
Structural Models for a Series of Allosteric Inhibitors of IGF1R Kinase

Abstract
1. Introduction
2. Results and Discussion
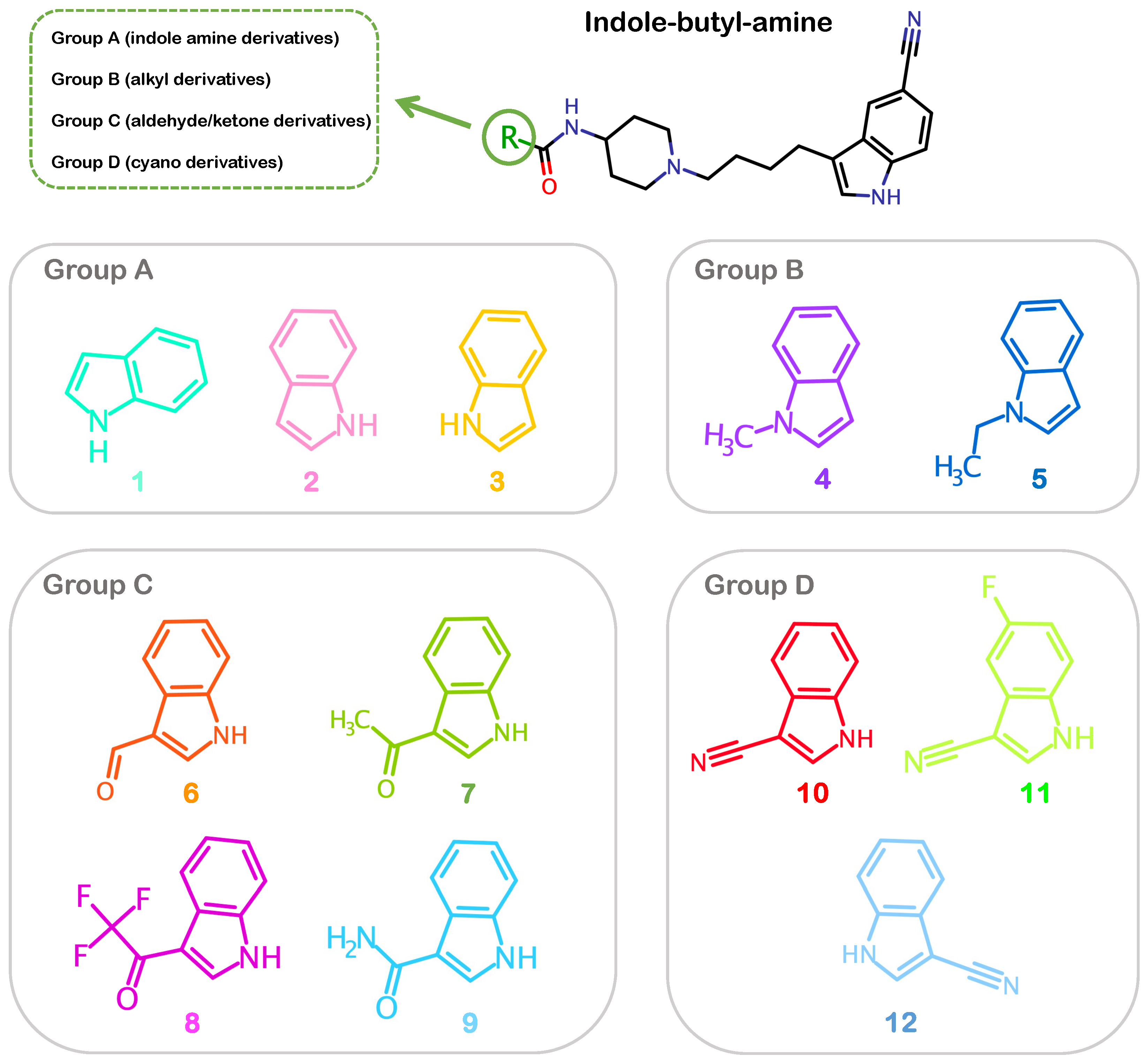
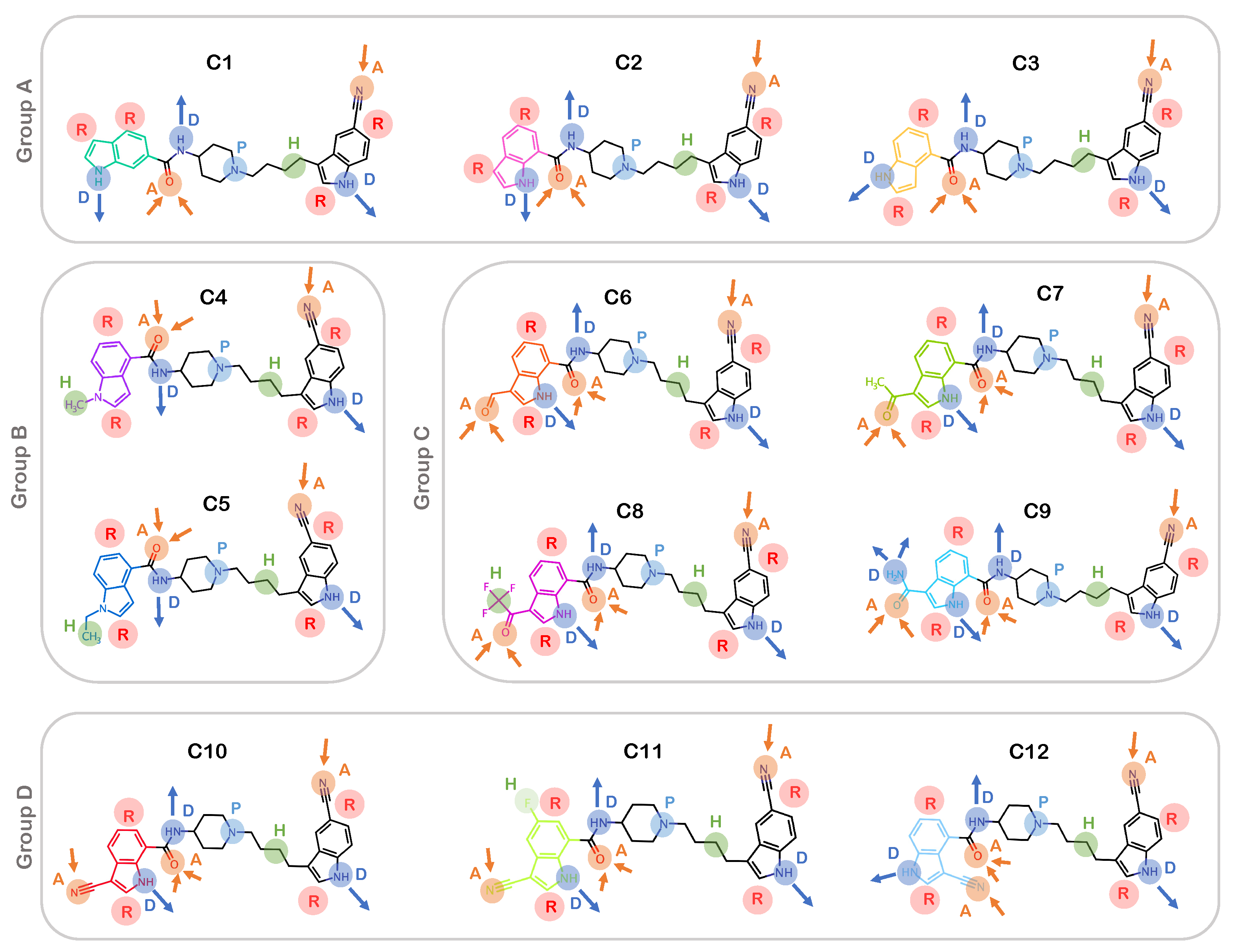
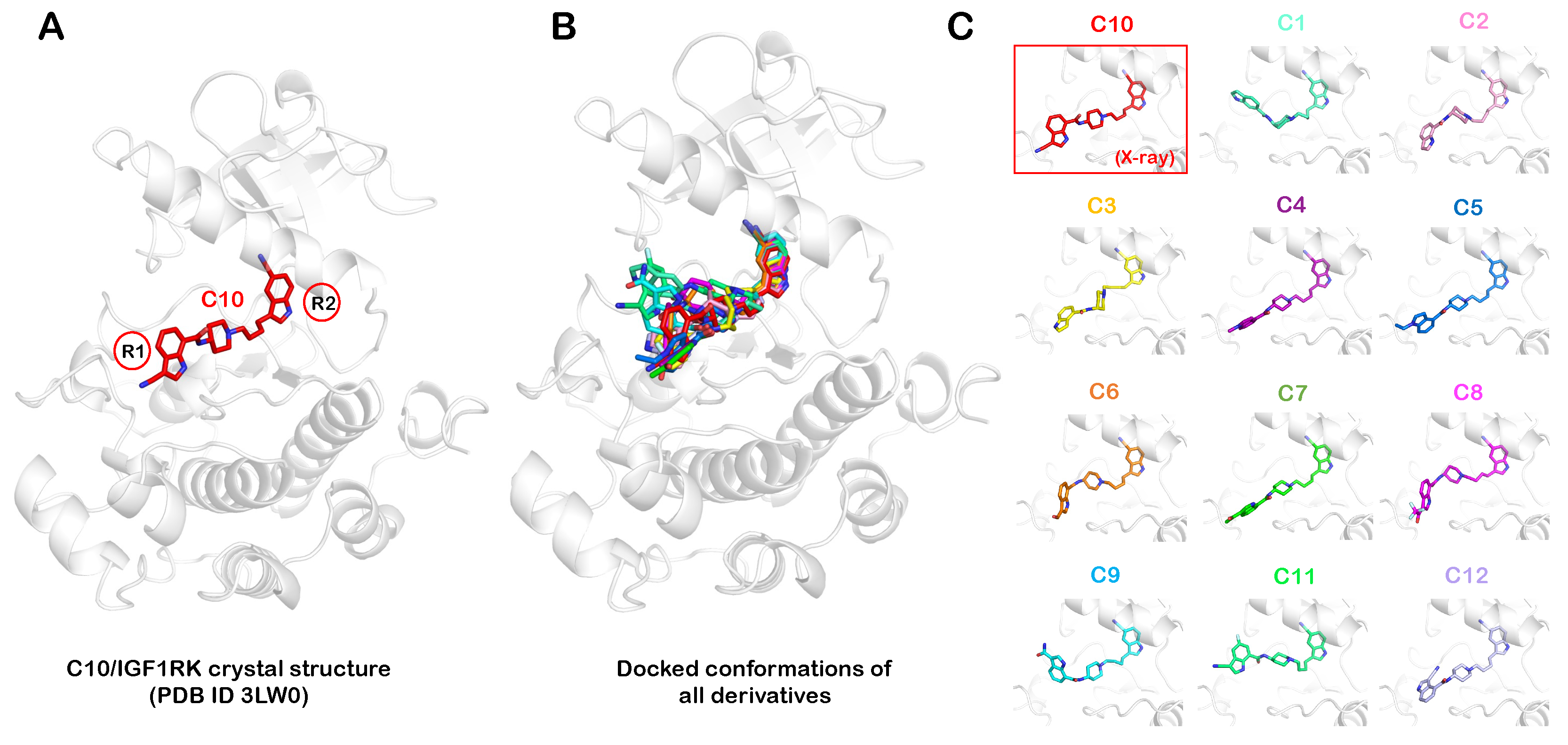
2.1. Pharmacophore Modeling and Molecular Docking
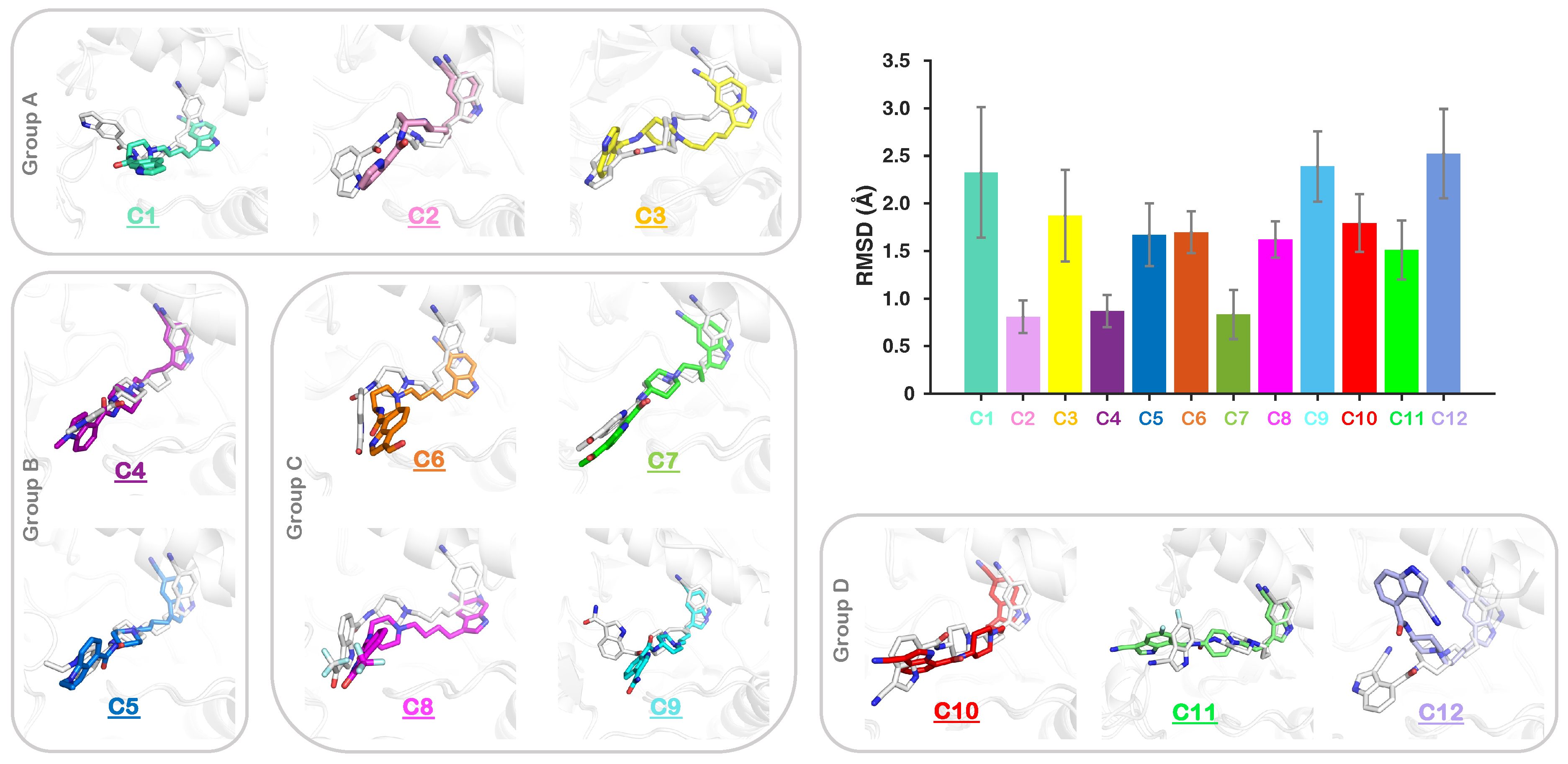
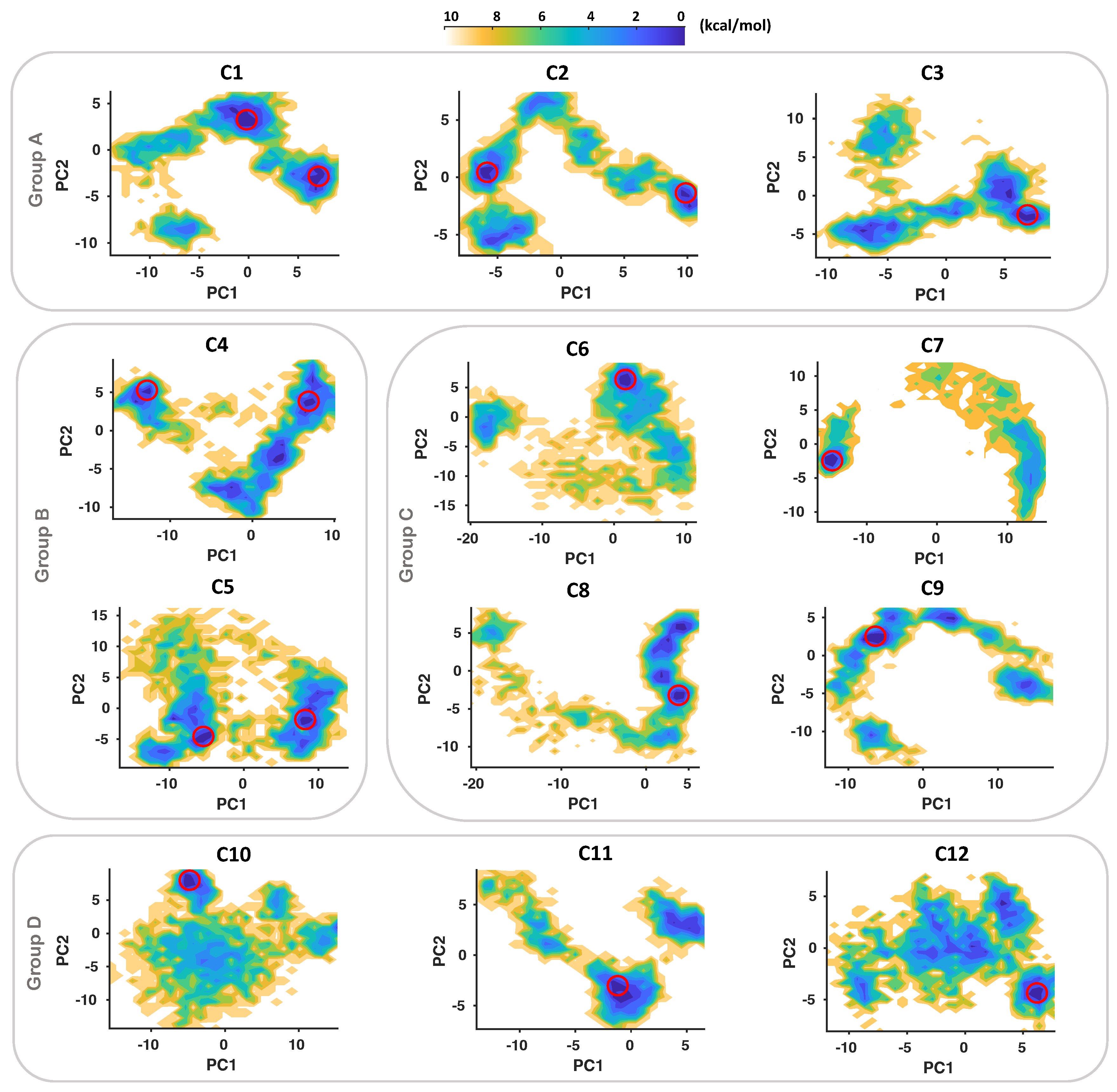
2.2. Conformational Dynamics of Complexes of IGF1RK/Inhibitors
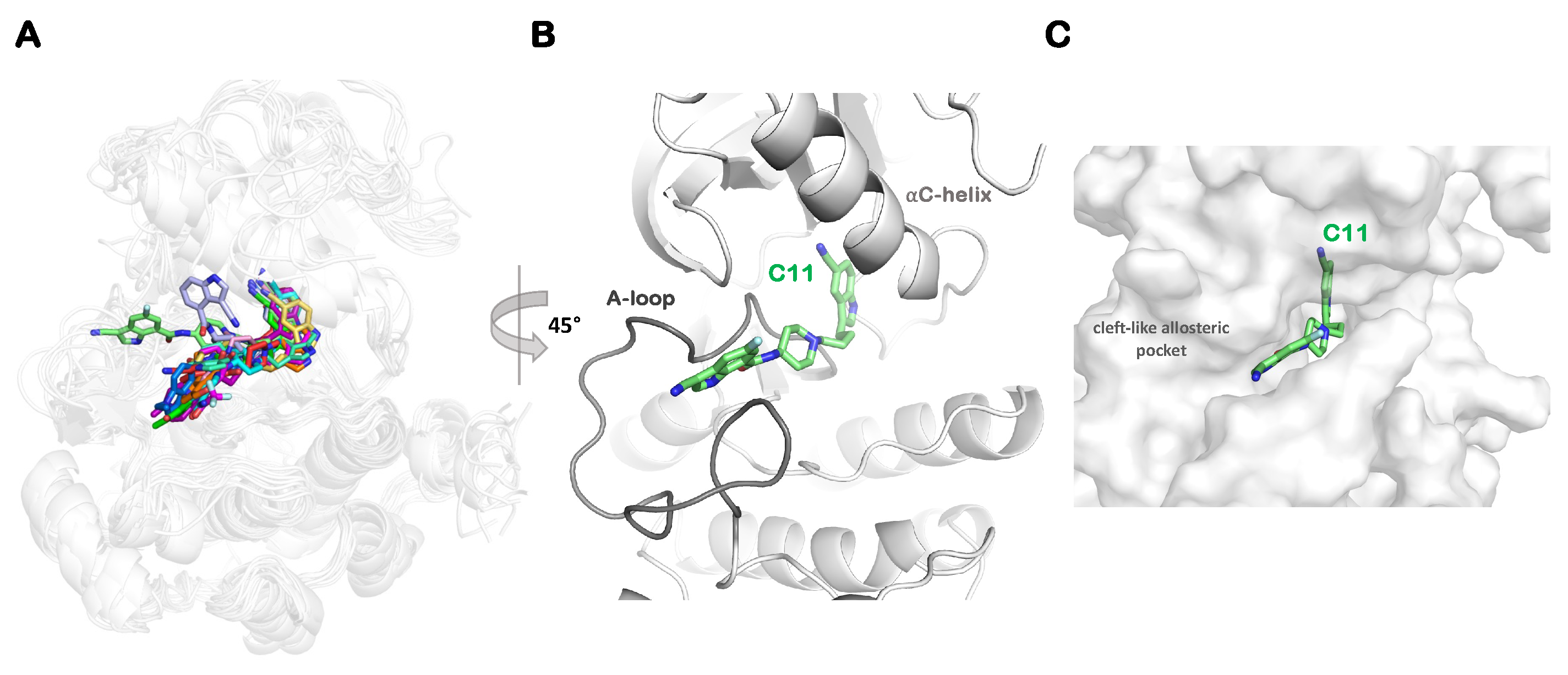
2.3. Dominant Conformation of Each Inhibitor in the Allosteric Pocket
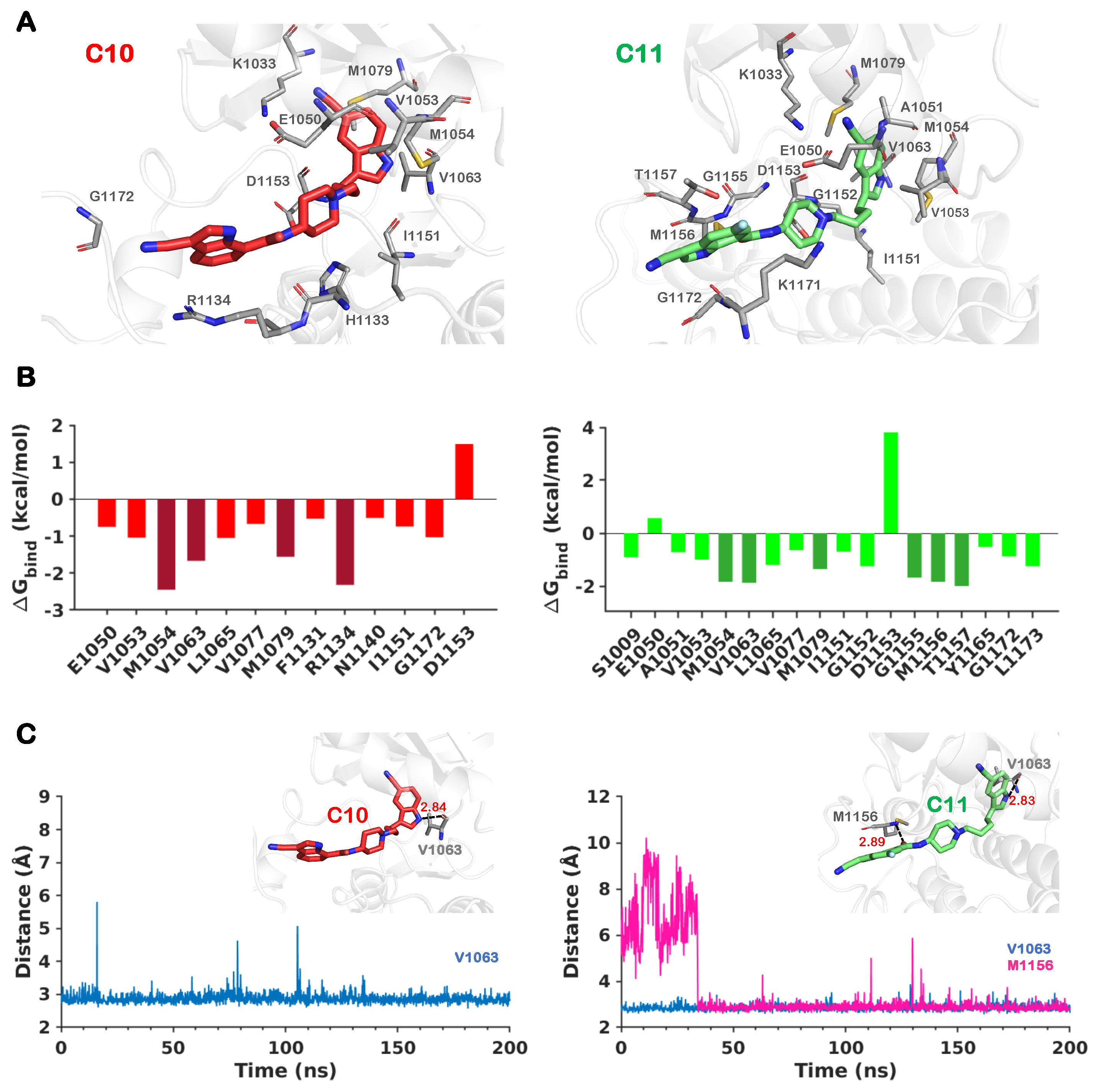
2.4. Effect of Substitutions at the R1 Indole Ring and Unique Binding Mode of C11
3. Materials and Methods
3.1. Generating 3D Chemical Structures and Pharmacophore Features of Allosteric Inhibitors
3.2. Molecular Docking of Allosteric Inhibitors with IGF1RK
3.3. Molecular Dynamics (MD) Simulation Setup
3.4. Binding Free-Energy Calculations
3.5. Conformational and Interaction Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CR | Cysteine-Rich Domain |
| EGFR | Epidermal Growth Factor Receptor |
| HER-2 | Human Epidermal Growth Factor Receptor 2 |
| IGF1R | Insulin-like Growth Factor 1 Receptor |
| IGF-1 | Insulin-like Growth Factor 1 |
| IGF-2 | Insulin-like Growth Factor 2 |
| IR | Insulin Receptor |
| RTK | Receptor Tyrosine Kinase |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Adams, T.; Epa, V.; Garrett, T.; Ward, C. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Cabail, M.Z.; Li, S.; Lemmon, E.; Bowen, M.E.; Hubbard, S.R.; Miller, W.T. The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 2015, 6, 6406. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Frenkel, M.J.; Bentley, J.D.; Lovrecz, G.O.; Elleman, T.C.; Cosgrove, L.J.; Ward, C.W. Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 1998, 394, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Choi, E.; Yu, H.; Bai, X.c. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed]
- Larsson, O.; Girnita, A.; Girnita, L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br. J. Cancer 2005, 92, 2097–2101. [Google Scholar] [CrossRef]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Li, M.; Zhang, S.; Lu, Y.; Liang, Y.; Zhao, K.; Li, Y. Insulin-like growth factor 1/insulin-like growth factor 1 receptor signaling protects against cell apoptosis through the PI3K/AKT pathway in glioblastoma cells. Exp. Ther. Med. 2018, 16, 1477–1482. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, X.; Shen, B. Molecular imaging of IGF-1R in cancer. Mol. Imaging 2017, 16, 1536012117736648. [Google Scholar] [CrossRef]
- Dricu, A.; Kanter, L.; Wang, M.; Nilsson, G.; Hjertman, M.; Wejde, J.; Larsson, O. Expression of the insulin-like growth factor 1 receptor (IGF-1R) in breast cancer cells: Evidence for a regulatory role of dolichyl phosphate in the transition from an intracellular to an extracellular IGF-1 pathway. Glycobiology 1999, 9, 571–579. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, C.; Cao, F.; Wu, Y.; Chen, S.; Han, X.; Mo, J.; Qiu, Z.; Fan, W.; Zhou, P.; et al. Pan-cancer analysis of IGF-1 and IGF-1R as potential prognostic biomarkers and immunotherapy targets. Front. Oncol. 2021, 11, 755341. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mak, V.C.; Cheung, L.W. Drugging IGF-1R in cancer: New insights and emerging opportunities. Genes Dis. 2023, 10, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Ofer, P.; Heidegger, I.; Eder, I.E.; Schöpf, B.; Neuwirt, H.; Geley, S.; Klocker, H.; Massoner, P. Both IGF1R and INSR knockdown exert antitumorigenic effects in prostate cancer in vitro and in vivo. Mol. Endocrinol. 2015, 29, 1694–1707. [Google Scholar] [CrossRef] [PubMed]
- Waraky, A.; Aleem, E.; Larsson, O. Downregulation of IGF-1 receptor occurs after hepatic linage commitment during hepatocyte differentiation from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2016, 478, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Jameson, M.J. Downregulation of IGF1R expression inhibits growth and enhances cisplatin sensitivity of head and neck squamous cell carcinoma cells in vitro. Horm. Cancer 2019, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pourpak, A.; Morris, S.W. Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J. Med. Chem. 2009, 52, 4981–5004. [Google Scholar] [CrossRef]
- Press, M.F.; Lenz, H.J. EGFR, HER2 and VEGF pathways: Validated targets for cancer treatment. Drugs 2007, 67, 2045–2075. [Google Scholar] [CrossRef]
- Jentzsch, V.; Osipenko, L.; Scannell, J.W.; Hickman, J.A. Costs and causes of oncology drug attrition with the example of insulin-like growth factor-1 receptor inhibitors. JAMA Netw. Open 2023, 6, e2324977. [Google Scholar] [CrossRef]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 2014, 32, 2059. [Google Scholar] [CrossRef]
- Sclafani, F.; Kim, T.Y.; Cunningham, D.; Kim, T.W.; Tabernero, J.; Schmoll, H.J.; Roh, J.K.; Kim, S.Y.; Park, Y.S.; Guren, T.K.; et al. A randomized phase II/III study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild-type, metastatic colorectal cancer. J. Natl. Cancer Inst. 2015, 107, djv258. [Google Scholar] [CrossRef]
- DuBois, S.G.; Krailo, M.D.; Glade-Bender, J.; Buxton, A.; Laack, N.; Randall, R.L.; Chen, H.X.; Seibel, N.L.; Boron, M.; Terezakis, S.; et al. Randomized phase III trial of ganitumab with interval-compressed chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2023, 41, 2098–2107. [Google Scholar] [CrossRef]
- Fuchs, C.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.; Riess, H.; Szczylik, C.; Moore, M.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The GAMMA trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef]
- de Bono, J.; Lin, C.C.; Chen, L.T.; Corral, J.; Michalarea, V.; Rihawi, K.; Ong, M.; Lee, J.H.; Hsu, C.H.; Yang, J.C.H.; et al. Two first-in-human studies of xentuzumab, a humanised insulin-like growth factor (IGF)-neutralising antibody, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1324–1332. [Google Scholar] [CrossRef]
- Chen, H.X.; Sharon, E. IGF-1R as an anti-cancer target—trials and tribulations. Chin. J. Cancer 2013, 32, 242. [Google Scholar] [CrossRef]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef]
- Gulbins, A.; Horstmann, M.; Flögel, U.; Oeverhaus, M.; Banga, J.P.; Krause, G.; Hammer, G.D.; Eckstein, A.; Görtz, G.E. Linsitinib, an IGF-1R inhibitor, attenuates disease development and progression in a model of thyroid eye disease. Front. Endocrinol. 2023, 14, 1211473. [Google Scholar] [CrossRef]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Larsson, O.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501. [Google Scholar] [CrossRef]
- Buck, E.; Mulvihill, M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert. Opin. Investig. Drugs 2011, 20, 605–621. [Google Scholar] [CrossRef]
- Baserga, R. The decline and fall of the IGF-I receptor. J. Cell. Physiol. 2013, 228, 675–679. [Google Scholar] [CrossRef]
- King, H.; Aleksic, T.; Haluska, P.; Macaulay, V.M. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat. Rev. 2014, 40, 1096–1105. [Google Scholar] [CrossRef]
- Boone, D.N.; Lee, A.V. Targeting the insulin-like growth factor receptor: Developing biomarkers from gene expression profiling. Crit. Rev. Oncog. 2012, 17, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Wang, B.; Wu, H.; Hu, C.; Wang, H.; Liu, J.; Wang, W.; Liu, Q. An overview of kinase downregulators and recent advances in discovery approaches. Signal Transduct. Target. Ther. 2021, 6, 423. [Google Scholar] [CrossRef]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Grädler, U.; Böttcher, H.; Blaukat, A.; Shutes, A. Allosteric IGF-1R inhibitors. ACS Med. Chem. Lett. 2010, 1, 199–203. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein- ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Modell. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Chester, C.; Friedman, B.; Ursell, F. An extension of the method of steepest descents. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1957; Volume 53, pp. 599–611. [Google Scholar]
- Berendsen, H.J.; Postma, J.V.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The MathWorks Inc. MATLAB, version: 9.13. 0 (R2022b); The MathWorks Inc.: Natick, MA, USA, 2022. [Google Scholar]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. In Protein Dynamics: Methods and Protocols; Livesay, D.R., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 193–226. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System; Schrödinger, Inc.: New York, NY, USA, 2015. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Compound | Name | IC50 (µM) |
|---|---|---|---|
| A | C1 | 1H-Indole-6-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 10 ± 0.09 |
| C2 | 1H-Indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl) butyl]-piperidin-4-yl-amide | 2.5 ± 0.002 | |
| C3 | 1H-Indole-4-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 6 ± 0.20 | |
| B | C4 | 1-Methyl-1H-indole-4-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 2.7 ± 1.10 |
| C5 | 1-Ethyl-1H-indole-4-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 3.6 ± 1.10 | |
| C | C6 | 3-Formyl-1H-indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 0.9 ± 0.01 |
| C7 | 3-Acetyl-1H-indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 4.6 ± 0.08 | |
| C8 | 3-(2,2,2-Trifluoro-acetyl)-1H-indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 4.0 ± 0.90 | |
| C9 | N7-1-[4-(5-cyano-1H-indol-3-yl)butyl]piperidin-4-yl-1H-indole-3,7-dicarboxamide | 5.8 ± 0.40 | |
| D | C10 | 3-Cyano-1H-indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 0.4 ± 0.07 |
| C11 | 3-Cyano-5-fluoro-1H-indole-7-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 0.2 ± 0.07 | |
| C12 | 3-Cyano-1H-indole-4-carboxylic acid 1-[4-(5-cyano-1H-indol-3-yl)-butyl]-piperidin-4-yl-amide | 3.5 ± 2.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, J.; Vashisth, H. Structural Models for a Series of Allosteric Inhibitors of IGF1R Kinase. Int. J. Mol. Sci. 2024, 25, 5368. https://doi.org/10.3390/ijms25105368
Verma J, Vashisth H. Structural Models for a Series of Allosteric Inhibitors of IGF1R Kinase. International Journal of Molecular Sciences. 2024; 25(10):5368. https://doi.org/10.3390/ijms25105368
Chicago/Turabian StyleVerma, Jyoti, and Harish Vashisth. 2024. "Structural Models for a Series of Allosteric Inhibitors of IGF1R Kinase" International Journal of Molecular Sciences 25, no. 10: 5368. https://doi.org/10.3390/ijms25105368
APA StyleVerma, J., & Vashisth, H. (2024). Structural Models for a Series of Allosteric Inhibitors of IGF1R Kinase. International Journal of Molecular Sciences, 25(10), 5368. https://doi.org/10.3390/ijms25105368