| NSP4 | 0.49 | 0.63 | 0.54 | 0.33 | 0.57 | 0.94 | 4.94 |
| NSP5 | 0 | 0.4 | 0.31 | 0.48 | 0.2 | 0.29 | 0.099 |
| NSP6 | 0.4 | 1.13 | 0.98 | 0.43 | 0.79 | 0.41 | 0.66 |
| NSP7a | 0 | 0 | 0.5 | 0 | 0.54 | 0 | 0.55 |
| NSP8 | 0 | 0.16 | 1 | 0 | 0.51 | 0.76 | 0.1 |
| NSP9 | 0 | 0.33 | 0.29 | 0 | 0.39 | 0.34 | 0.29 |
| NSP10 | 0 | 0.08 | 0.73 | 0.68 | 0.3 | 0 | 0 |
| NSP12/RDRP | 0.27 | 0.68 | 0.42 | 0.72 | 0.6 | 0.46 | 0.6 |
| NSP13 | 0.17 | 0.59 | 0.42 | 0.48 | 1.06 | 0.24 | 0.26 |
| NSP14 | 0.33 | 0.3 | 0.18 | 0.83 | 0.54 | 0.39 | 1.23 |
| NSP15 | 1.07 | 0.34 | 1.4 | 3.56 | 0.2 | 0.81 | 0.27 |
| NSP16 | 0.093 | 2.37 | 0.15 | 0.56 | 0.15 | 0.44 | 0.15 |
| ORF3A | 1.85 | 1.69 | 1.55 | 3.33 | 0.39 | 1.39 | 0.3 |
| ORF4 | 0 | 0 | 1.04 | 0 | 0 | 1.15 | 0.57 |
| ORF5 | 0 | 0.85 | 0.16 | 0.64 | 0.3 | 0.23 | 0.4 |
| ORF6 | 0.4 | 1.64 | 0.98 | −67.7 | 0.55 | 1.54 | 0.55 |
| ORF7aA | 0 | 0 | 2.8 | 0.33 | 0 | 2.76 | 0 |
| ORF7B | 0 | 0.56 | 0.56 | 0 | 0.84 | 1.29 | 1.31 |
| ORF8 | 1.86 | 5.48 | 4.15 | 0.24 | 2.21 | 2.7 | 0 |
| ORF9 | 0.79 | 0.47 | 0.7 | 1.47 | 1.47 | 0.6 | 1 |
| ORF10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, A.K. Progressive Evolutionary Dynamics of Gene-Specific ω Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization. Int. J. Mol. Sci. 2024, 25, 6306. https://doi.org/10.3390/ijms25126306
Maiti AK. Progressive Evolutionary Dynamics of Gene-Specific ω Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization. International Journal of Molecular Sciences. 2024; 25(12):6306. https://doi.org/10.3390/ijms25126306
Chicago/Turabian StyleMaiti, Amit K. 2024. "Progressive Evolutionary Dynamics of Gene-Specific ω Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization" International Journal of Molecular Sciences 25, no. 12: 6306. https://doi.org/10.3390/ijms25126306
APA StyleMaiti, A. K. (2024). Progressive Evolutionary Dynamics of Gene-Specific ω Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization. International Journal of Molecular Sciences, 25(12), 6306. https://doi.org/10.3390/ijms25126306