

Two Enterococcus faecium Isolates Demonstrated Modulating Effects on the Dysbiosis of Mice Gut Microbiota Induced by Antibiotic Treatment
 ,
,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Isolation and Characterization of E. faecium DC-K7 and DC-K9
2.2. General Features of E. faecium DC-K7 and DC-K9 Genome
2.3. Analyses of CAZyme-Encoding Genes
2.4. Examinations of SCFA-Producing Abilities
2.5. Bacteriocin Prediction
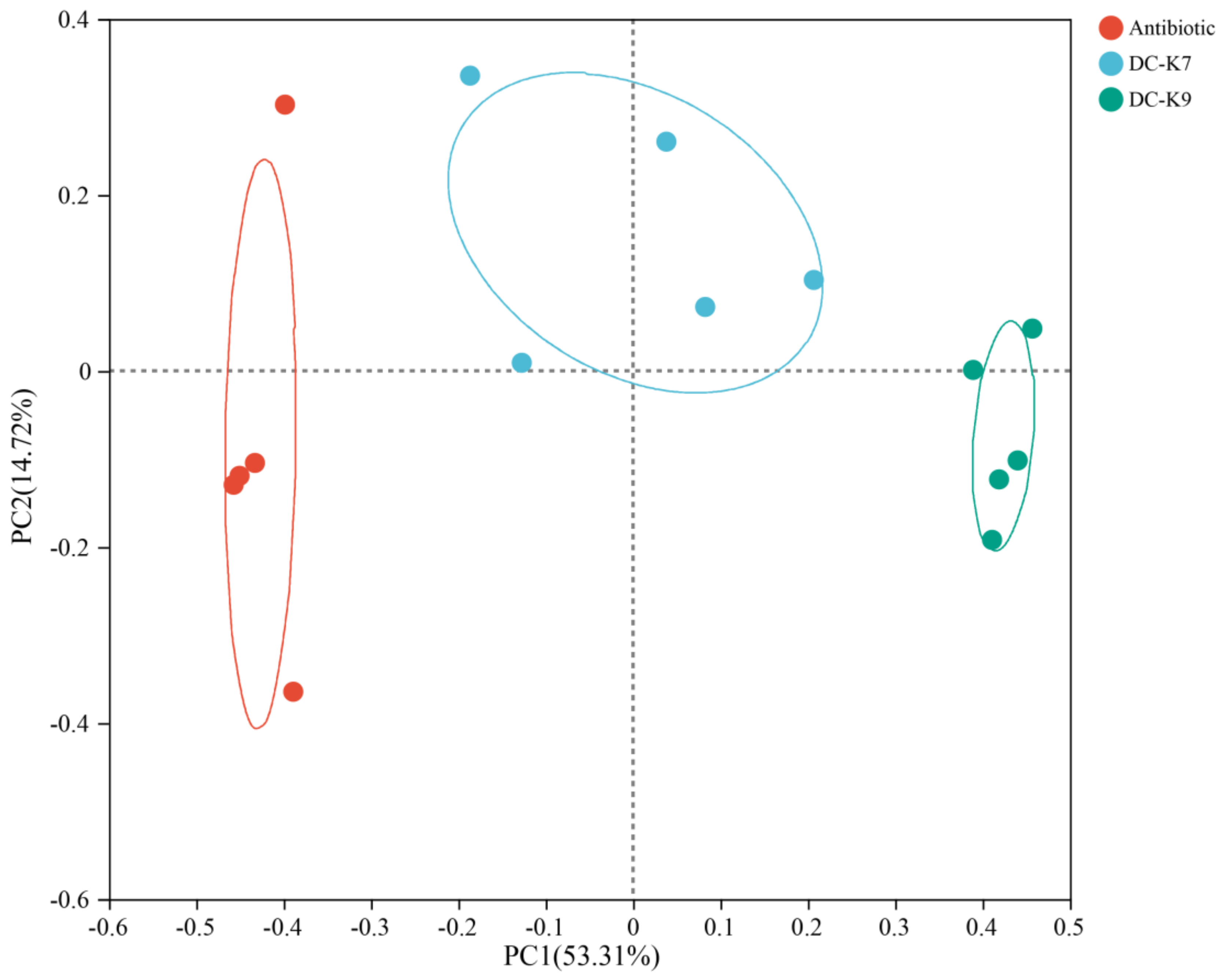
2.6. Microbial Diversity Analysis of the Mice Gut Microbiota
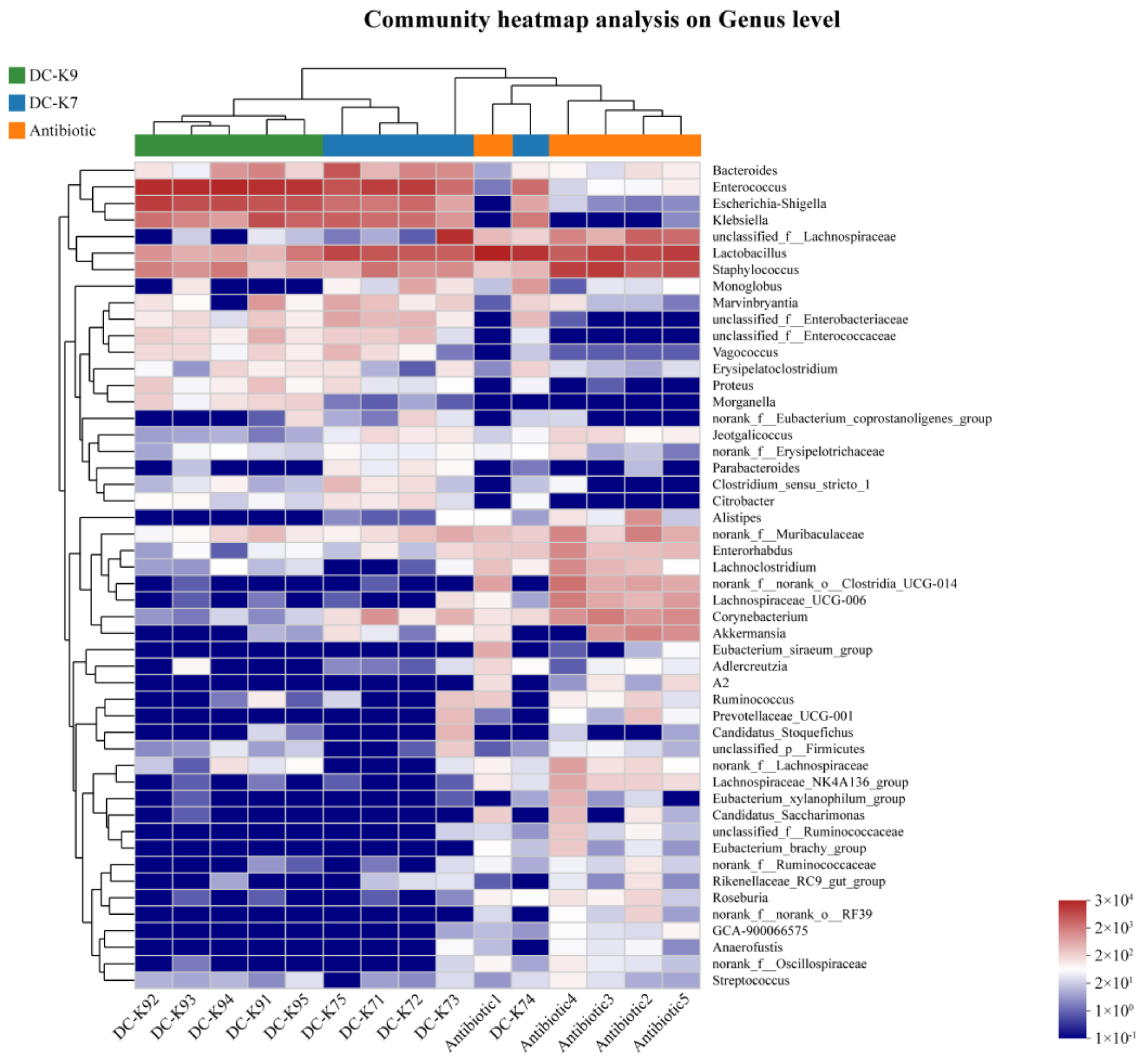
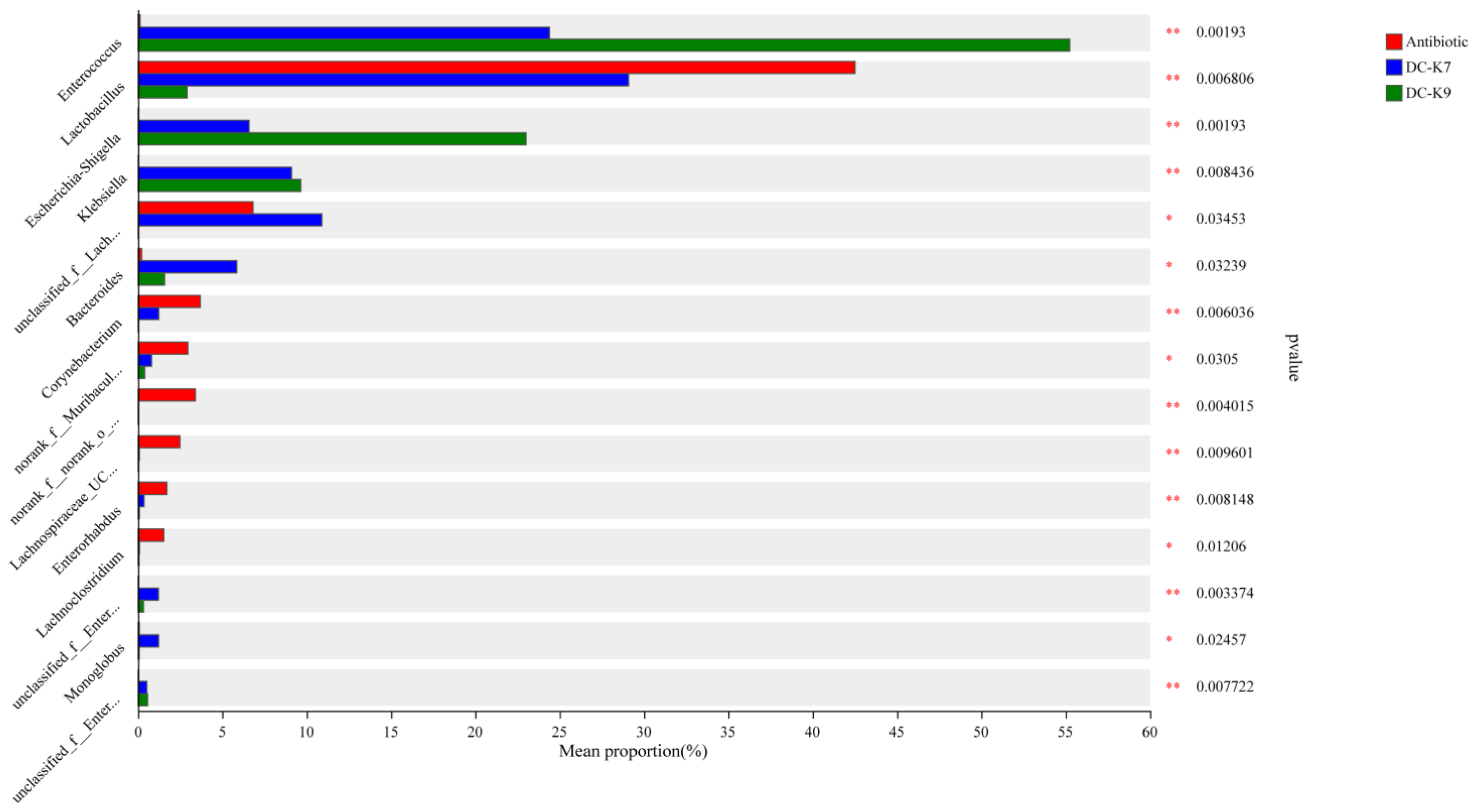
2.7. Alterations of the Gut Microbial Compositions
2.8. Comparisons of the Gut Microbial Communities
3. Discussion
4. Materials and Methods
4.1. Isolation and Characterization of E. faecium DC-K7 and DC-K9
4.2. Genomic Features of E. faecium DC-K7 and DC-K9
4.2.1. Genome Sequencing, Assembly, CDS Prediction and Annotation
4.2.2. Prediction of CAZyme-Encoding Genes
4.2.3. Prediction of Bacteriocin
4.3. Examination of SCFA-Producing Abilities
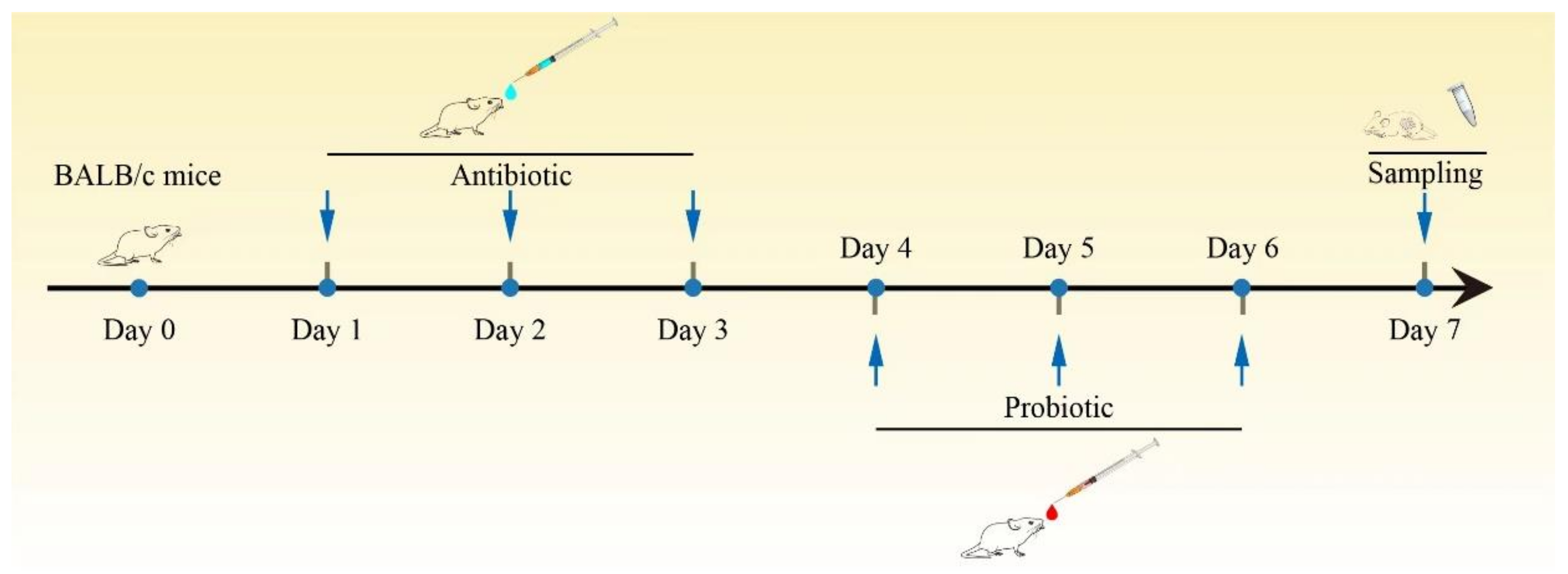
4.4. Animal Studies
4.5. Microbial Community Profiling
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; D Lieber, A.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016, 8, 343ra382. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hämäläinen, A.-M.; Härkönen, T.; Ryhänen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016, 8, 343ra381. [Google Scholar] [CrossRef] [PubMed]
- Larcombe, S.; Hutton, M.L.; Lyras, D. Involvement of Bacteria Other Than Clostridium difficile in Antibiotic-Associated Diarrhoea. Trends Microbiol. 2016, 24, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.K.; Wang, B.; Ahmadi, S.; Burnham, C.-A.D.; Tarr, P.I.; Warner, B.B.; Dantas, G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 2016, 1, 16024. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Virta, L.J.; Kekkonen, R.A.; Forslund, K.; Bork, P.; de Vos, W.M. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat. Commun. 2016, 7, 10410. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.P.; Russell, S.L.; Finlay, B.B. Shifting the balance: Antibiotic effects on host-microbiota mutualism. Nat. Reviews. Microbiol. 2011, 9, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Butts, C.A.; Paturi, G.; Hedderley, D.I.; Martell, S.; Dinnan, H.; Stoklosinski, H.; Carpenter, E.A. Goat and cow milk differ in altering microbiota composition and fermentation products in rats with gut dysbiosis induced by amoxicillin. Food Funct. 2021, 12, 3104–3119. [Google Scholar] [CrossRef]
- Mekonnen, S.A.; Merenstein, D.; Fraser, C.M.; Marco, M.L. Molecular mechanisms of probiotic prevention of antibiotic-associated diarrhea. Curr. Opin. Biotechnol. 2020, 61, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.J. Role of colonic short-chain fatty acid transport in diarrhea. Annu. Rev. Physiol. 2010, 72, 297–313. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2013, 62, 1591–1601. [Google Scholar] [CrossRef]
- Kumar, A.; Alrefai, W.A.; Borthakur, A.; Dudeja, P.K. Lactobacillus acidophilus counteracts enteropathogenic E. coli-induced inhibition of butyrate uptake in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G602–G607. [Google Scholar] [CrossRef]
- Cresci, G.; Nagy, L.E.; Ganapathy, V. Lactobacillus GG and tributyrin supplementation reduce antibiotic-induced intestinal injury. JPEN J. Parenter. Enter. Nutr. 2013, 37, 763–774. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Pattani, R.; Palda, V.A.; Hwang, S.W.; Shah, P.S. Probiotics for the prevention of antibiotic-associated diarrhea and Clostridium difficile infection among hospitalized patients: Systematic review and meta-analysis. Open Med. 2013, 7, e56–e67. [Google Scholar]
- Guo, Q.; Goldenberg, J.Z.; Humphrey, C.; El Dib, R.; Johnston, B.C. Probiotics for the prevention of pediatric antibiotic-associated diarrhea. Cochrane Database Syst. Rev. 2019, 4, CD004827. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.B.M.; van de Steeg, E.; van Bilsen, J.; Meijerink, M. Mechanisms and immunomodulatory properties of pre- and probiotics. Benef. Microbes 2019, 10, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Hardy, H.; Harris, J.; Lyon, E.; Beal, J.; Foey, A.D. Probiotics, prebiotics and immunomodulation of gut mucosal defences: Homeostasis and immunopathology. Nutrients 2013, 5, 1869–1912. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, B.; Xu, J.; Liu, Y.; Qiu, E.; Li, Z.; Li, Z.; He, Y.; Zhou, H.; Bai, Y.; et al. Bacteroides fragilis Protects Against Antibiotic-Associated Diarrhea in Rats by Modulating Intestinal Defenses. Front. Immunol. 2018, 9, 1040. [Google Scholar] [CrossRef]
- Du, W.; Xu, H.; Mei, X.; Cao, X.; Gong, L.; Wu, Y.; Li, Y.; Yu, D.; Liu, S.; Wang, Y.; et al. Probiotic Bacillus enhance the intestinal epithelial cell barrier and immune function of piglets. Benef. Microbes 2018, 9, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Heeney, D.D.; Zhai, Z.; Bendiks, Z.; Barouei, J.; Martinic, A.; Slupsky, C.; Marco, M.L. Lactobacillus plantarum bacteriocin is associated with intestinal and systemic improvements in diet-induced obese mice and maintains epithelial barrier integrity in vitro. Gut Microbes 2019, 10, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, M.; Nuccio, S.-P.; Liu, H.; Hernandez, D.; Vu, C.T.; Takahashi, A.A.; Edwards, R.A.; Raffatellu, M. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 2016, 540, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Popović, N.; Dinić, M.; Tolinački, M.; Mihajlović, S.; Terzić-Vidojević, A.; Bojić, S.; Djokić, J.; Golić, N.; Veljović, K. New Insight into Biofilm Formation Ability, the Presence of Virulence Genes and Probiotic Potential of Enterococcus sp. Dairy Isolates. Front. Microbiol. 2018, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Niu, Q.; Wei, Q.; Zhang, Y.; Ma, X.; Kim, S.W.; Lin, M.; Huang, R. Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus Faecalis as alternatives to antibiotics. Sci. Rep. 2017, 7, 41395. [Google Scholar] [CrossRef]
- Pedicord, V.A.; Lockhart, A.A.K.; Rangan, K.J.; Craig, J.W.; Loschko, J.; Rogoz, A.; Hang, H.C.; Mucida, D. Exploiting a host-commensal interaction to promote intestinal barrier function and enteric pathogen tolerance. Sci. Immunol. 2016, 1, eaai7732. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Wang, Y.-C.; Hespen, C.W.; Espinosa, J.; Salje, J.; Rangan, K.J.; Oren, D.A.; Kang, J.Y.; Pedicord, V.A.; Hang, H.C. Enterococcus faecium secreted antigen A generates muropeptides to enhance host immunity and limit bacterial pathogenesis. eLife 2019, 8, e45343. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pasolli, E.; Ercolini, D. The food-gut axis: Lactic acid bacteria and their link to food, the gut microbiome and human health. FEMS Microbiol. Rev. 2020, 44, 454–489. [Google Scholar] [CrossRef]
- Wu, J.; Gan, T.; Zhang, Y.; Xia, G.; Deng, S.; Lv, X.; Zhang, B.; Lv, B. The prophylactic effects of BIFICO on the antibiotic-induced gut dysbiosis and gut microbiota. Gut Pathog. 2020, 12, 41. [Google Scholar] [CrossRef]
- Wang, P.; Yin, X.; Chen, G.; Li, L.; Le, Y.; Xie, Z.; Ouyang, W.; Tong, J. Perioperative probiotic treatment decreased the incidence of postoperative cognitive impairment in elderly patients following non-cardiac surgery: A randomised double-blind and placebo-controlled trial. Clin. Nutr. 2021, 40, 64–71. [Google Scholar] [CrossRef]
- Ahmadi, S.; Wang, S.; Nagpal, R.; Wang, B.; Jain, S.; Razazan, A.; Mishra, S.P.; Zhu, X.; Wang, Z.; Kavanagh, K.; et al. A human-origin probiotic cocktail ameliorates aging-related leaky gut and inflammation via modulating the microbiota/taurine/tight junction axis. JCI Insight 2020, 5, e132055. [Google Scholar] [CrossRef]
- Derrien, M.; Alvarez, A.S.; de Vos, W.M. The Gut Microbiota in the First Decade of Life. Trends Microbiol. 2019, 27, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Sprockett, D.; Fukami, T.; Relman, D.A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhya, I.; Martin, J.C.; Shaw, S.; McKinley, A.J.; Gratz, S.W.; Scott, K.P. Comparison of microbial signatures between paired faecal and rectal biopsy samples from healthy volunteers using next-generation sequencing and culturomics. Microbiome 2022, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Xiao, C.; Yan, Z.; Pan, R.; Gao, Y.; Li, B.; Wei, J.; Qiu, Y.; Liu, K.; et al. Genomic and metabolic features of the Lactobacillus sakei JD10 revealed potential probiotic traits. Microbiol. Res. 2022, 256, 126954. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Xu, Z.; Xia, P.; Feng, Y.; Liu, B.; Liu, H.; Chen, Y.; Yan, G.; Lv, B.; Yan, Z.; et al. The Short-Term Efficacy of Bifidobacterium Quadruple Viable Tablet in Patients With Diarrhea-Predominant Irritable Bowel Syndrome: Potentially Mediated by Metabolism Rather Than Diversity Regulation. Am. J. Gastroenterol. 2023, 118, 1256–1267. [Google Scholar] [CrossRef]
- Andresen, V.; Gschossmann, J.; Layer, P. Heat-inactivated Bifidobacterium bifidum MIMBb75 (SYN-HI-001) in the treatment of irritable bowel syndrome: A multicentre, randomised, double-blind, placebo-controlled clinical trial. Lancet Gastroenterol. Hepatol. 2020, 5, 658–666. [Google Scholar] [CrossRef]
- Ford, S.A.; King, K.C. In Vivo Microbial Coevolution Favors Host Protection and Plastic Downregulation of Immunity. Mol. Biol. Evol. 2021, 38, 1330–1338. [Google Scholar] [CrossRef]
- Nilsen, T.; Nes, I.F.; Holo, H. Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 2003, 69, 2975–2984. [Google Scholar] [CrossRef]
- Enuh, B.M.; Gedikli, S.; Aytar Çelik, P.; Çabuk, A. Genome sequence and probiotic potential of newly isolated Enterococcus durans strain MN187066. Lett. Appl. Microbiol. 2023, 76, ovad035. [Google Scholar] [CrossRef]
- Brunse, A.; Offersen, S.M.; Mosegaard, J.J.; Deng, L.; Damborg, P.; Nielsen, D.S.; Sangild, P.T.; Thymann, T.; Nguyen, D.N. Enteral broad-spectrum antibiotics antagonize the effect of fecal microbiota transplantation in preterm pigs. Gut Microbes 2021, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zeise, K.D.; Woods, R.J.; Huffnagle, G.B. Interplay between Candida albicans and Lactic Acid Bacteria in the Gastrointestinal Tract: Impact on Colonization Resistance, Microbial Carriage, Opportunistic Infection, and Host Immunity. Clin. Microbiol. Rev. 2021, 34, e0032320. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.; Savio, V.; Cammarota, M.; Alfano, A.; Schiraldi, C.; Donnarumma, G. Beta-Defensin-2 and Beta-Defensin-3 Reduce Intestinal Damage Caused by Salmonella typhimurium Modulating the Expression of Cytokines and Enhancing the Probiotic Activity of Enterococcus faecium. J. Immunol. Res. 2017, 2017, 6976935. [Google Scholar] [CrossRef]
- Nagpal, R.; Wang, S.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 12649. [Google Scholar] [CrossRef]
- Li, P.; Gu, Q.; Wang, Y.; Yu, Y.; Yang, L.; Chen, J.V. Novel vitamin B12-producing Enterococcus spp. and preliminary in vitro evaluation of probiotic potentials. Appl. Microbiol. Biotechnol. 2017, 101, 6155–6164. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, Q.; Yang, L.; Yu, Y.; Wang, Y. Characterization of extracellular vitamin B12 producing Lactobacillus plantarum strains and assessment of the probiotic potentials. Food Chem. 2017, 234, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinform. 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Lin, L.; Li, Z.; Liu, F.; Zhu, L.; Chen, J.; Zhang, N.; Cao, X.; Ran, S.; et al. Layer-Specific BTX-A Delivery to the Gastric Muscularis Achieves Effective Weight Control and Metabolic Improvement. Adv. Sci. 2023, 10, e2300822. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, Q.; Li, Y.; Guan, Z.; Wei, J.; Li, B.; Liu, K.; Shao, D.; Mi, R.; Liu, H.; et al. Analysis and Comparison of Gut Microbiome in Young Detection Dogs. Front. Microbiol. 2022, 13, 872230. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, B.; Ren, L.; Du, H.; Fei, C.; Qian, C.; Li, B.; Zhang, R.; Liu, H.; Li, Z.; et al. High-fiber diet ameliorates gut microbiota, serum metabolism and emotional mood in type 2 diabetes patients. Front. Cell. Infect. Microbiol. 2023, 13, 1069954. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Di, D.; Sun, Q.; Yao, X.; Wei, J.; Li, B.; Liu, K.; Shao, D.; Qiu, Y.; Liu, H.; et al. Comparative Analyses of the Gut Microbiota in Growing Ragdoll Cats and Felinae Cats. Animals 2022, 12, 2467. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | DC-K7 | DC-K9 | |
|---|---|---|---|
| Size (bp) | 2,748,310 | 2,748,609 | |
| GC content (%) | 38.09 | 38.09 | |
| ORFs | Protein-coding genes (CDSs) | 2707 | 2704 |
| Gene density (gene per kb) | 0.985 | 0.984 | |
| Average gene length (bases per gene) | 869 | 870 | |
| ORF/Genome (%) | 85.63 | 85.60 | |
| RNAs | rRNAs (16S-23S-5S) | 5 | 6 |
| tRNAs | 57 | 57 |
| CAZymes | DC-K7 | DC-K9 |
|---|---|---|
| Glycosyl Transferase (GT) | 20 | 20 |
| Polysaccharide Lyase (PL) | 4 | 4 |
| Carbohydrate Esterase (CE) | 16 | 16 |
| Auxiliary Activities (AAs) | 4 | 4 |
| Carbohydrate-Binding Module (CBM) | 8 | 8 |
| Glycoside Hydrolase (GH) | 56 | 56 |
| SCFAs | DC-K7 (µM) | DC-K9 (µM) |
|---|---|---|
| Acetic acid | 4327.02 ± 287.02 | 2329.69 ± 161.42 |
| Propionic acid | 37.12 ± 3.57 | 30.70 ± 2.69 |
| Isobutyric acid | 13.41 ± 2.07 | 11.93 ± 2.93 |
| Butyric acid | 2.96 ± 0.24 | 2.47 ± 0.28 |
| Isovaleric acid | 2.04 ± 0.12 | 1.24 ± 0.14 |
| Valeric acid | 1.10 ± 0.45 | 1.73 ± 0.25 |
| 4-Methylvaleric acid | 0.13 ± 0.03 | 0.08 ± 0.04 |
| Caproic acid | 6.16 ± 1.45 | 5.38 ± 1.04 |
| Phyla | Antibiotics | DC-K7 | DC-K9 |
|---|---|---|---|
| Firmicutes | 86.68% | 73.22% | 63.72% |
| Proteobacteria | 0.03% | 17.37% | 34.05% |
| Bacteroidota | 4.51% | 7.56% | 2.06% |
| Actinobacteriota | 5.75% | 1.66% | 0.13% |
| Verrucomicrobiota | 2.50% | 0.14% | 0.01% |
| others | 0.54% | 0.04% | 0.02% |
| Genera | Antibiotics | DC-K7 | DC-K9 |
|---|---|---|---|
| Enterococcus | 0.12% | 24.20% | 55.07% |
| Lactobacillus | 42.23% | 27.69% | 2.77% |
| Escherichia-Shigella | 0.01% | 6.63% | 23.17% |
| Staphylococcus | 24.13% | 3.87% | 3.42% |
| Klebsiella | 0.00% | 9.05% | 9.64% |
| unclassified_f__Lachnospiraceae | 6.58% | 12.11% | 0.03% |
| Bacteroides | 0.20% | 6.24% | 1.65% |
| Corynebacterium | 3.69% | 1.23% | 0.02% |
| norank_f__Muribaculaceae | 2.92% | 0.82% | 0.40% |
| norank_f__norank_o__Clostridia_UCG-014 | 3.50% | 0.00% | 0.00% |
| Akkermansia | 2.50% | 0.14% | 0.01% |
| Lachnospiraceae_UCG-006 | 2.54% | 0.08% | 0.00% |
| Enterorhabdus | 1.78% | 0.34% | 0.07% |
| Lachnoclostridium | 1.61% | 0.08% | 0.05% |
| unclassified_f__Enterobacteriaceae | 0.00% | 1.20% | 0.32% |
| Monoglobus | 0.07% | 1.12% | 0.06% |
| others | 8.11% | 5.20% | 3.33% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, X.; Nie, W.; Chen, X.; Zhang, J.; Wei, J.; Qiu, Y.; Liu, K.; Shao, D.; Liu, H.; Ma, Z.; et al. Two Enterococcus faecium Isolates Demonstrated Modulating Effects on the Dysbiosis of Mice Gut Microbiota Induced by Antibiotic Treatment. Int. J. Mol. Sci. 2024, 25, 5405. https://doi.org/10.3390/ijms25105405
Yao X, Nie W, Chen X, Zhang J, Wei J, Qiu Y, Liu K, Shao D, Liu H, Ma Z, et al. Two Enterococcus faecium Isolates Demonstrated Modulating Effects on the Dysbiosis of Mice Gut Microbiota Induced by Antibiotic Treatment. International Journal of Molecular Sciences. 2024; 25(10):5405. https://doi.org/10.3390/ijms25105405
Chicago/Turabian StyleYao, Xiaohui, Wansen Nie, Xi Chen, Junjie Zhang, Jianchao Wei, Yafeng Qiu, Ke Liu, Donghua Shao, Haixia Liu, Zhiyong Ma, and et al. 2024. "Two Enterococcus faecium Isolates Demonstrated Modulating Effects on the Dysbiosis of Mice Gut Microbiota Induced by Antibiotic Treatment" International Journal of Molecular Sciences 25, no. 10: 5405. https://doi.org/10.3390/ijms25105405
APA StyleYao, X., Nie, W., Chen, X., Zhang, J., Wei, J., Qiu, Y., Liu, K., Shao, D., Liu, H., Ma, Z., Li, Z., & Li, B. (2024). Two Enterococcus faecium Isolates Demonstrated Modulating Effects on the Dysbiosis of Mice Gut Microbiota Induced by Antibiotic Treatment. International Journal of Molecular Sciences, 25(10), 5405. https://doi.org/10.3390/ijms25105405