
Genome-Wide Analysis of BBX Gene Family in Three Medicago Species Provides Insights into Expression Patterns under Hormonal and Salt Stresses

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification of BBX Genes
2.2. Phylogenetic Analysis of BBX Proteins
2.3. Conserved Motif, Domains, and Gene Structure Analysis of BBX Genes
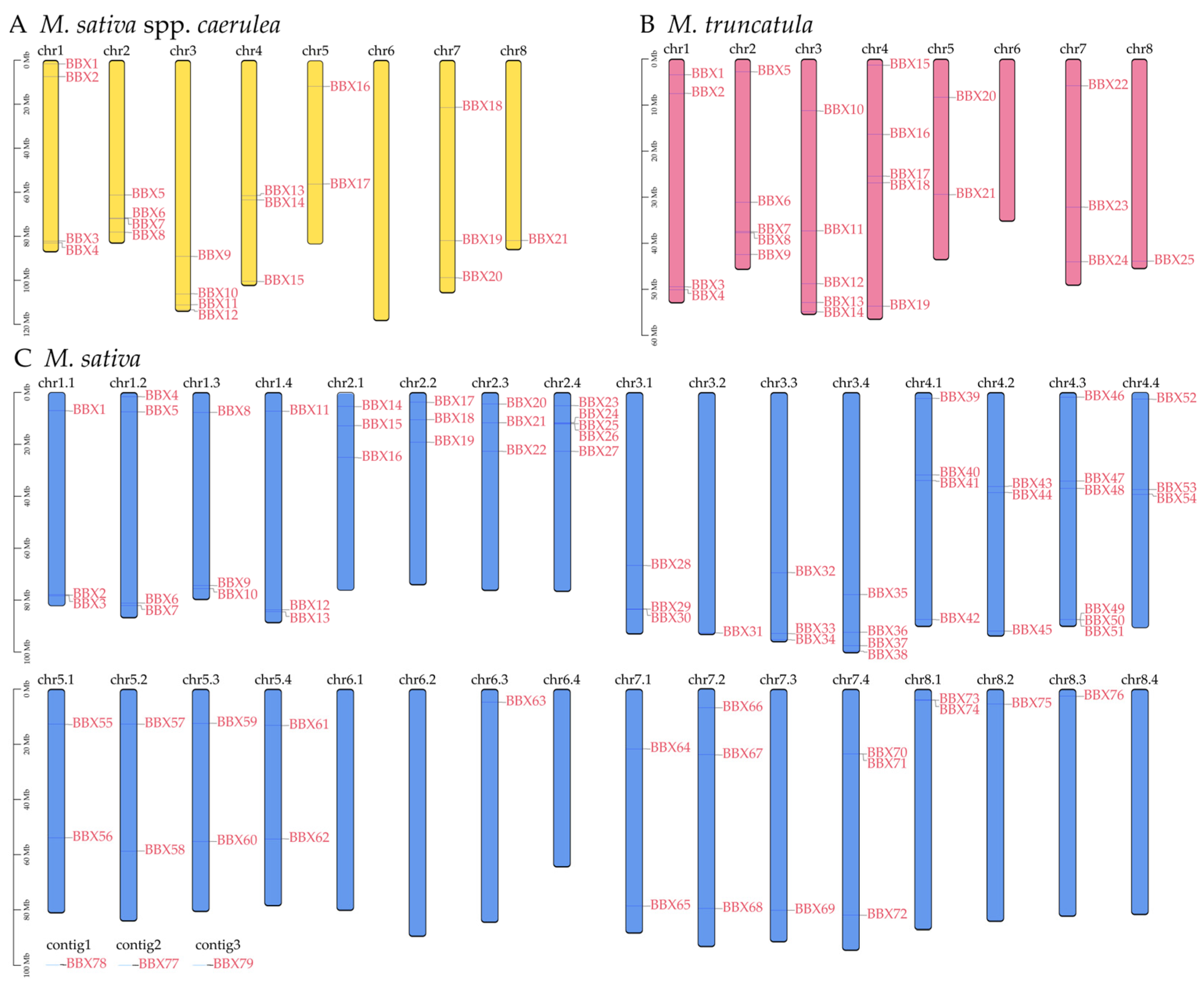
2.4. Chromosomal Localization Analysis of BBX
2.5. Analysis of the Synteny and Gene Duplication Event of the BBXs
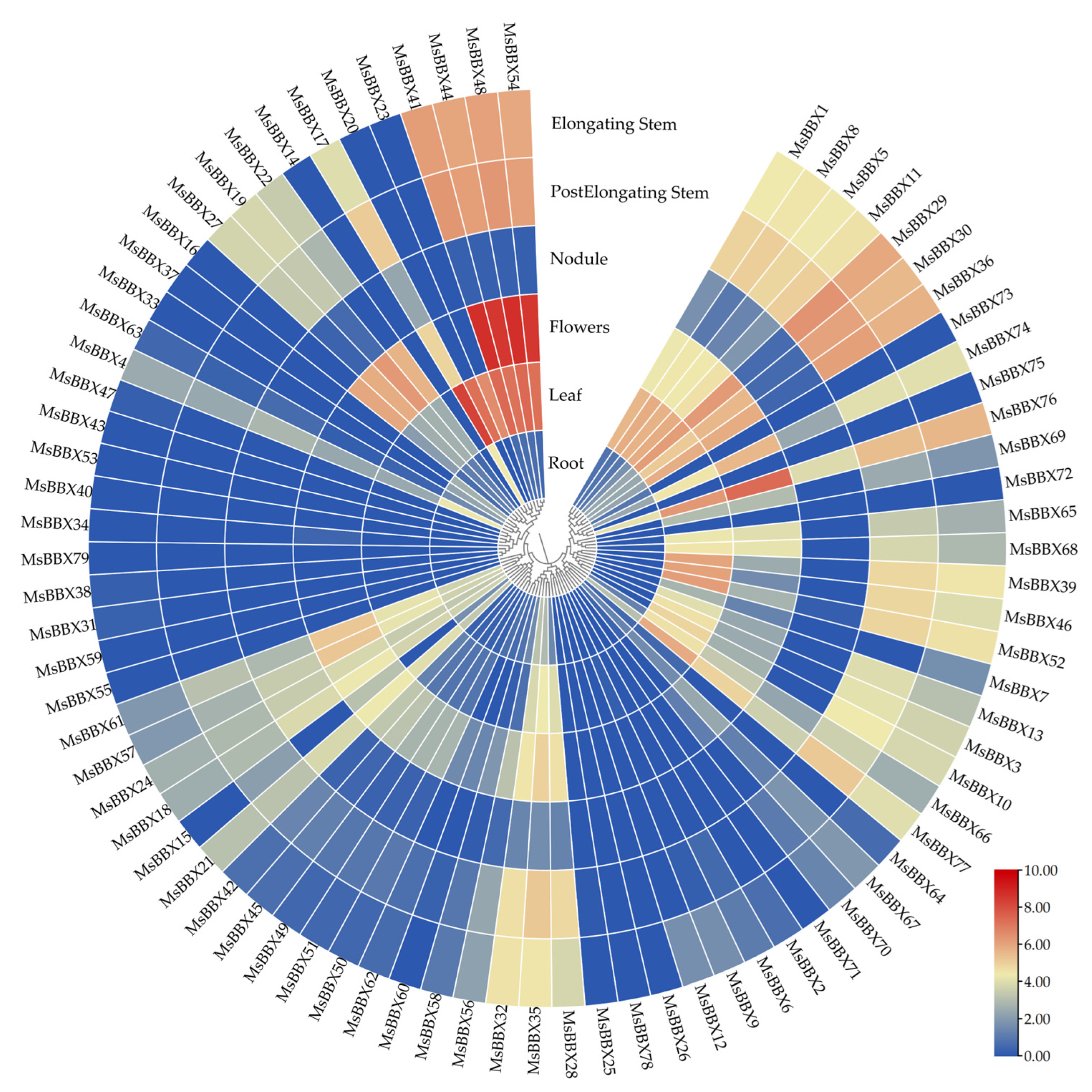
2.6. Expression Patterns of BBX Genes in Different Tissues
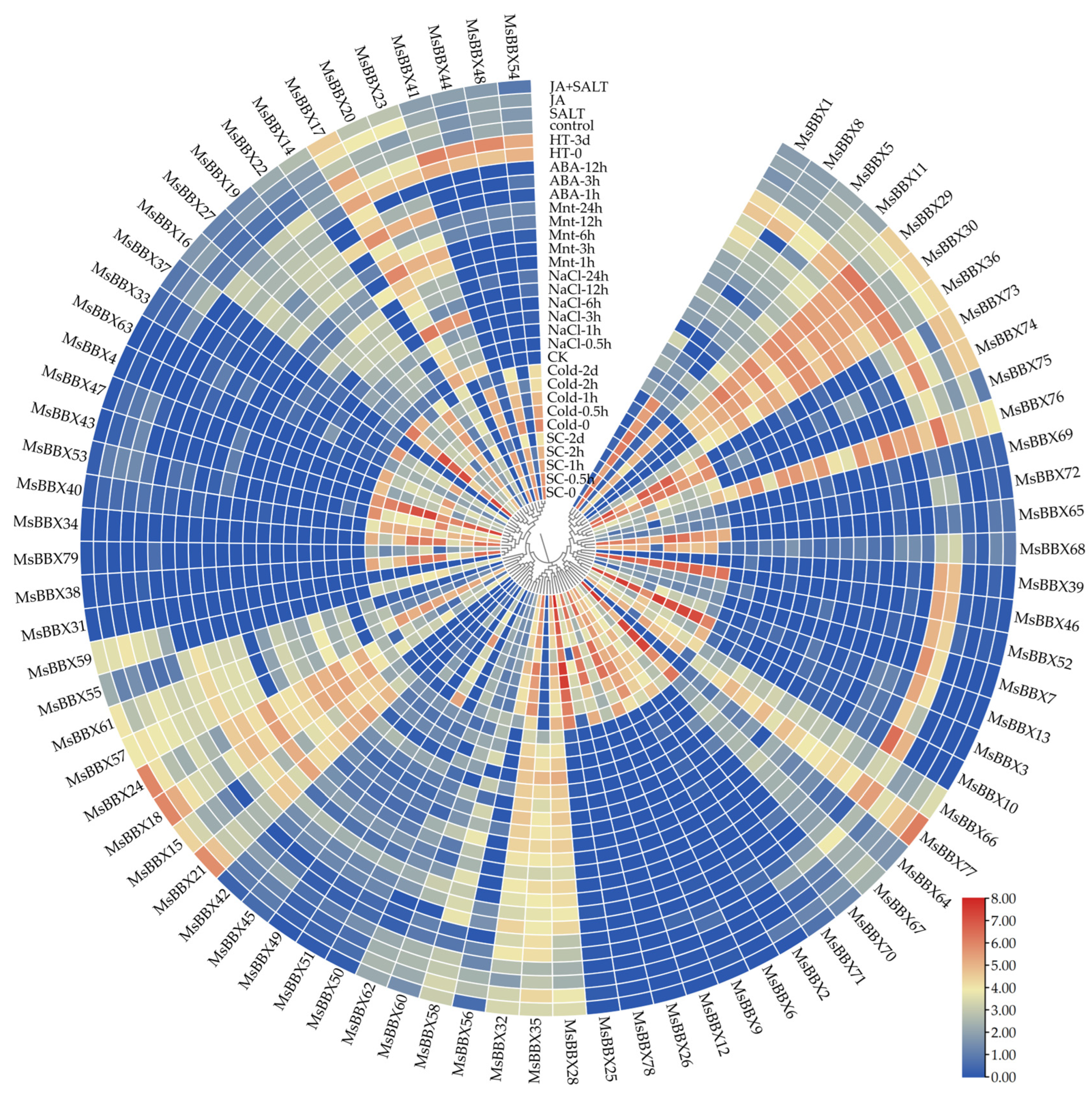
2.7. Expression Patterns of BBX Genes under Abiotic Stresses
2.8. qRT-PCR Analysis of BBX Genes under Hormone and Salt Stresses
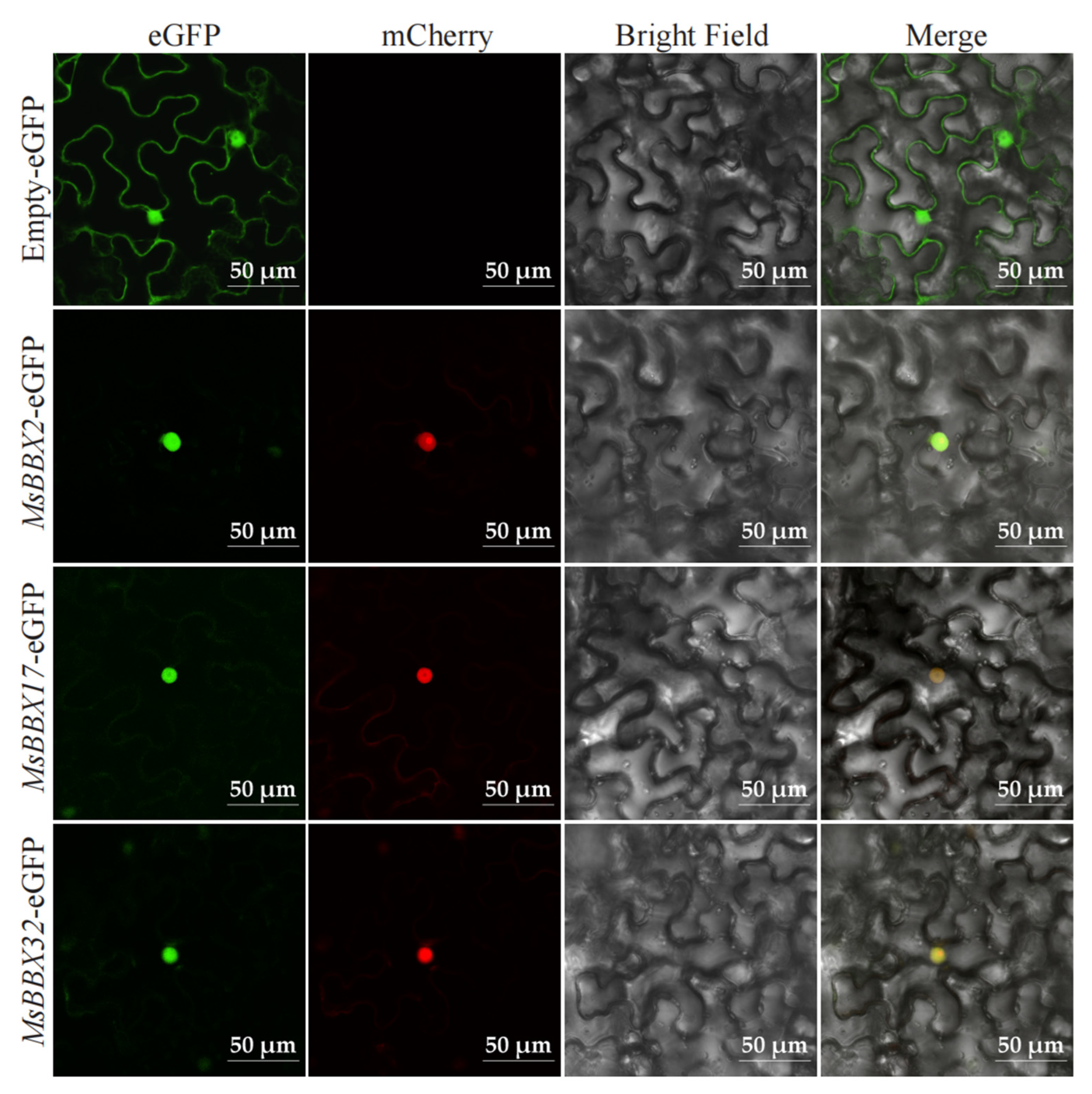
2.9. Subcellular Localization of BBX Genes
3. Discussion
4. Materials and Methods
4.1. Genome-Wide Identification of MaBBXs, MtBBXs, and MsBBXs
4.2. Phylogenetic Analysis of BBX Genes
4.3. Gene Structure, Conserved Domains, and Motif Composition of BBXs
4.4. Chromosomal Localization and Collinearity analysis of BBX
4.5. Analysis of Expression Level of MsBBX Gene
4.6. Plant Materials
4.7. Total RNA Extraction and qRT-PCR Analysis
4.8. Subcellular Localization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays News Rev. Mol. Cell. Dev. Biol. 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Min, J.-H.; Chung, J.-S.; Lee, K.-H.; Kim, C.S. The CONSTANS-like 4 transcription factor, AtCOL4, positively regulates abiotic stress tolerance through an abscisic acid-dependent manner in Arabidopsis. J. Integr. Plant Biol. 2015, 57, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wenkel, S.; Turck, F.; Singer, K.; Gissot, L.; Le Gourrierec, J.; Samach, A.; Coupland, G. CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis. Plant Cell 2006, 18, 2971–2984. [Google Scholar] [CrossRef] [PubMed]
- Massiah, M.A.; Matts, J.A.B.; Short, K.M.; Simmons, B.N.; Singireddy, S.; Yi, Z.; Cox, T.C. Solution structure of the MID1 B-box2 CHC(D/C)C2H2 zinc-binding domain: Insights into an evolutionarily conserved RING fold. J. Mol. Biol. 2007, 369, 1–10. [Google Scholar] [CrossRef]
- Crocco, C.D.; Botto, J.F. BBX proteins in green plants: Insights into their evolution, structure, feature and functional diversification. Gene 2013, 531, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Shalmani, A.; Fan, S.; Jia, P.; Li, G.; Muhammad, I.; Li, Y.; Sharif, R.; Dong, F.; Zuo, X.; Li, K.; et al. Genome Identification of B-BOX Gene Family Members in Seven Rosaceae Species and Their Expression Analysis in Response to Flower Induction in Malus domestica. Molecules 2018, 23, 1763. [Google Scholar] [CrossRef]
- Gangappa, S.N.; Botto, J.F. The BBX family of plant transcription factors. Trends Plant Sci. 2014, 19, 460–470. [Google Scholar] [CrossRef]
- Chu, Z.; Wang, X.; Li, Y.; Yu, H.; Li, J.; Lu, Y.; Li, H.; Ouyang, B. Genomic Organization, Phylogenetic and Expression Analysis of the B-BOX Gene Family in Tomato. Front. Plant Sci. 2016, 7, 1552. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, X.; Weng, X.; Wang, L.; Xie, W. The rice B-box zinc finger gene family: Genomic identification, characterization, expression profiling and diurnal analysis. PLoS ONE 2012, 7, e48242. [Google Scholar] [CrossRef]
- Feng, Z.; Li, M.; Li, Y.; Yang, X.; Wei, H.; Fu, X.; Ma, L.; Lu, J.; Wang, H.; Yu, S. Comprehensive identification and expression analysis of B-Box genes in cotton. BMC Genom. 2021, 22, 439. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Ji, M.; Wu, Y.; Zhang, S.; Zhu, Y.; Yao, J.; Li, Z.; Gao, H.; Wang, X. Genome-wide identification and expression analysis of the B-box transcription factor gene family in grapevine (Vitis vinifera L.). BMC Genom. 2021, 22, 221. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hettiarachchi, G.H.C.M.; Deng, X.-W.; Holm, M. Arabidopsis CONSTANS-LIKE3 is a positive regulator of red light signaling and root growth. Plant Cell 2006, 18, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Ando, E.; Ohnishi, M.; Wang, Y.; Matsushita, T.; Watanabe, A.; Hayashi, Y.; Fujii, M.; Ma, J.F.; Inoue, S.-i.; Kinoshita, T. TWIN SISTER OF FT, GIGANTEA, and CONSTANS have a positive but indirect effect on blue light-induced stomatal opening in Arabidopsis. Plant Physiol. 2013, 162, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Mandaokar, A.; Thines, B.; Shin, B.; Lange, B.M.; Choi, G.; Koo, Y.J.; Yoo, Y.J.; Choi, Y.D.; Choi, G.; Browse, J. Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. Cell Mol. Biol. 2006, 46, 984–1008. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, H.; Ping, Q.; Zhang, Z.; Guan, Z.; Fang, W.; Chen, S.; Chen, F.; Jiang, J.; Zhang, F. The heterologous expression of CmBBX22 delays leaf senescence and improves drought tolerance in Arabidopsis. Plant Cell Rep. 2019, 38, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.; Takano, T. Salt tolerance-related protein STO binds to a Myb transcription factor homologue and confers salt tolerance in Arabidopsis. J. Exp. Bot. 2003, 54, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Gangappa, S.N.; Crocco, C.D.; Johansson, H.; Datta, S.; Hettiarachchi, C.; Holm, M.; Botto, J.F. The Arabidopsis B-BOX protein BBX25 interacts with HY5, negatively regulating BBX22 expression to suppress seedling photomorphogenesis. Plant Cell 2013, 25, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Soitamo, A.J.; Piippo, M.; Allahverdiyeva, Y.; Battchikova, N.; Aro, E.-M. Light has a specific role in modulating Arabidopsis gene expression at low temperature. BMC Plant Biol. 2008, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Kirik, V.; Bäumlein, H. A novel leaf-specific myb-related protein with a single binding repeat. Gene 1996, 183, 109–113. [Google Scholar] [CrossRef]
- Salanoubat, M.; Lemcke, K.; Rieger, M.; Ansorge, W.; Unseld, M.; Fartmann, B.; Valle, G.; Blöcker, H.; Perez-Alonso, M.; Obermaier, B.; et al. Sequence and analysis of chromosome 3 of the plant Arabidopsis thaliana. Nature 2000, 408, 820–822. [Google Scholar]
- Datta, S.; Hettiarachchi, C.; Johansson, H.; Holm, M. SALT TOLERANCE HOMOLOG2, a B-box protein in Arabidopsis that activates transcription and positively regulates light-mediated development. Plant Cell 2007, 19, 3242–3255. [Google Scholar] [CrossRef]
- An, J.-P.; Wang, X.-F.; Espley, R.V.; Lin-Wang, K.; Bi, S.-Q.; You, C.-X.; Hao, Y.-J. An Apple B-Box Protein MdBBX37 Modulates Anthocyanin Biosynthesis and Hypocotyl Elongation Synergistically with MdMYBs and MdHY5. Plant Cell Physiol. 2020, 61, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ouhibi, C.; Attia, H.; Rebah, F.; Msilini, N.; Chebbi, M.; Aarrouf, J.; Urban, L.; Lachaal, M. Salt stress mitigation by seed priming with UV-C in lettuce plants: Growth, antioxidant activity and phenolic compounds. Plant Physiol. Biochem. PPB 2014, 83, 126–133. [Google Scholar] [CrossRef]
- Yu, Z.; Duan, X.; Luo, L.; Dai, S.; Ding, Z.; Xia, G. How Plant Hormones Mediate Salt Stress Responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef]
- Liu, X.; Li, R.; Dai, Y.; Chen, X.; Wang, X. Genome-wide identification and expression analysis of the B-box gene family in the Apple (Malus domestica Borkh.) genome. Mol. Genet. Genom. 2018, 293, 303–315. [Google Scholar] [CrossRef]
- Liu, X.; Li, R.; Dai, Y.; Yuan, L.; Sun, Q.; Zhang, S.; Wang, X. A B-box zinc finger protein, MdBBX10, enhanced salt and drought stresses tolerance in Arabidopsis. Plant Mol. Biol. 2019, 99, 437–447. [Google Scholar] [CrossRef]
- An, J.-P.; Wang, X.-F.; Zhang, X.-W.; You, C.-X.; Hao, Y.-J. Apple B-box protein BBX37 regulates jasmonic acid mediated cold tolerance through the JAZ-BBX37-ICE1-CBF pathway and undergoes MIEL1-mediated ubiquitination and degradation. New Phytol. 2021, 229, 2707–2729. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zheng, L.; Xie, R. Effect of pre-harvest application of ethephon on colouration and expression of ripening related genes in citrus fruit. J. Hortic. Sci. Biotechnol. 2020, 96, 514–526. [Google Scholar] [CrossRef]
- Gharbi, E.; Martínez, J.-P.; Benahmed, H.; Lepoint, G.; Vanpee, B.; Quinet, M.; Lutts, S. Inhibition of ethylene synthesis reduces salt-tolerance in tomato wild relative species Solanum chilense. J. Plant Physiol. 2017, 210, 24–37. [Google Scholar] [CrossRef]
- Chang, C.; Wang, B.; Shi, L.; Li, Y.; Duo, L.; Zhang, W. Alleviation of salt stress-induced inhibition of seed germination in cucumber (Cucumis sativus L.) by ethylene and glutamate. J. Plant Physiol. 2010, 167, 1152–1156. [Google Scholar] [CrossRef]
- Jayakannan, M.; Bose, J.; Babourina, O.; Rengel, Z.; Shabala, S. Salicylic acid in plant salinity stress signalling and tolerance. Plant Growth Regul. 2015, 76, 25–40. [Google Scholar] [CrossRef]
- Khoury, C.K.; Bjorkman, A.D.; Dempewolf, H.; Ramirez-Villegas, J.; Guarino, L.; Jarvis, A.; Rieseberg, L.H.; Struik, P.C. Increasing homogeneity in global food supplies and the implications for food security. Proc. Natl. Acad. Sci. USA 2014, 111, 4001–4006. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zeng, Y.; Yang, Y.; Huang, L.; Tang, B.; Zhang, H.; Hao, F.; Liu, W.; Li, Y.; Liu, Y.; et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 2020, 11, 2494. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, A.; Du, X.; Chen, J.-Y.; Yin, M.; Hu, H.-Y.; Shrestha, N.; Wu, S.-D.; Wang, H.-Q.; Dou, Q.-W.; et al. A chromosome-scale genome assembly of a diploid alfalfa, the progenitor of autotetraploid alfalfa. Hortic. Res. 2020, 7, 194. [Google Scholar] [CrossRef]
- Pecrix, Y.; Staton, S.E.; Sallet, E.; Lelandais-Brière, C.; Moreau, S.; Carrère, S.; Blein, T.; Jardinaud, M.-F.; Latrasse, D.; Zouine, M.; et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 2018, 4, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Shen, Y.-P.; Ma, L.-G.; Pan, Y.; Du, Y.-L.; Wang, D.-H.; Yang, J.-Y.; Hu, L.-D.; Liu, X.-F.; Dong, C.-X.; et al. Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol. 2004, 135, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Chien, T.-C.; Chen, T.-Y.; Chiang, M.-H.; Lai, M.-H.; Chang, M.-C. Overexpression of a Novel ERF-X-Type Transcription Factor, OsERF106MZ, Reduces Shoot Growth and Tolerance to Salinity Stress in Rice. Rice 2021, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Valverde, F. CONSTANS and the evolutionary origin of photoperiodic timing of flowering. J. Exp. Bot. 2011, 62, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Ledger, S.; Strayer, C.; Ashton, F.; Kay, S.A.; Putterill, J. Analysis of the function of two circadian-regulated CONSTANS-LIKE genes. Plant J. Cell Mol. Biol. 2001, 26, 15–22. [Google Scholar] [CrossRef]
- Wang, Q.; Zeng, J.; Deng, K.; Tu, X.; Zhao, X.; Tang, D.; Liu, X. DBB1a, involved in gibberellin homeostasis, functions as a negative regulator of blue light-mediated hypocotyl elongation in Arabidopsis. Planta 2011, 233, 13–23. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Z.; Luo, S.; Li, X.; Lyu, J.; Liu, Z.; Wan, Z.; Yu, J. Genome-wide identification and expression analysis of the cucumber PP2C gene family. BMC Genom. 2022, 23, 563. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Sverdlov, A.V.; Babenko, V.N.; Koonin, E.V. Analysis of evolution of exon-intron structure of eukaryotic genes. Brief Bioinform. 2005, 6, 118–134. [Google Scholar] [CrossRef]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Karpinska, B.; Razak, N.; James, E.K.; Morris, J.A.; Verrall, S.R.; Hedley, P.E.; Hancock, R.D.; Foyer, C.H. WHIRLY1 functions in the nucleus to regulate barley leaf development and associated metabolite profiles. Biochem. J. 2022, 479, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, T.; Li, M.; Dong, X.; Han, Y.; Xu, C.; Li, S.; Zhang, J.; He, X.; Zhou, Q.; et al. MODMS: A multi-omics database for facilitating biological studies on alfalfa (Medicago sativa L.). Hortic. Res. 2024, 11, uhad245. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, gkae268. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Meng, Z.; He, H.; Du, P.; Dijkwel, P.P.; Shi, S.; Li, H.; Xie, Q. Genome-Wide Analysis of BBX Gene Family in Three Medicago Species Provides Insights into Expression Patterns under Hormonal and Salt Stresses. Int. J. Mol. Sci. 2024, 25, 5778. https://doi.org/10.3390/ijms25115778
Wang J, Meng Z, He H, Du P, Dijkwel PP, Shi S, Li H, Xie Q. Genome-Wide Analysis of BBX Gene Family in Three Medicago Species Provides Insights into Expression Patterns under Hormonal and Salt Stresses. International Journal of Molecular Sciences. 2024; 25(11):5778. https://doi.org/10.3390/ijms25115778
Chicago/Turabian StyleWang, Jiayin, Zhuang Meng, Huan He, Pingping Du, Paul P. Dijkwel, Shandang Shi, Hongbin Li, and Quanliang Xie. 2024. "Genome-Wide Analysis of BBX Gene Family in Three Medicago Species Provides Insights into Expression Patterns under Hormonal and Salt Stresses" International Journal of Molecular Sciences 25, no. 11: 5778. https://doi.org/10.3390/ijms25115778
APA StyleWang, J., Meng, Z., He, H., Du, P., Dijkwel, P. P., Shi, S., Li, H., & Xie, Q. (2024). Genome-Wide Analysis of BBX Gene Family in Three Medicago Species Provides Insights into Expression Patterns under Hormonal and Salt Stresses. International Journal of Molecular Sciences, 25(11), 5778. https://doi.org/10.3390/ijms25115778