
Adenosine A3 Receptor: From Molecular Signaling to Therapeutic Strategies for Heart Diseases

 , ,
, ,
Abstract
1. Introduction
2. The Distribution of A3ARs in the Cardiovascular System
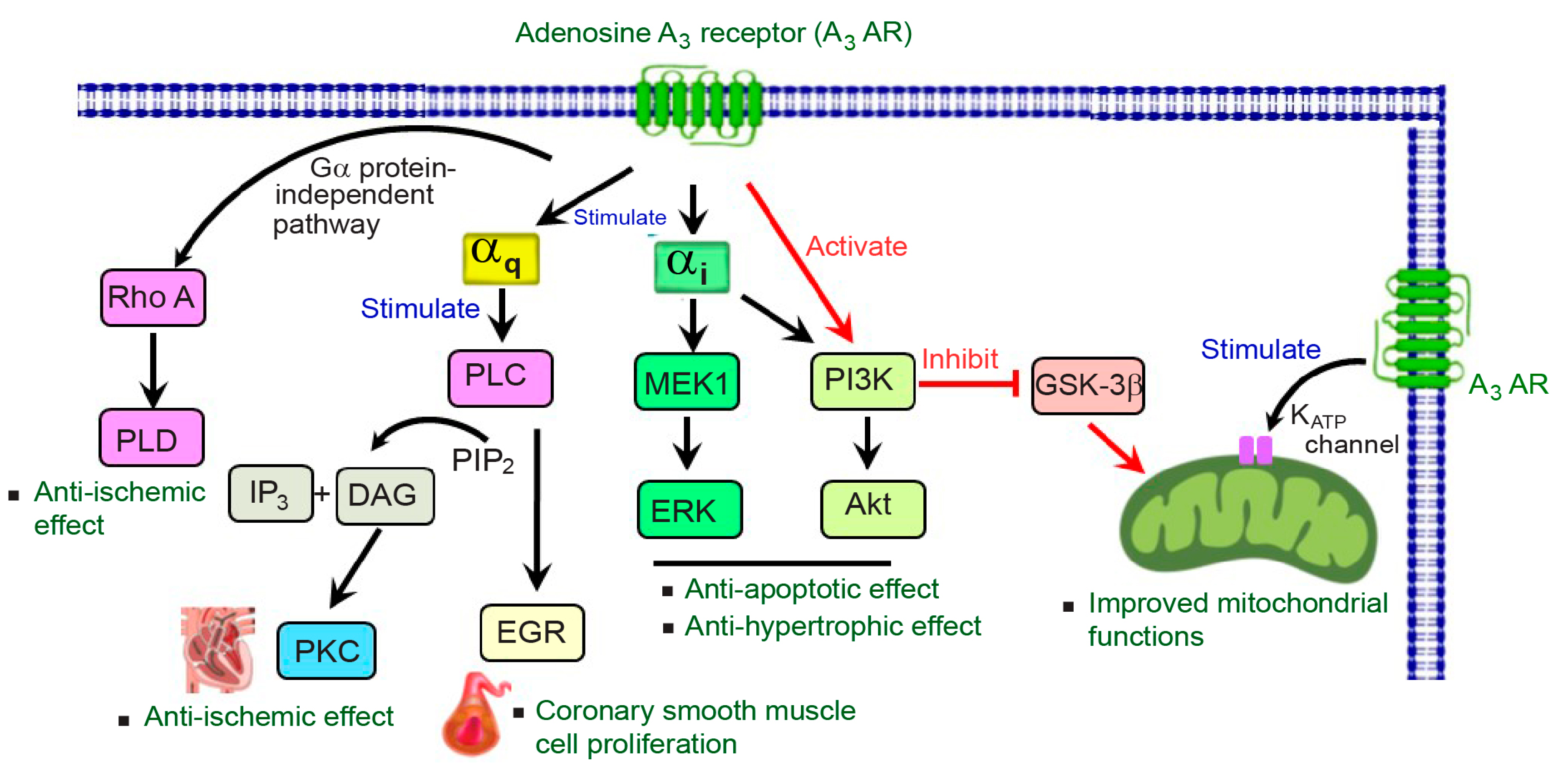
3. A3AR Signaling in the Cardiovascular System
3.1. Gi Protein-Dependent Signaling
3.2. Gq Protein-Dependent Signaling
3.3. ATP-Sensitive Potassium Channels (KATP)
3.4. G Protein-Independent Signaling
4. The Role of A3ARs in Heart Diseases
4.1. Contributions of A3ARs in Ischemic Heart Disease

{kind=link}
| Agents | Models | Main Findings |
|---|---|---|
| Ischemia and myocardial infarction | ||
| CP-532903 [57] | Mice model of ischemia-reperfusion (I/R) injury |
|
| CP-532903 [60] | Isolated rat hearts and isolated cardiomyocytes |
|
| CP-532903 and CP-608039 [61] | Rabbit model of I/R injury |
|
| Namodenoson (2Cl-IB-MECA; CF-102) [65] | Mice model of I/R injury |
|
| Namodenoson (2Cl-IB-MECA; CF-102) [19] | Isolated rat hearts with I/R injury |
|
| Namodenoson (2Cl-IB-MECA; CF-102) [18] | Mice model of I/R injury |
|
| Piclidenoson (IB-MECA; CF-101) [62] | Rabbit model of I/R injury |
|
| Piclidenoson (IB-MECA; CF-101) [4] | Isolated human atrial muscle |
|
| Piclidenoson (IB-MECA; CF-101) [68] | Rabbit model of I/R injury |
|
| Heart failure | ||
| N6-cyclopentyl-adenosine (CPA; full agonist) [28] | Wild-type and A3AR-knockout mice with transverse aortic constriction-induced pressure overload |
|
4.1.1. Anti-Inflammatory Effects
4.1.2. Protection of Oxidative Stress, Apoptosis, and Mitochondrial Dysfunction
4.2. Impacts of A3ARs on Heart Failure
4.2.1. Hypertrophic Effects
4.2.2. Fibrotic Effects
| Agents | Models | Main Findings |
|---|---|---|
| 2-Chloroadenosine (adenosine analog) [28] | Wild-type and A3AR-knockout mice with transverse aortic constriction-induced pressure overload |
|
| Cl-IB-MECA (2Cl-IB-MECA; CF-102) [78] | Wild-type and A3AR-knockout mice aorta |
|
4.2.3. Effects on Cardiac Contractility and Heart Rate
4.3. Involvement of A3ARs in Hypertension
5. The Role of A3AR Agonists in the Treatment of Heart Diseases
6. Clinical Studies of A3AR Agonists for the Treatment of Non-Cardiac Diseases
6.1. Namodenoson (2Cl-IB-MECA; CF-102)
| Drug | Study Population | Treatment | Primary Endpoints | Main Findings |
|---|---|---|---|---|
| Namodenoson (Phase II) [89] | NAFLD patients with or without NASH (N = 60) | Namodenoson (12.5 or 25 mg) or placebo, BID, for 12 weeks | Serum ALT level |
|
| Namodenoson (Phase II) [91] | Patients with advanced HCC and Child-Pugh B cirrhosis (N = 78) | Namodenoson (25 mg) or placebo, BID | Overall survival |
|
| Piclidenoson (CF-101) (Phase I) [92] | Healthy men (N = 43) | CF-101 (1, 5, or 10 mg) or placebo, single dose and repeated doses of up to 4 mg BID, for 7 days | Safety, tolerability, pharmacokinetics, and hemodynamic profiles |
|
| Piclidenoson (CF-101) (Phase II/III) [93] | Patients with moderate-to-severe chronic plaque psoriasis (N = 293) | CF-101 (1 or 2 mg) or placebo, BID, followed up to 32 weeks | Proportion of patients achieving ≥ 75% improvement in PASI |
|
| Piclidenoson (Phase III; COMFORT-1) [94] | Patients with moderate-to-severe chronic plaque psoriasis (N = 529) | Piclidenoson (2 or 3 mg), apremilast (30 mg), or placebo, BID, for 16 weeks | Proportion of patients achieving ≥ 75% improvement in PASI |
|
6.2. Piclidenoson (IB-MECA; CF-101)
7. Limitations and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Reiss, A.B.; Grossfeld, D.; Kasselman, L.J.; Renna, H.A.; Vernice, N.A.; Drewes, W.; Konig, J.; Carsons, S.E.; DeLeon, J. Adenosine and the cardiovascular system. Am. J. Cardiovasc. Drugs 2019, 19, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.T.; Swierkosz, T.A.; Herrmann, H.C.; Kimmel, S.; Jacobson, K.A. Adenosine and ischemic preconditioning. Curr. Pharm. Des. 1999, 5, 1029. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.S.; Hill, R.J.; Masamune, H.; Kennedy, S.P.; Knight, D.R.; Tracey, W.R.; Yellon, D.M. Evidence for a role for both the adenosine A1 and A3 receptors in protection of isolated human atrial muscle against simulated ischaemia. Cardiovasc. Res. 1997, 36, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Albrecht-Küpper, B.E.; Leineweber, K.; Nell, P.G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2012, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.J.; Sabbah, H.N.; Butler, J.; Voors, A.A.; Albrecht-Küpper, B.E.; Düngen, H.D.; Dinh, W.; Gheorghiade, M. Partial adenosine A1 receptor agonism: A potential new therapeutic strategy for heart failure. Heart Fail. Rev. 2016, 21, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Nishat, S.; Khan, L.A.; Ansari, Z.M.; Basir, S.F. Adenosine A3 receptor: A promising therapeutic target in cardiovascular disease. Curr. Cardiol. Rev. 2016, 12, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Rothermel, B.A.; Hill, J.A. Adenosine A3 receptor and cardioprotection: Enticing, enigmatic, elusive. Circulation 2008, 118, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; Ashton, K.J.; Rose’Meyer, R.B.; Peart, J.N. Cardiovascular adenosine receptors: Expression, actions and interactions. Pharmacol. Ther. 2013, 140, 92–111. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Varani, K.; Leung, E.; Mac Lennan, S.; Borea, P.A. The A3 adenosine receptor: An enigmatic player in cell biology. Pharmacol. Ther. 2008, 117, 123–140. [Google Scholar] [CrossRef]
- Garcia-Garcia, L.; Olle, L.; Martin, M.; Roca-Ferrer, J.; Muñoz-Cano, R. Adenosine signaling in mast cells and allergic diseases. Int. J. Mol. Sci. 2021, 22, 5203. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; Contri, C.; Borea, P.A.; Vincenzi, F.; Varani, K. Adenosine and inflammation: Here, there and everywhere. Int. J. Mol. Sci. 2021, 22, 7685. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, C.A.; Jacobson, M.A.; Taylor, H.E.; Linden, J.; Johnson, R.G. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10365–10369. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [PubMed]
- Brandon, C.I.; Vandenplas, M.; Dookwah, H.; Murray, T.F. Cloning and pharmacological characterization of the equine adenosine A3 receptor. J. Vet. Pharmacol. Ther. 2006, 29, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.R.; Townsend-Nicholson, A.; Nicholl, J.K.; Sutherland, G.R.; Schofield, P.R. Cloning, characterisation and chromosomal assignment of the human adenosine A3 receptor (ADORA3) gene. Neurosci. Res. 1997, 29, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.D.; van der Hoeven, D.; Maas, J.E.; Wan, T.C.; Auchampach, J.A. A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J. Mol. Cell. Cardiol. 2010, 49, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Gharanei, A.M.; Nagra, A.S.; Maddock, H.L. Caspase inhibition via A3 adenosine receptors: A new cardioprotective mechanism against myocardial infarction. Cardiovasc. Drugs Ther. 2014, 28, 19–32. [Google Scholar] [CrossRef]
- Germack, R.; Dickenson, J.M. Adenosine triggers preconditioning through MEK/ERK1/2 signalling pathway during hypoxia/reoxygenation in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 429–442. [Google Scholar] [CrossRef]
- Sandhu, H.; Cooper, S.; Hussain, A.; Mee, C.; Maddock, H. Attenuation of Sunitinib-induced cardiotoxicity through the A3 adenosine receptor activation. Eur. J. Pharmacol. 2017, 814, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Emanuelov, A.K.; Shainberg, A.; Chepurko, Y.; Kaplan, D.; Sagie, A.; Porat, E.; Arad, M.; Hochhauser, E. Adenosine A3 receptor-mediated cardioprotection against doxorubicin-induced mitochondrial damage. Biochem. Pharmacol. 2010, 79, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Talukder, M.H.; Morrison, R.R.; Mustafa, S.J. Comparison of the vascular effects of adenosine in isolated mouse heart and aorta. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H49–H57. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.K.; Linden, J.; Duling, B.R. Adenosine-induced vasoconstriction in vivo: Role of the mast cell and A3 adenosine receptor. Circ. Res. 1996, 78, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Chen, F.; Li, Q.; Hu, D.Y.; Liu, L.M. Stimulation of the adenosine A3 receptor reverses vascular hyporeactivity after hemorrhagic shock in rats. Acta Pharmacol. Sin. 2010, 31, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Cross, H.R.; Murphy, E.; Black, R.G.; Auchampach, J.; Steenbergen, C. Overexpression of A3 adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1562–H1568. [Google Scholar] [CrossRef] [PubMed]
- Black, R.G., Jr.; Guo, Y.; Ge, Z.D.; Murphree, S.S.; Prabhu, S.D.; Jones, W.K.; Bolli, R.; Auchampach, J.A. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ. Res. 2002, 91, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Fassett, J.; Xu, X.; Hu, X.; Zhu, G.; French, J.; Zhang, P.; Schnermann, J.; Bache, R.J.; Chen, Y. Adenosine A3 receptor deficiency exerts unanticipated protective effects on the pressure-overloaded left ventricle. Circulation 2008, 118, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.F.; Low, L.M.; Rose’Meyer, R.B. Pharmacology of the adenosine A3 receptor in the vasculature and essential hypertension. PLoS ONE 2016, 11, e0150021. [Google Scholar] [CrossRef]
- Linden, J. Cloned adenosine A3 receptors: Pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306. [Google Scholar] [CrossRef]
- Hinze, A.V.; Mayer, P.; Harst, A.; von Kügelgen, I. Adenosine A(3) receptorinduced proliferation of primary human coronary smooth muscle cells involving the induction of early growth response genes. J. Mol. Cell. Cardiol. 2012, 53, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Grandoch, M.; Hoffmann, J.; Röck, K.; Wenzel, F.; Oberhuber, A.; Schelzig, H.; Fischer, J.W. Novel effects of adenosine receptors on pericellular hyaluronan matrix: Implications for human smooth muscle cell phenotype and interactions with monocytes during atherosclerosis. Basic. Res. Cardiol. 2013, 108, 340. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Makaritsis, K.; Francis, C.E.; Gavras, H.; Ravid, K. A role for the A3 adenosine receptor in determining tissue levels of cAMP and blood pressure: Studies in knock-out mice. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2000, 1500, 280–290. [Google Scholar] [CrossRef]
- Tarifa, C.; Jiménez-Sábado, V.; Franco, R.; Montiel, J.; Guerra, J.; Ciruela, F.; Hove-Madsen, L. Expression and impact of adenosine A3 receptors on calcium homeostasis in human right atrium. Int. J. Mol. Sci. 2023, 24, 4404. [Google Scholar] [CrossRef] [PubMed]
- Tracey, W.R.; Magee, W.; Masamune, H.; Kennedy, S.P.; Knight, D.R.; Buchholz, R.A.; Hill, R.J. Selective adenosine A3 receptor stimulation reduces ischemic myocardial injury in the rabbit heart. Cardiovasc. Res. 1997, 33, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Headrick, J.P. Adenosinergic cardioprotection: Multiple receptors, multiple pathways. Pharmacol. Ther. 2007, 114, 208–221. [Google Scholar] [CrossRef]
- Vincenzi, F.; Pasquini, S.; Contri, C.; Cappello, M.; Nigro, M.; Travagli, A.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K. Pharmacology of adenosine receptors: Recent advancements. Biomolecules 2023, 13, 1387. [Google Scholar] [CrossRef] [PubMed]
- Koscsó, B.; Csóka, B.; Pacher, P.; Haskó, G. Investigational A3 adenosine receptor targeting agents. Expert. Opin. Investig. Drugs. 2011, 20, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Redfern, C.H.; Degtyarev, M.Y.; Kwa, A.T.; Salomonis, N.; Cotte, N.; Nanevicz, T.; Fidelman, N.; Desai, K.; Vranizan, K.; Lee, E.K.; et al. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 4826–4831. [Google Scholar] [CrossRef]
- Redfern, C.H.; Coward, P.; Degtyarev, M.Y.; Lee, E.K.; Kwa, A.T.; Hennighausen, L.; Bujard, H.; Fishman, G.I.; Conklin, B.R. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat. Biotechnol. 1999, 17, 165–169. [Google Scholar] [CrossRef]
- Germack, R.; Griffin, M.; Dickenson, J.M. Activation of protein kinase B by adenosine A1 and A3 receptors in newborn rat cardiomyocytes. J. Mol. Cell. Cardiol. 2004, 37, 989–999. [Google Scholar] [CrossRef]
- Das, S.; Cordis, G.A.; Maulik, N.; Das, D.K. Pharmacological preconditioning with resveratrol: Role of CREB-dependent Bcl-2 signaling via adenosine A3 receptor activation. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H328–H335. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Tosaki, A.; Bagchi, D.; Maulik, N.; Das, D.K. Resveratrol-mediated activation of cAMP response element-binding protein through adenosine A3 receptor by Akt-dependent and-independent pathways. J. Pharmacol. Exp. Ther. 2005, 314, 762–769. [Google Scholar] [CrossRef]
- Schulte, G.; Fredholm, B.B. Human adenosine A1, A2A, A2B, and A3 receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol. Pharmacol. 2000, 58, 477–482. [Google Scholar] [CrossRef]
- Schulte, G.; Fredholm, B.B. Signaling pathway from the human adenosine A3 receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol. Pharmacol. 2002, 62, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Salie, R.; Moolman, J.A.; Lochner, A. The mechanism of beta-adrenergic preconditioning: Roles for adenosine and ROS during triggering and mediation. Basic Res. Cardiol. 2012, 107, 281. [Google Scholar] [CrossRef] [PubMed]
- Leshem-Lev, D.; Hochhauser, E.; Chanyshev, B.; Isak, A.; Shainberg, A. Adenosine A1 and A3 receptor agonists reduce hypoxic injury through the involvement of p38 MAPK. Mol. Cell Biochem. 2010, 345, 153–160. [Google Scholar] [CrossRef]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J. Biol. Chem. 2005, 280, 19516–19526. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Vincenzi, F.; Tosi, A.; Targa, M.; Masieri, F.F.; Ongaro, A.; de Mattei, M.; Massari, L.; Borea, P.A. Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br. J. Pharmacol. 2010, 160, 101–115. [Google Scholar] [CrossRef]
- Park, S.S.; Zhao, H.; Jang, Y.; Mueller, R.A.; Xu, Z. N6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3β. J. Pharmacol. Exp. Ther. 2006, 318, 124–131. [Google Scholar] [CrossRef]
- Kutryb-Zając, B.; Kawecka, A.; Nasadiuk, K.; Braczko, A.; Stawarska, K.; Caiazzo, E.; Koszałka, P.; Cicala, C. Drugs targeting adenosine signaling pathways: A current view. Biomed. Pharmacother. 2023, 165, 115184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.C.; Kukreja, R.C. Protein kinase C-δ mediates adenosine A3 receptor-induced delayed cardioprotection in mouse. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H434–H441. [Google Scholar] [CrossRef] [PubMed]
- Ghelardoni, S.; Frascarelli, S.; Carnicelli, V.; Ronca-Testoni, S.; Zucchi, R. Modulation of cardiac sarcoplasmic reticulum calcium release by aenosine: A protein kinase C-dependent pathway. Mol. Cell. Biochem. 2006, 288, 59–64. [Google Scholar] [CrossRef]
- Koda, K.; Salazar-Rodriguez, M.; Corti, F.; Chan, N.Y.K.; Estephan, R.; Silver, R.B.; Mochly-Rosen, D.; Levi, R. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 2010, 122, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.H.; Jaafari, N.; Cimarosti, H.; Hanley, J.G.; Henley, J.M.; Mellor, J.R. Oxygen/glucose deprivation induces a reduction in synaptic AMPA receptors on hippocampal CA3 neurons mediated by mGluR1 and adenosine A3 receptors. J. Neurosci. 2011, 31, 11941–11952. [Google Scholar] [CrossRef] [PubMed]
- Tracey, W.R.; Magee, W.; Masamune, H.; Oleynek, J.J.; Hill, R.J. Selective activation of adenosine A3 receptors with N 6-(3-chlorobenzyl)-5′-N-methylcarboxamidoadenosine (CB-MECA) provides cardioprotection via KATP channel activation. Cardiovasc. Res. 1998, 40, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.C.; Ge, Z.D.; Tampo, A.; Mio, Y.; Bienengraeber, M.W.; Tracey, W.R.; Gross, G.J.; Kwok, W.M.; Auchampach, J.A. The A3 adenosine receptor agonist CP-532,903 [N6-(2, 5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J. Pharmacol. Exp. Ther. 2008, 324, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Mozzicato, S.; Joshi, B.V.; Jacobson, K.A.; Liang, B.T. Role of direct RhoA-phospholipase D interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J. 2004, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef]
- Wan, T.C.; Tampo, A.; Kwok, W.M.; Auchampach, J.A. Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem. Pharmacol. 2019, 163, 21–31. [Google Scholar] [CrossRef]
- Tracey, W.R.; Magee, W.P.; Oleynek, J.J.; Hill, R.J.; Smith, A.H.; Flynn, D.M.; Knight, D.R. Novel N 6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2780–H2787. [Google Scholar] [CrossRef] [PubMed]
- Auchampach, J.A.; Rizvi, A.; Qiu, Y.; Tang, X.L.; Maldonado, C.; Teschner, S.; Bolli, R. Selective activation of A3 adenosine receptors with N 6-(3-iodobenzyl) adenosine-5′-N-methyluronamide protects against myocardial stunning and infarction without hemodynamic changes in conscious rabbits. Circ. Res. 1997, 80, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Deninno, M.P.; Masamune, H.; Chenard, L.K.; DiRico, K.J.; Eller, C.; Etienne, J.B.; Tickner, J.E.; Kennedy, S.P.; Knight, D.R.; Kong, J.; et al. 3‘-Aminoadenosine-5‘-uronamides: Discovery of the first highly selective agonist at the human adenosine A3 receptor. J. Med. Chem. 2003, 46, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Richards, S.C.; Olsson, R.A.; Mullane, K.; Walsh, R.S.; Downey, J.M. Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovas. Res. 1994, 28, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.D.; Peart, J.N.; Kreckler, L.M.; Wan, T.C.; Jacobson, M.A.; Gross, G.J.; Auchampach, J.A. Cl-IB-MECA [2-Chloro-N6-(3-iodobenzyl) adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J. Pharmacol. Exp. Ther. 2006, 319, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, R.; Out, M.; Maas, W.J.; De Jong, J.W. Preconditioning of rat hearts by adenosine A1 or A3 receptor activation. Eur. J. Pharmacol. 2002, 441, 165–172. [Google Scholar] [CrossRef]
- Procopio, M.C.; Lauro, R.; Nasso, C.; Carerj, S.; Squadrito, F.; Bitto, A.; Di, B.G.; Micari, A.; Irrera, N.; Costa, F. Role of adenosine and purinergic receptors in myocardial infarction: Focus on different signal transduction pathways. Biomedicines 2021, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Kodani, E.; Bolli, R.; Tang, X.L.; Auchampach, J.A. Protection of IB-MECA against myocardial stunning in conscious rabbits is not mediated by the A1 adenosine receptor. Basic. Res. Cardiol. 2001, 96, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.C. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 427–436. [Google Scholar] [CrossRef]
- Harrison, G.J.; Cerniway, R.J.; Peart, J.; Berr, S.S.; Ashton, K.; Regan, S.; Matherne, P.G.; Headrick, J.P. Effects of A3 adenosine receptor activation and gene knock-out in ischemic-reperfused mouse heart. Cardiovas. Res. 2002, 53, 147–155. [Google Scholar] [CrossRef]
- Guo, Y.; Bolli, R.; Bao, W.; Wu, W.J.; Black Jr, R.G.; Murphree, S.S.; Salvatore, C.A.; Jacobson, M.A.; Auchampach, J.A. Targeted deletion of the A3 adenosine receptor confers resistance to myocardial ischemic injury and does not prevent early preconditioning. J. Mol. Cell Cardiol. 2001, 33, 825–830. [Google Scholar] [CrossRef]
- Cerniway, R.J.; Yang, Z.; Jacobson, M.A.; Linden, J.; Matherne, G.P. Targeted deletion of A3 adenosine receptors improves tolerance to ischemia-reperfusion injury in mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1751–H1758. [Google Scholar] [CrossRef]
- Shneyvays, V.; Mamedova, L.; Zinman, T.; Jacobson, K.; Shainberg, A. Activation of A3 adenosine receptor protects against doxorubicin-induced cardiotoxicity. J. Mol. Cell. Cardiol. 2001, 33, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Campos-Martins, A.; Bragança, B.; Correia-de-Sá, P.; Fontes-Sousa, A.P. Pharmacological tuning of adenosine signal nuances underlying heart failure with preserved ejection fraction. Front. pharmacol. 2021, 12, 724320. [Google Scholar] [CrossRef]
- Xu, X.; Fassett, J.; Hu, X.; Zhu, G.; Lu, Z.; Li, Y.; Schnermann, J.; Bache, R.J.; Chen, Y. Ecto-5′-nucleotidase deficiency exacerbates pressure-overload–induced left ventricular hypertrophy and dysfunction. Hypertension 2008, 51, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Kirchhof, P.; Fortmüller, L.; Auchampach, J.A.; Baba, H.A.; Breithardt, G.; Neumann, J.; Boknik, P.; Schmitz, W. Gene dose-dependent atrial arrhythmias, heart block, and brady-cardiomyopathy in mice overexpressing A3 adenosine receptors. Cardiovas. Res. 2004, 62, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zollbrecht, C.; Winerdal, M.E.; Zhuge, Z.; Zhang, X.M.; Terrando, N.; Checa, A.; Sallstrom, J.; Wheelock, C.E.; Wingvist, O.; et al. Genetic abrogation of adenosine A3 receptor prevents uninephrectomy and high salt–induced hypertension. J. Am. Heart Assoc. 2016, 5, e003868. [Google Scholar] [CrossRef]
- Ansari, H.R.; Nadeem, A.; Tilley, S.L.; Mustafa, S.J. Involvement of COX-1 in A3 adenosine receptor-mediated contraction through endothelium in mice aorta. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3448–H3455. [Google Scholar] [CrossRef]
- El-Awady, M.S.; Ansari, H.R.; Fil, D.; Tilley, S.L.; Mustafa, S.J. NADPH oxidase pathway is involved in aortic contraction induced by A3 adenosine receptor in mice. J. Pharmacol. Exp. Ther. 2011, 338, 711–717. [Google Scholar] [CrossRef]
- Jones, M.R.; Zhao, Z.; Sullivan, C.P.; Schreiber, B.M.; Stone, P.J.; Toselli, P.A.; Kagan, H.M.; Cohen, R.A.; Ravid, K. A3 adenosine receptor deficiency does not influence atherogenesis. J. Cell. Biochem. 2004, 92, 1034–1043. [Google Scholar] [CrossRef]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Beikoghli Kalkhoran, S.; Davidson, S.M. The RISK pathway leading to mitochondria and cardioprotection: How everything started. Basic. Res. Cardiol. 2023, 118, 22. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Madi, L.; Cohn, I. A3 adenosine receptor as a target for cancer therapy. Anti-Cancer Drugs 2002, 13, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Signhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P. Drugs targeting the A3 adenosine receptor: Human clinical study data. Molecules 2022, 27, 3680. [Google Scholar] [CrossRef]
- Safadi, R.; Braun, M.; Francis, A.; Milgrom, Y.; Massarwa, M.; Hakimian, D.; Hazou, W.; Issachar, A.; Harpaz, Z.; Farbstein, M.; et al. Randomised clinical trial: A phase 2 double-blind study of namodenoson in non-alcoholic fatty liver disease and steatohepatitis. Aliment. Pharmacol. Ther. 2021, 54, 1405–1415. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in advanced hepatocellular carcinoma and Child–Pugh B cirrhosis: Randomized placebo-controlled clinical trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- van Troostenburg, A.R.; Clark, E.V.; Carey, W.D.H.; Warrington, S.J.; Kerns, W.D.; Cohn, I.; Silverman, M.H.; Bar-Yehuda, S.; Fong, K.L.L.; Fishman, P. Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 534–542. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Gospodinov, D.K.; Gheorghe, N.; Mateev, G.S.; Rusinova, M.V.; Hristakieva, E.; Solovastru, L.G.; Fishman, P. Treatment of plaque-type psoriasis with oral CF101: Data from a phase II/III multicenter, randomized, controlled trial. J. Drugs Dermatol. 2016, 15, 931–938. [Google Scholar] [PubMed]
- Papp, K.; Beyska-Rizova, S.; Gantcheva, M.; Slavcheva Simeonova, E.; Brezoev, P.; Celic, M.; Groppa, L.; Blicharski, T.; Selmanagic, A.; Kalicka-Dudzik, M.; et al. Efficacy and safety of piclidenoson in plaque psoriasis: Results from a randomized phase 3 clinical trial (COMFORT-1). J. Eur. Acad. Dermatol. Venereol. 2024, 38, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Duangrat, R.; Parichatikanond, W.; Kurose, H. New therapeutics for heart failure: Focusing on cGMP signaling. Int. J. Mol. Sci. 2023, 24, 12866. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: Focusing on TGF-β signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duangrat, R.; Parichatikanond, W.; Chanmahasathien, W.; Mangmool, S. Adenosine A3 Receptor: From Molecular Signaling to Therapeutic Strategies for Heart Diseases. Int. J. Mol. Sci. 2024, 25, 5763. https://doi.org/10.3390/ijms25115763
Duangrat R, Parichatikanond W, Chanmahasathien W, Mangmool S. Adenosine A3 Receptor: From Molecular Signaling to Therapeutic Strategies for Heart Diseases. International Journal of Molecular Sciences. 2024; 25(11):5763. https://doi.org/10.3390/ijms25115763
Chicago/Turabian StyleDuangrat, Ratchanee, Warisara Parichatikanond, Wisinee Chanmahasathien, and Supachoke Mangmool. 2024. "Adenosine A3 Receptor: From Molecular Signaling to Therapeutic Strategies for Heart Diseases" International Journal of Molecular Sciences 25, no. 11: 5763. https://doi.org/10.3390/ijms25115763
APA StyleDuangrat, R., Parichatikanond, W., Chanmahasathien, W., & Mangmool, S. (2024). Adenosine A3 Receptor: From Molecular Signaling to Therapeutic Strategies for Heart Diseases. International Journal of Molecular Sciences, 25(11), 5763. https://doi.org/10.3390/ijms25115763