
The Characteristics of Structural Properties and Diffusion Pathway of Alkali in Sodium Trisilicate: Nanoarchitectonics and Molecular Dynamic Simulation


Abstract
1. Introduction
2. Results and Discussion
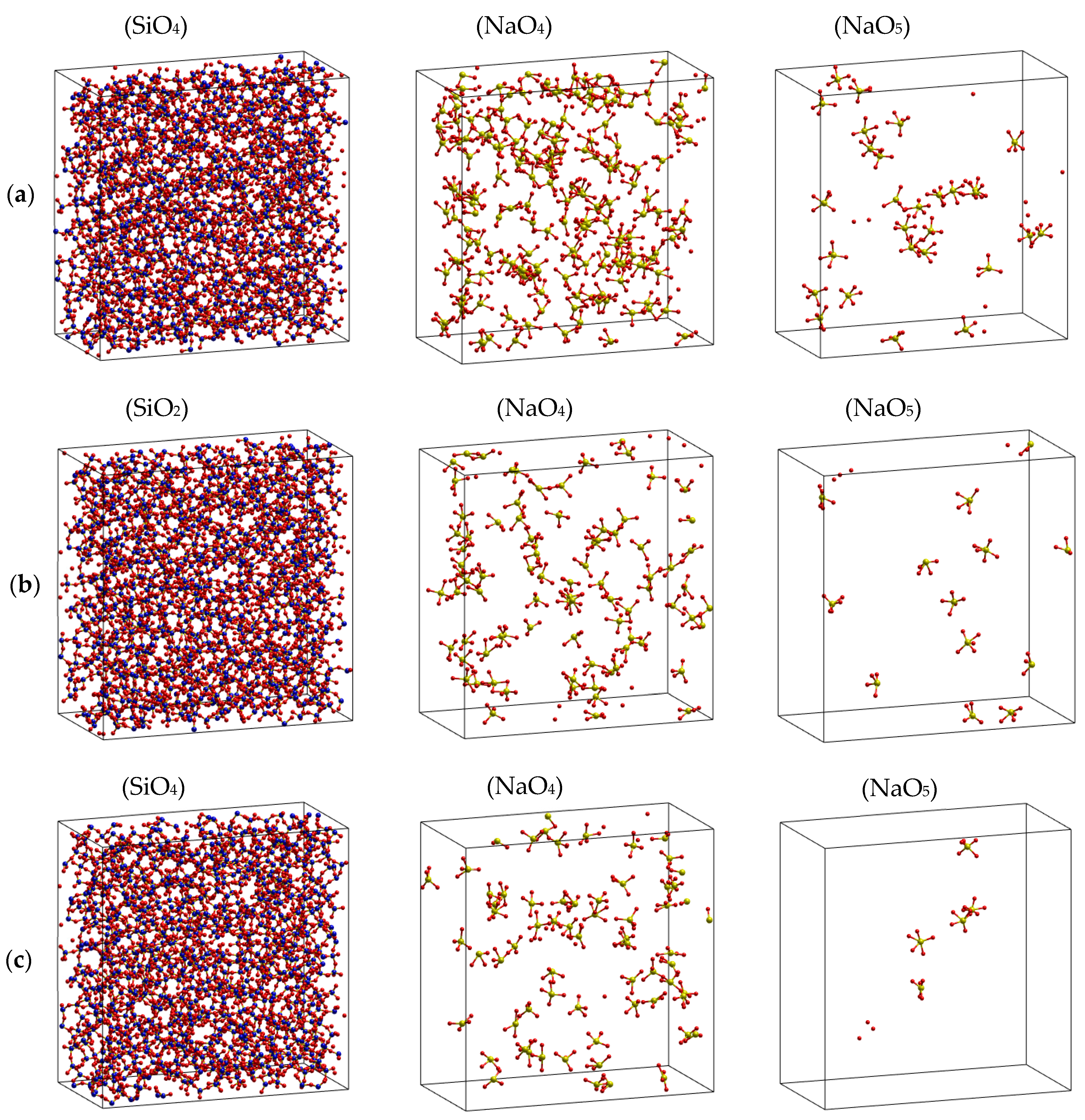
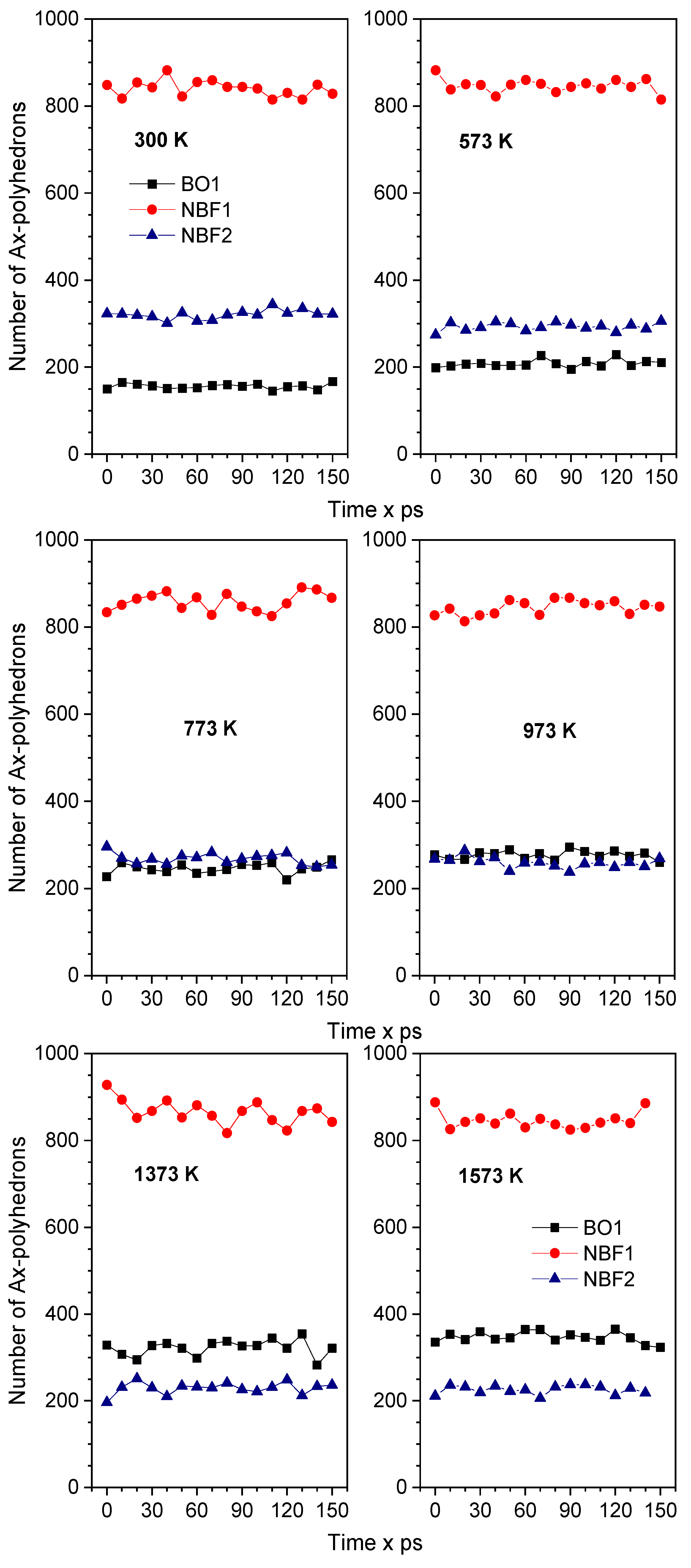
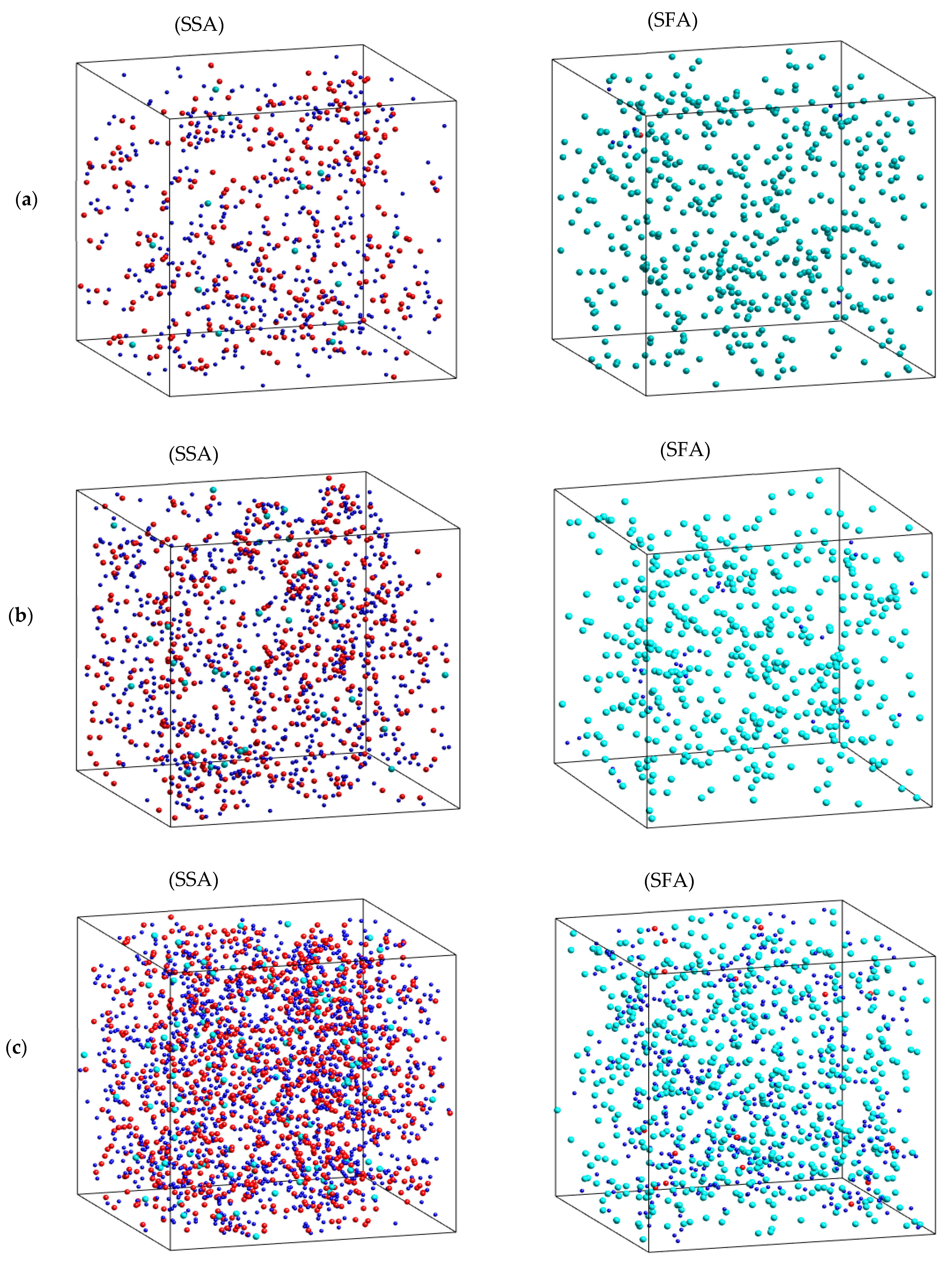
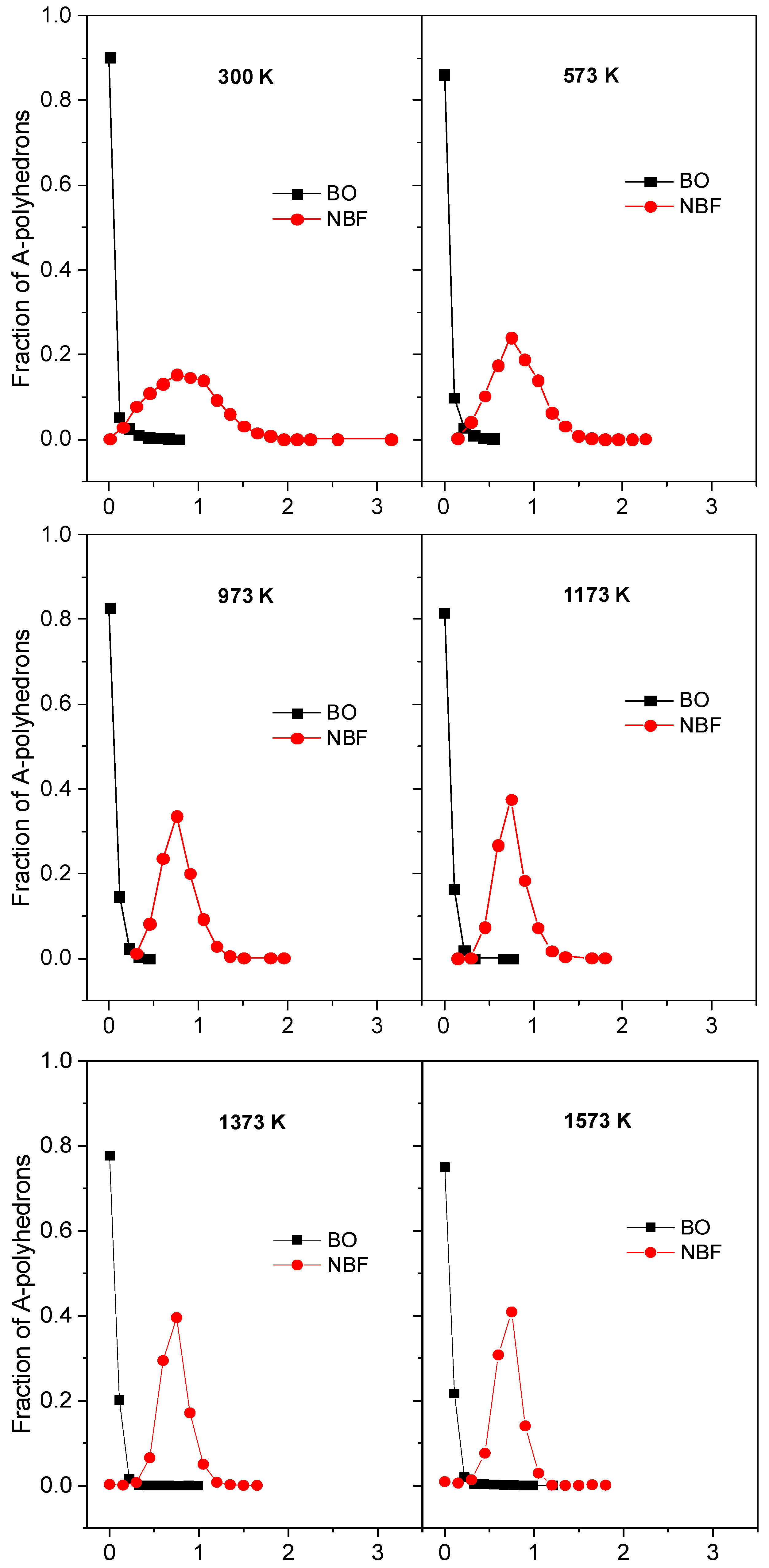
2.1. Characteristics of Structural Na2O∙3SiO2
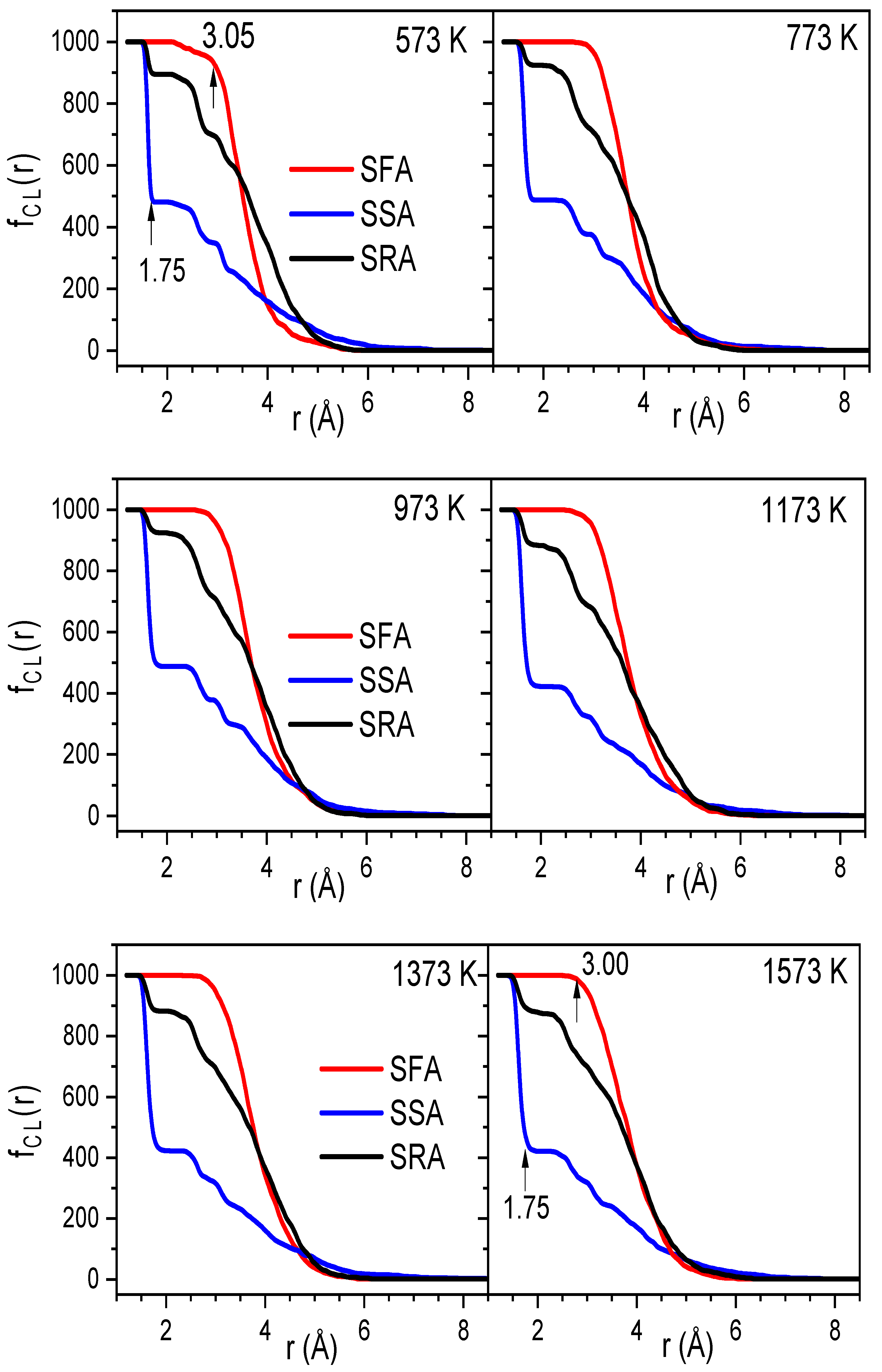
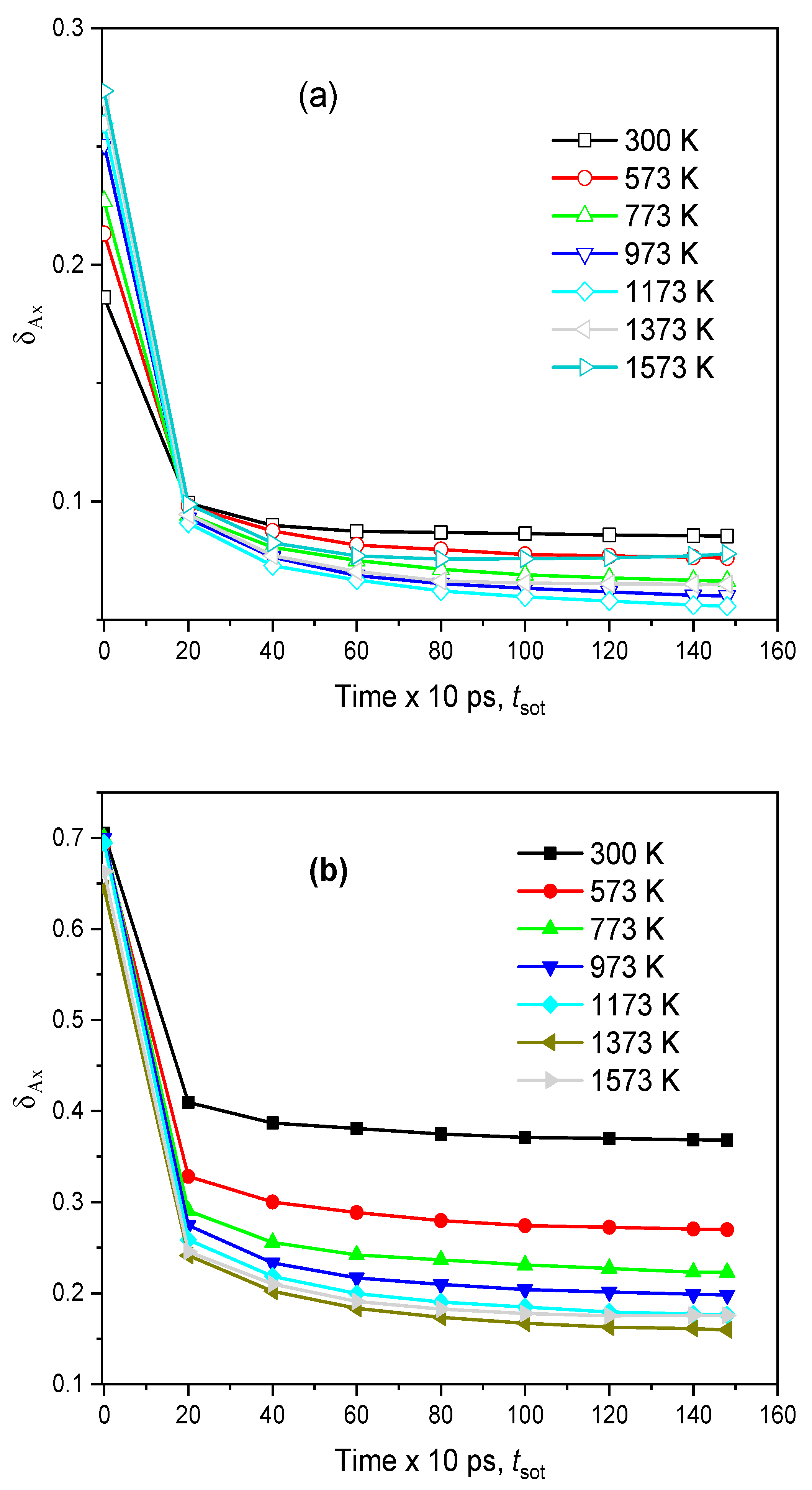
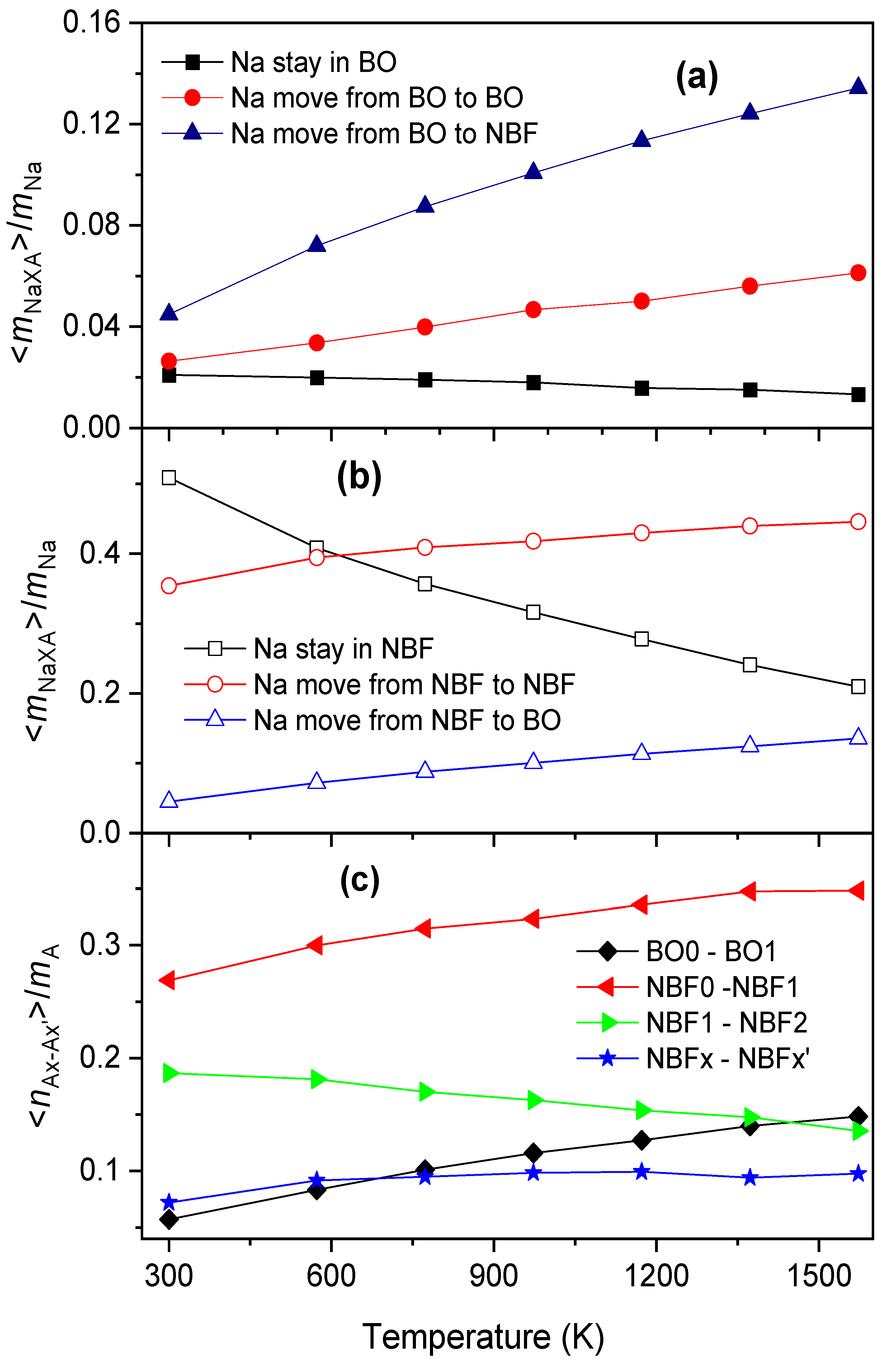
2.2. The Diffusion Pathway of Na Atoms in Na2O∙3SiO2
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fábián, M.; Jóvári, P.; Sváb, E.; Mészáros, G.; Proffen, T.; Veress, E. Network structure of 0.7SiO2-0.3Na2O glass from neutron and X-ray diffraction and RMC modelling. J. Phys. Condens. Matter 2007, 19, 335209. [Google Scholar] [CrossRef]
- Mozzi, R.L.; Warren, B.E. The structure of vitreous silica. J. Appl. Crystallogr. 1969, 2, 164–172. [Google Scholar] [CrossRef]
- Benmore, C.J.; Weber, J.K.R.; Wilding, M.C.; Du, J.; Parise, J.B. Temperature-dependent structural heterogeneity in calcium silicate liquids. Phys. Rev. B 2010, 82, 224202. [Google Scholar] [CrossRef]
- Saito, Y.; Takumi, Y.; Atsunobu, M.; Hiroyuki, I.; Koji, O.; Shinji, K. Structural change of Na2O-doped SiO2 glasses by melting. J. Ceram. Soc. Jpn. 2016, 124, 717–720. [Google Scholar] [CrossRef]
- Li, X.; Weiying, S.; Morten, M.S.; John, C.M.; Mathieu, B. Quantifying the internal stress in over-constrained glasses by molecular dynamics simulations. J. Non-Cryst. Solids X 2019, 1, 100013. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Henderson, G.S.; Bancroft, G.M.; Ho, R. Experimental evidence for Na coordination to bridging oxygen in Na-silicate glasses: Implications for spectroscopic studies and for the modified random network model. J. Non-Cryst. Solids 2015, 409, 139–148. [Google Scholar] [CrossRef]
- Davidenko, A.O.; Sokol’skii, V.E.; Roik, A.S.; Goncharov, I.A. Structural study of sodium silicate glasses and melts. Inorg. Mater. 2014, 50, 1289–1296. [Google Scholar] [CrossRef]
- Zhao, Q.; Guerette, M.; Scannell, G.; Huang, L. In-situ high temperature Raman and Brillouin light scattering studies of sodium silicate glasses. J. Non-Cryst. Solids 2012, 358, 3418–3426. [Google Scholar] [CrossRef]
- Smith, W.; Greaves, G.N.; Gillan, M.J. Computer simulation of Na disilicate glass. J. Chem. Phys. 1995, 103, 3091–3097. [Google Scholar] [CrossRef]
- Yen, N.V.; Dung, M.V.; Hung, P.K.; Van, T.B.; Vinh, L.T. Spatial distribution of cations through Voronoi polyhedrons and their exchange between polyhedrons in sodium silicate liquids. J. Non-Cryst. Solids 2021, 566, 120898. [Google Scholar] [CrossRef]
- Jabraoui, H.; Achhal, E.M.; Hasnaoui, A.; Garden, J.L.; Vaills, Y.; Ouaskit, S. Molecular dynamics simulation of thermodynamic and structural properties of silicate glass: Effect of the alkali oxide modifiers. J. Non-Cryst. Solids 2016, 448, 16–26. [Google Scholar] [CrossRef]
- Mountjoy, G.; Al-Hasni, B.M.; Storey, C. Structural organisation in oxide glasses from molecular dynamics modelling. J. Non-Cryst. Solids 2011, 357, 2522–2529. [Google Scholar] [CrossRef]
- Ojovan, M.I. The Modified Random Network (MRN) Model within the Configuron Percolation Theory (CPT) of Glass Transition. Ceramics 2021, 4, 121–134. [Google Scholar] [CrossRef]
- Jabraoui, H.; Vaills, Y.; Hasnaoui, A.; Badawi, M.; Ouaskit, S. Effect of Na oxide modifier on structural and elastic properties of silicate glass. J. Phys. Chem. B 2016, 120, 13193–13205. [Google Scholar] [CrossRef]
- Noritake, F. Structural transformations in sodium silicate liquids under pressure: New static and dynamic structure analyses. J. Non-Cryst. Solids 2017, 473, 102–107. [Google Scholar] [CrossRef]
- Voigtmann, T.; Horbach, J. Slow dynamics in ion-conducting sodium silicate melts: Simulation and mode-coupling theory. Europhys. Lett. 2006, 74, 459. [Google Scholar] [CrossRef]
- Hung, P.K.; Noritake, F.; San, L.T.; Van, T.B. Study of diffusion and local structure of sodium-silicate liquid: The molecular dynamic simulation. Eur. Phys. J. B 2017, 90, 185. [Google Scholar] [CrossRef]
- Gupta, Y.P.; King, T.B. Self-diffusion of sodium in sodium silicate liquids. Trans. Metall. Soc. AIME 1967, 239, 1701. [Google Scholar]
- Braedt, M.; Frischat, G.H. Sodium self diffusion in glasses and melts of the system Na2O-Rb2O-SiO2. Phys. Chem. Glas. 1988, 29, 214–218. [Google Scholar]
- Knoche, R.; Dingwell, D.B.; Seifert, F.A.; Webb, S.L. Non-linear properties of supercooled liquids in the system Na2O∙SiO2. Chem. Geol. 1994, 116, 1–16. [Google Scholar] [CrossRef]
- Meyer, A.; Horbach, J.; Kob, W.; Kargl, F.; Schober, H. Channel formation and intermediate range order in sodium silicate melts and glasses. Phys. Rev. Lett. 2004, 93, 027801. [Google Scholar] [CrossRef]
- Kargl, F.; Weis, H.; Unruh, T.; Meyer, A. Self diffusion in liquid aluminium. In J. Phys. Conf. Ser. 2012, 340, 012077. [Google Scholar] [CrossRef]
- Philippe, J.; Kob, W.; Jullien, R. Channel diffusion of Na in a silicate glass. Phys. Rev. B 2001, 64, 134303. [Google Scholar]
- Van, T.B.; Hung, P.K.; Vinh, L.T.; Ha, N.T.T.; San, L.T.; Noritake, F. Network cavity, spatial distribution of Na and dynamics in sodium silicate melts. J. Mater. Sci. 2020, 55, 2870–2880. [Google Scholar] [CrossRef]
- Konstantinou, K.; Duffy, D.M.; Shluger, A.L. Structure and luminescence of intrinsic localized states in sodium silicate glasses. Phys. Rev. B 2016, 94, 174202. [Google Scholar] [CrossRef]
- Greaves, G.N. Structure and ionic transport in disordered silicates. Mineral. Mag. 2000, 64, 441–446. [Google Scholar] [CrossRef]
- Greaves, G.N.; Sen, S. Inorganic glasses, glass-forming liquids and amorphizing solids. Adv. Phys. 2007, 56, 1–166. [Google Scholar] [CrossRef]
- Bauchy, M.; Micoulaut, M. From pockets to channels: Density-controlled diffusion in sodium silicates. Phys. Rev. B 2011, 83, 184118. [Google Scholar]
- Noritake, F.; Katsuyuki, K.; Takashi, Y.; Eiichi, T. Molecular dynamics simulation and electrical conductivity measurement of Na2O∙3SiO2 melt under high pressure; relationship between its structure and properties. J. Non-Cryst. Solids 2012, 358, 3109–3118. [Google Scholar] [CrossRef]
- Sunyer, E.; Philippe, J.; Rémi, J. Characterization of channel diffusion in a Na tetrasilicate glass via molecular-dynamics simulations. Phys. Rev. B 2002, 65, 214203. [Google Scholar] [CrossRef]
- Horbach, J.; Walter, K.; Kurt, B. Structural and dynamical properties of sodium silicate melts: An investigation by molecular dynamics computer simulation. Chem. Geol. 2001, 174, 87–101. [Google Scholar] [CrossRef]
- Cormack, A.N.; Du, J.; Zeitler, T.R. Alkali ion migration mechanisms in silicate glasses probed by molecular dynamics simulations. Phys. Chem. Chem. Phys. 2002, 4, 3193–3197. [Google Scholar] [CrossRef]
- Cormack, A.N.; Du, J.; Zeitler, T.R. Sodium ion migration mechanisms in silicate glasses probed by molecular dynamics simulations. J. Non-Cryst. Solids 2003, 323, 147–154. [Google Scholar] [CrossRef]
- Noritake, F. Diffusion mechanism of network-forming elements in silicate liquids. J. Non-Cryst. Solids 2021, 553, 120512. [Google Scholar] [CrossRef]
- Habasaki, J.; Yasuaki, H. Molecular dynamics study of the mechanism of ion transport in lithium silicate glasses: Characteristics of the potential energy surface and structures. Phys. Rev. B 2004, 69, 144207. [Google Scholar] [CrossRef]
- Morab, S.; Sundaram, M.M.; Pivrikas, A. Influence of traps and Lorentz Force on charge transport in organic semiconductors. Materials 2023, 16, 4691. [Google Scholar] [CrossRef]
- Van Yen, N.; Plan, E.L.V.M.; Kien, P.H.; Nguyen, A.T.; Van Hong, N.; Phan, H. Topological structural analysis and dynamical properties in MgSiO3 liquid under compression. Eur. Phys. J. B 2022, 95, 62. [Google Scholar] [CrossRef]
- Hung, P.K.; Vinh, L.T.; Hong, N.V.; Thu Ha, N.T.; Toshiaki, I. Two-domain structure and dynamics heterogeneity in a liquid SiO2. J. Non-Cryst. Solids 2018, 484, 124–131. [Google Scholar] [CrossRef]
- Ha, N.T.T.; Hong, N.V.; Hung, P.K. Distribution of Na and dynamical heterogeneity in sodium silicate liquid. Int. J. Mod. Phys. B 2019, 33, 1950013. [Google Scholar] [CrossRef]
- Sakuma, H.; Kawamura, K. Structure and dynamics of water on muscovite mica surfaces. Geochim. Cosmochim. Acta 2009, 73, 4100–4110. [Google Scholar] [CrossRef]
- Glaze, F.W.; Young, J.C.; Finn, A.N. The density of some soda-lime-silica glasses as a function of the composition. Bur. Stand. J. Res. 1932, 9, 799. [Google Scholar] [CrossRef]
- Noritake, F.; Naito, S. Mechanism of mixed alkali effect in silicate glass/liquid: Pathway and network analysis. J. Non-Cryst. Solids 2023, 610, 122321. [Google Scholar] [CrossRef]
- Ariga, K. Nanoarchitectonics: Method for everything in material science. Bull. Chem. Soc. Jpn. 2023, 97, uoad001. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | mBO/mO | mBO0/mO | mBO1/mO | mNBF/mO | mNBF0/mO | mNBF1/mO | mNBF2/mO |
|---|---|---|---|---|---|---|---|
| 300 | 0.7151 | 0.6884 | 0.0268 | 0.2849 | 0.0842 | 0.1441 | 0.0550 |
| 573 | 0.7151 | 0.6794 | 0.0357 | 0.2849 | 0.0880 | 0.1452 | 0.0502 |
| 773 | 0.7151 | 0.6729 | 0.0422 | 0.2849 | 0.0903 | 0.1471 | 0.0460 |
| 973 | 0.7151 | 0.6676 | 0.0475 | 0.2849 | 0.0941 | 0.1448 | 0.0445 |
| 1173 | 0.7151 | 0.6640 | 0.0511 | 0.2849 | 0.0955 | 0.1454 | 0.0429 |
| 1373 | 0.7145 | 0.6591 | 0.0552 | 0.2855 | 0.0967 | 0.1485 | 0.0393 |
| 1573 | 0.7145 | 0.6591 | 0.0552 | 0.2855 | 0.0967 | 0.1485 | 0.0393 |
| T (K) | <vSi> | <vBO> | <vNBF> | VSi/VSB | VBO/VSB | VNBF/VSB | mNaBO/mNa | mNaNBF/mNa |
|---|---|---|---|---|---|---|---|---|
| 300 | 7.33 | 19.37 | 29.40 | 0.1237 | 0.5461 | 0.3302 | 0.0990 | 0.9010 |
| 573 | 7.43 | 19.51 | 29.72 | 0.1244 | 0.5446 | 0.3310 | 0.1218 | 0.8782 |
| 773 | 7.52 | 19.64 | 30.10 | 0.1246 | 0.5428 | 0.3326 | 0.1555 | 0.8445 |
| 973 | 7.61 | 19.75 | 30.47 | 0.1250 | 0.5420 | 0.3330 | 0.1603 | 0.8397 |
| 1173 | 7.70 | 19.93 | 30.88 | 0.1254 | 0.5417 | 0.3329 | 0.1771 | 0.8229 |
| 1373 | 7.82 | 20.11 | 31.41 | 0.1255 | 0.5380 | 0.3365 | 0.1843 | 0.8157 |
| 1573 | 7.94 | 20.37 | 31.96 | 0.1260 | 0.5374 | 0.3366 | 0.2119 | 0.7881 |
| Atomic Parameters | Z | a (nm) | b (nm) | c (kJ/mol∙nm3) |
|---|---|---|---|---|
| Si | - | 0.099759 | 0.00830 | 0.000 |
| O | - | 0.181819 | 0.01539 | 27,400 |
| Na | 1.00000 | 0.139500 | 0.01150 | 10,000 |
| Pair parameters | D1 (kJ/mol) | β1 (1/nm) | D2 (kJ/mol) | β2 (1/nm) |
| SiO | 668,428 | 59.636 | −105,335 | 45.514 |
| 3-body parameters | f (kJ/mol) | θ0 (degree) | rm (nm) | gr (1/nm) |
| Si-O-Si | 0.0006 | 147.0 | 0.170 | 168.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kien, P.H.; Trang, G.T.T. The Characteristics of Structural Properties and Diffusion Pathway of Alkali in Sodium Trisilicate: Nanoarchitectonics and Molecular Dynamic Simulation. Int. J. Mol. Sci. 2024, 25, 5628. https://doi.org/10.3390/ijms25115628
Kien PH, Trang GTT. The Characteristics of Structural Properties and Diffusion Pathway of Alkali in Sodium Trisilicate: Nanoarchitectonics and Molecular Dynamic Simulation. International Journal of Molecular Sciences. 2024; 25(11):5628. https://doi.org/10.3390/ijms25115628
Chicago/Turabian StyleKien, Pham Huu, and Giap Thi Thuy Trang. 2024. "The Characteristics of Structural Properties and Diffusion Pathway of Alkali in Sodium Trisilicate: Nanoarchitectonics and Molecular Dynamic Simulation" International Journal of Molecular Sciences 25, no. 11: 5628. https://doi.org/10.3390/ijms25115628
APA StyleKien, P. H., & Trang, G. T. T. (2024). The Characteristics of Structural Properties and Diffusion Pathway of Alkali in Sodium Trisilicate: Nanoarchitectonics and Molecular Dynamic Simulation. International Journal of Molecular Sciences, 25(11), 5628. https://doi.org/10.3390/ijms25115628