

Current Advancements in Anti-Cancer Chimeric Antigen Receptor T Cell Immunotherapy and How Nanotechnology May Change the Game


Abstract
1. Introduction
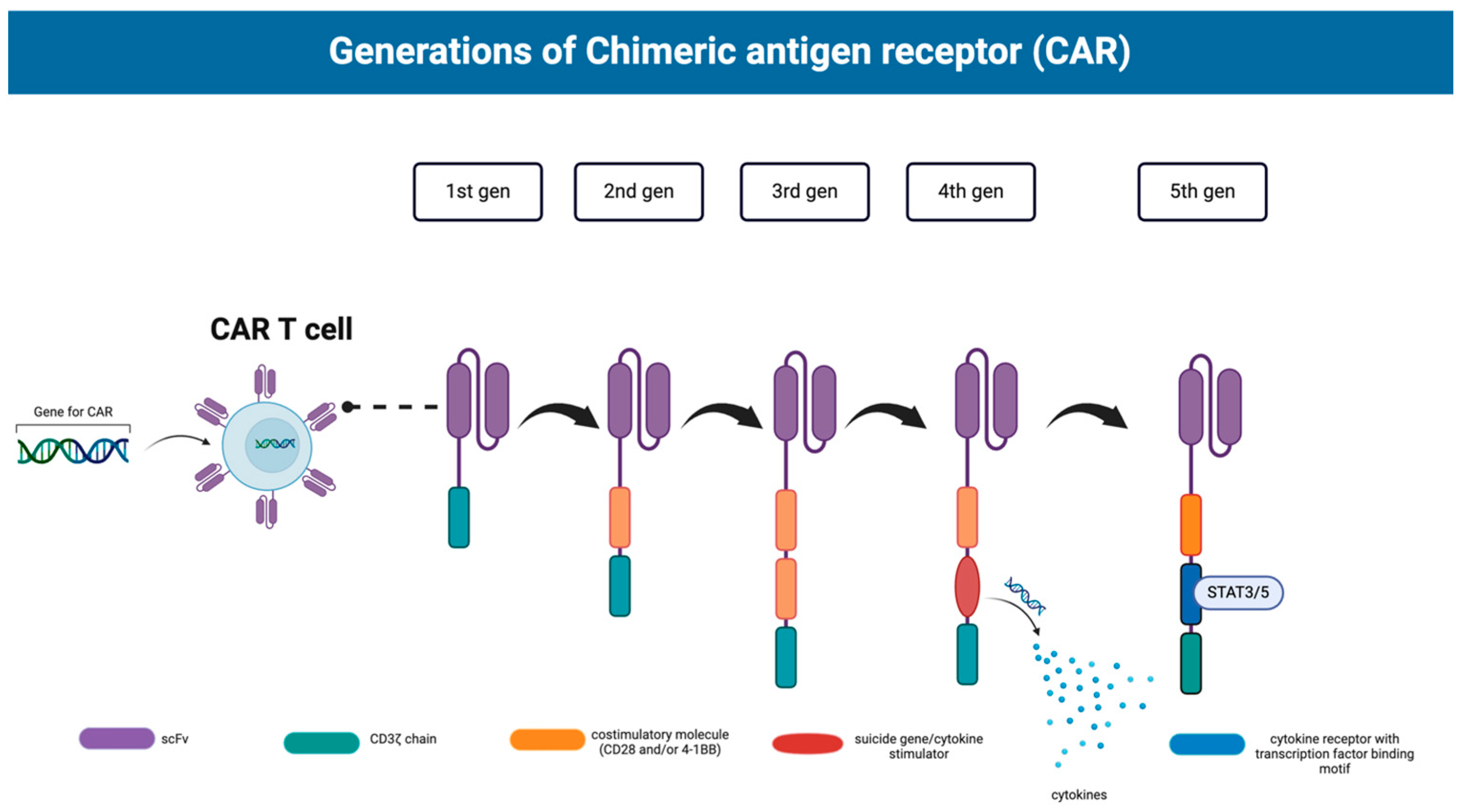
2. Current Status of (CAR)-T Cell Therapy
2.1. Hematological Malignancies

{kind=link}
{kind=link}
{kind=link}
| Generic Name | Brand Name | Target | Year of Approval | Disease |
|---|---|---|---|---|
| Tisagenlecleucel | Kymriah® | CD19 | 2017 | Acute lymphocytic leukemia |
| Axicabtagene | Yescarta® | CD19 | 2017 | Diffuse large B-cell lymphoma |
| Brexucabtagene autoleucel | Tecartus® | CD19 | 2020 | Mantle cell lymphoma |
| Lisocabtagene maraleuce | Breyanzi® | CD19 | 2022 | Adult large B-cell lymphoma |
| Ciltacabtagene autoleucel | Abecma® | B-cell maturation antigen (BCMA) | 2022 | Multiple myeloma |
| Ciltacabtagene autoleucel | Carvykti® | BCMA | 2022 | Relapsed or refractory Multiple myeloma |
2.2. Solid Tumors
| Target Antigen | Tumor Type |
|---|---|
| EGFRvIII | GB, NSCLC |
| IL13Ralpha2 | GB |
| HLA-G | CRC, GC, OC, thyroid cancer, cervical cancer, endometrial cancer [43] |
| Mesothelin | MPM, PDAC, OC |
| HER2 | GB, pancreatic, bile duct |
| PSMA | PCa |
| Mucin-1 | Pancreatic, NSCLC |
| GD2 | Osteosarcoma, neuroblastoma, glioma |
| NKG2D | CRC |
| Claudin18.2 (CLDN18.2) | GC |
| CEA | CRC, other CEA-positive tumors |
| EpCAM | pancreatic |
| GPC3 | HCC |
| B7H3 | CNS malignancies and others |
| FAP | CRC, OC, lung cancer, PDAC |
| Clinical Trial | Target | Status |
|---|---|---|
| NCT04348643 | CEA | Recruiting |
| NCT06006390 | CEA | Recruiting |
| NCT05538195 | CEA | Recruiting |
| NCT05947487 | CD70 | Recruiting |
| NCT05944185 | MSLN | Not yet recruiting |
| NCT03179007 | MUC1 | Unknown |
| NCT05748938 | ROR1 | Recruiting |
| NCT03182816 | EGFR | Unknown |
| NCT05812326 | MUC1 | Completed |
| NCT03706326 | MUC1 | Unknown |
| NCT03030001 | Mesothelin | Unknown |
| NCT03356795 | GD2, PSMA, Muc1, Mesothelin | Unknown |
| NCT05693844 | MSLN | Recruiting |
| NCT03182803 | Mesothelin | Unknown |
| NCT05437315 | GD2/PSMA | Recruiting |
| NCT03356782 | CD133, GD2, Muc1, CD117, and other sarcoma markers | Unknown |
| NCT05736731 | CEA | Recruiting |
| NCT06051695 | MSLN | Recruiting |
| NCT03615313 | Mesothelin | Unknown |
| NCT03373097 | GD2 | Recruiting |
| NCT04581473 | CLDN18.2 | Recruiting |
| NCT06150885 | HLA-G | Not yet recruiting |
| NCT05120271 | GPC3 | Recruiting |
| NCT02873390 | EGFR | Unknown |
| NCT02862028 | EGFR | Unknown |
| NCT02830724 | CD70 | Recruiting |
| NCT02992210 | GD2 | Unknown |
| NCT06082557 | TROP2 | Not yet recruiting |
2.3. Limitations
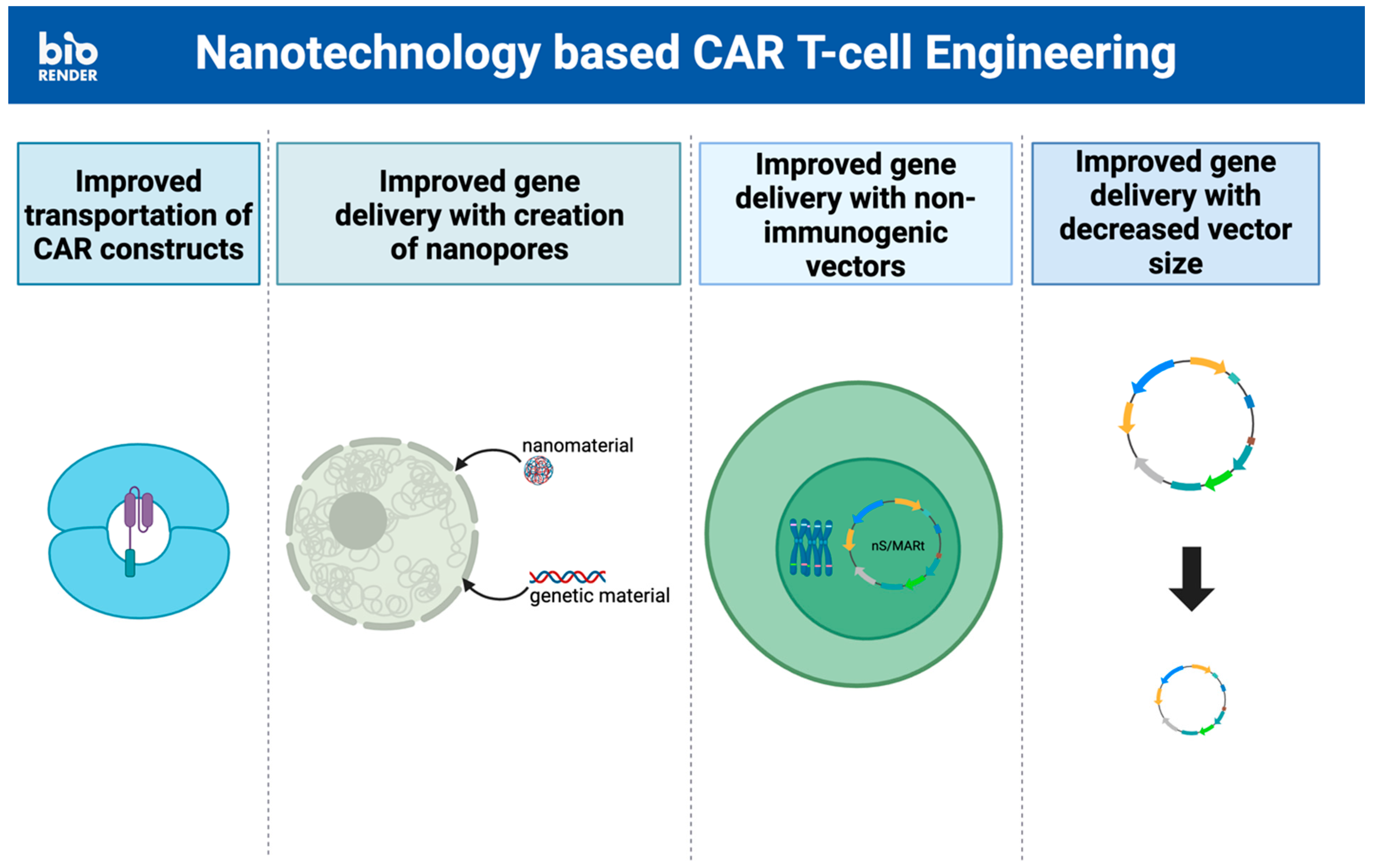
3. Incorporation of Nanotechnology into (CAR)-T Cell Therapy
3.1. T Cell Engineering
3.2. Avoiding Systemic Toxicity
3.3. Downregulating Immunosuppression by the Tumor Microenvironment
3.4. Nanobody-Based (CAR)-T Cells
4. Challenges of Incorporating Nanotherapy
4.1. Challenges in Manufacturing and Regulating Nanoparticles
4.2. Safety Challenges and Toxicity of Nanoparticles
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Herck, S.; De Geest, B.G. Nanomedicine-mediated alteration of the pharmacokinetic profile of small molecule cancer immunotherapeutics. Acta Pharmacol. Sin. 2020, 41, 881–894. [Google Scholar] [CrossRef]
- Sun, Q.; Bai, X.; Sofias, A.M.; van der Meel, R.; Ruiz-Hernandez, E.; Storm, G.; Hennink, W.E.; De Geest, B.; Kiessling, F.; Yu, H.-J.; et al. Cancer nanomedicine meets immunotherapy: Opportunities and challenges. Acta Pharmacol. Sin. 2020, 41, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Ye, Q.; Min, Y. Advances in Nanotechnology Development to Overcome Current Roadblocks in CAR-T Therapy for Solid Tumors. Front. Immunol. 2022, 13, 849759. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Y.; Low, M.H.; Chen, Y.; Lim, F.L.W.I. CAR T Cell Therapy in Hematological Malignancies: Implications of the Tumor Microenvironment and Biomarkers on Efficacy and Toxicity. Int. J. Mol. Sci. 2022, 23, 6931. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 2014, 4, 3571. [Google Scholar] [CrossRef] [PubMed]
- Chailyan, A.; Marcatili, P.; Tramontano, A. The association of heavy and light chain variable domains in antibodies: Implications for antigen specificity. FEBS J. 2011, 278, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharmacother. 2021, 146, 112512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Z.; Wang, T.; Wang, X.-F.; Zhang, Y.-Q.; Song, S.-X.; Ma, C.-Q. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol. Res. 2021, 175, 106036. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Ivica, N.A.; Young, C.M. Tracking the CAR-T Revolution: Analysis of Clinical Trials of CAR-T and TCR-T Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Victor, X.; Lu, P.D. Summary Basis for Regulatory Action; KYMRIAH: Kansas, MO, USA, 2017. [Google Scholar]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- BREYANZI (Lisocabtagene Maraleucel); Food and Drug Administration: Washington, DC, USA, 2022.
- Rodríguez-Vicente, A.E.; Díaz, M.G.; Hernández-Rivas, J.M. Chronic lymphocytic leukemia: A clinical and molecular heterogenous disease. Cancer Genet. 2013, 206, 49–62. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Ciltacabtagene Autoleucel for Relapsed or Refractory Multiple Myeloma; Food and Drug Administration: Washington, DC, USA, 2022.
- Abebe, E.C.; Shiferaw, M.Y.; Admasu, F.T.; Dejenie, T.A. Ciltacabtagene autoleucel: The second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Front. Immunol. 2022, 13, 991092. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Approves Lisocabtagene Maraleucel for Second-Line Treatment of Large B-Cell Lymphoma; Food and Drug Administration: Washington, DC, USA, 2022.
- Denlinger, N.; Bond, D.; Jaglowski, S. CAR T-cell therapy for B-cell lymphoma. Curr. Probl. Cancer 2021, 46, 100826. [Google Scholar] [CrossRef] [PubMed]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA + Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.X.; Ong, X.J.; D’souza, C.; Neeson, P.J.; Zhu, J.J. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front. Immunol. 2023, 14, 1140541. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Serganova, I.; Moroz, E.; Cohen, I.; Moroz, M.; Mane, M.; Zurita, J.; Shenker, L.; Ponomarev, V.; Blasberg, R. Enhancement of PSMA-Directed CAR Adoptive Immunotherapy by PD-1/PD-L1 Blockade. Mol. Ther. Oncolytics 2017, 4, 41–54. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, S.; Turtle, C.J. CAR-T Cell Therapy for Acute Myeloid Leukemia: Preclinical Rationale, Current Clinical Progress, and Barriers to Success. BioDrugs 2021, 35, 281–302. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Wang, R.; Li, W.; Zhang, Z.; Chang, Y.; Hu, Y.; Zhao, J.; Zheng, X.; Mao, Q.; Xia, H. A novel TanCAR targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy. Mol. Ther. Oncolytics 2022, 24, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Guzman, G.; Reed, M.R.; Bielamowicz, K.; Koss, B.; Rodriguez, A. CAR-T Therapies in Solid Tumors: Opportunities and Challenges. Curr. Oncol. Rep. 2023, 25, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; He, H.; Mao, Y.; Ding, X.; Zhang, X.; Xu, J. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark. Res. 2020, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Xia, Y.; Zhang, L.; Jiang, Y.; Liu, M.; Gao, Q.; Zhang, C. Harnessing the potential of HLA-G in cancer therapy: Advances, challenges, and prospects. J. Transl. Med. 2024, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.; Thekkat, P.; Loew, A.; Chen, T.J.; et al. Pre-clinical validation of a humanized anti-EGFR variant III chimeric antigen receptor and phase I trial of CART-EGFRvIII in glioblastoma. J. Immunother. Cancer 2014, 2, O1. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Castelletti, L.; Yeo, D.; van Zandwijk, N.; Rasko, J.E.J. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomark. Res. 2021, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-Specific Chimeric Antigen Receptor mRNA-Engineered T Cells Induce Antitumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2021, 148, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shi, D.; Chi, J.; Cui, D.; Tang, X.; Lin, Y.; Wang, S.; Li, Z.; Jin, H.; Zhai, B. Combined local therapy and CAR-GPC3 T-cell therapy in advanced hepatocellular carcinoma: A proof-of-concept treatment strategy. Cancer Commun. 2023, 43, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA+ liver metastases. Cancer Gene Ther. 2019, 27, 341–355. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, S.; Wei, C.; Hu, Y.; Xu, T.; Xin, X.; Zhu, T.; Shang, L.; Ke, S.; Zhou, J.; et al. Development of Nectin4/FAP-targeted CAR-T cells secreting IL-7, CCL19, and IL-12 for malignant solid tumors. Front. Immunol. 2022, 13, 958082. [Google Scholar] [CrossRef]
- Sorkhabi, A.D.; Khosroshahi, L.M.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Chi, X.; Luo, S.; Ye, P.; Hwang, W.-L.; Cha, J.-H.; Yan, X.; Yang, W.-H. T-cell exhaustion and stemness in antitumor immunity: Characteristics, mechanisms, and implications. Front. Immunol. 2023, 14, 1104771. [Google Scholar] [CrossRef]
- Kapinos, K.A.; Hu, E.; Trivedi, J.; Geethakumari, P.R.; Kansagra, A. Cost-Effectiveness Analysis of CAR T-Cell Therapies vs Antibody Drug Conjugates for Patients with Advanced Multiple Myeloma. Cancer Control. 2023, 30, 10732748221142945. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lammers, T. Combining Nanomedicine and Immunotherapy. Acc. Chem. Res. 2019, 52, 1543–1554. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Shokouhi, A.; Chen, Y.; Yoh, H.Z.; Brenker, J.; Alan, T.; Murayama, T.; Suu, K.; Morikawa, Y.; Voelcker, N.H.; Elnathan, R. Engineering Efficient CAR-T Cells via Electroactive Nanoinjection. Adv. Mater. 2023, 35, e2304122. [Google Scholar] [CrossRef]
- Kitte, R.; Rabel, M.; Geczy, R.; Park, S.; Fricke, S.; Koehl, U.; Tretbar, U.S. Lipid nanoparticles outperform electroporation in mRNA-based CAR T cell engineering. Mol. Ther. Methods Clin. Dev. 2023, 31, 101139. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Kim, H.; Kim, U.; Kim, G.-B.; Kim, J.; Joo, B.; Cho, D.; Lee, D.-S.; Chung, A.J. Genetically Stable and Scalable Nanoengineering of Human Primary T Cells via Cell Mechanoporation. Nano Lett. 2023, 23, 7341–7349. [Google Scholar] [CrossRef] [PubMed]
- Bozza, M.; De Roia, A.; Correia, M.P.; Berger, A.; Tuch, A.; Schmidt, A.; Zörnig, I.; Jäger, D.; Schmidt, P.; Harbottle, R.P. A nonviral, nonintegrating DNA nanovector platform for the safe, rapid, and persistent manufacture of recombinant T cells. Sci. Adv. 2021, 7, eabf1333. [Google Scholar] [CrossRef]
- Moretti, A.; Ponzo, M.; Nicolette, C.A.; Tcherepanova, I.Y.; Biondi, A.; Magnani, C.F. The Past, Present, and Future of Non-Viral CAR T Cells. Front. Immunol. 2022, 13, 867013. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Zhang, T.; Gu, H. Emerging advances in nanobiomaterials-assisted chimeric antigen receptor (CAR)-macrophages for tumor immunotherapy. Front. Bioeng. Biotechnol. 2023, 11, 1211687. [Google Scholar] [CrossRef]
- Fransen, M.F.; Sluijter, M.; Morreau, H.; Arens, R.; Melief, C.J. Local Activation of CD8 T Cells and Systemic Tumor Eradication without Toxicity via Slow Release and Local Delivery of Agonistic CD40 Antibody. Clin. Cancer Res. 2011, 17, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Sandin, L.C.; Orlova, A.; Gustafsson, E.; Ellmark, P.; Tolmachev, V.; Tötterman, T.H.; Mangsbo, S.M. Locally Delivered CD40 Agonist Antibody Accumulates in Secondary Lymphoid Organs and Eradicates Experimental Disseminated Bladder Cancer. Cancer Immunol. Res. 2014, 2, 80–90. [Google Scholar] [CrossRef]
- Park, W.; Heo, Y.-J.; Han, D.K. New opportunities for nanoparticles in cancer immunotherapy. Biomater. Res. 2018, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Gong, X.; Li, J.; Wen, J.; Li, Y.; Zhang, Z. Current approaches of nanomedicines in the market and various stage of clinical translation. Acta Pharm. Sin. B 2022, 12, 3028–3048. [Google Scholar] [CrossRef]
- Khawar, M.B.; Afzal, A.; Abbasi, M.H.; Sheikh, N.; Sun, H. Nano-immunoengineering of CAR-T cell therapy against tumor microenvironment: The way forward in combating cancer. OpenNano 2023, 10, 100124. [Google Scholar] [CrossRef]
- Sparreboom, A.; Scripture, C.D.; Trieu, V.; Williams, P.J.; De, T.; Yang, A.; Beals, B.; Figg, W.D.; Hawkins, M.; Desai, N. Comparative Preclinical and Clinical Pharmacokinetics of a Cremophor-Free, Nanoparticle Albumin-Bound Paclitaxel (ABI-007) and Paclitaxel Formulated in Cremophor (Taxol). Clin. Cancer Res. 2005, 11, 4136–4143. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.G.; Swingle, K.L.; Joseph, R.A.; Mai, D.; Gong, N.; Billingsley, M.M.; Alameh, M.; Weissman, D.; Sheppard, N.C.; June, C.H.; et al. Ionizable Lipid Nanoparticles with Integrated Immune Checkpoint Inhibition for mRNA CAR T Cell Engineering. Adv. Healthc. Mater. 2023, 12, e2301515. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, J.; Zhou, G.; Liu, T.; Dai, Y.; Nie, G.; Zhao, Q. Remodeling of Tumor Microenvironment by Tumor-Targeting Nanozymes Enhances Immune Activation of CAR T Cells for Combination Therapy. Small 2021, 17, 2102624. [Google Scholar] [CrossRef] [PubMed]
- Grosskopf, A.K.; Labanieh, L.; Klysz, D.D.; Roth, G.A.; Xu, P.; Adebowale, O.; Gale, E.C.; Jons, C.K.; Klich, J.H.; Yan, J.; et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors. Sci. Adv. 2022, 8, eabn8264. [Google Scholar] [CrossRef] [PubMed]
- Grosskopf, A.K.; Roth, G.A.; Smith, A.A.A.; Gale, E.C.; Hernandez, H.L.; Appel, E.A. Injectable supramolecular polymer–nanoparticle hydrogels enhance human mesenchymal stem cell delivery. Bioeng. Transl. Med. 2019, 5, e10147. [Google Scholar] [CrossRef]
- Dinakaran, D.; Azad, A.K.; Wilson, B.C. Chapter 13—Photodynamic and photothermal therapy using PLGA nanoparticles. In Poly(lactic-co-glycolic acid) (PLGA) Nanoparticles for Drug Delivery; Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2023; pp. 357–391. [Google Scholar]
- Kozani, P.S.; Naseri, A.; Mirarefin, S.M.J.; Salem, F.; Nikbakht, M.; Bakhshi, S.E.; Kozani, P.S. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark. Res. 2022, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Duan, S.; Jiang, X.; Yang, X.; Hou, X.; Shi, W.; Carlos, C.J.J.; Liu, A.; Yin, S.; Wang, W.; et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Gorovits, B.; Koren, E. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs 2019, 33, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xie, J.; Lin, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr. Purif. 2012, 83, 21–29. [Google Scholar] [CrossRef]
- Li, D.; Wang, R.; Liang, T.; Ren, H.; Park, C.; Tai, C.-H.; Ni, W.; Zhou, J.; Mackay, S.; Edmondson, E.; et al. Camel nanobody-based B7-H3 CAR-T cells show high efficacy against large solid tumours. Nat. Commun. 2023, 14, 5920. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, D.; Luo, H.; Shi, W.; Xie, S.; Qiang, H.; Zhu, L.; Gao, L.; Liu, J.; Sun, S.; et al. Nanobody-based CAR T cells targeting intracellular tumor antigens. Biomed. Pharmacother. 2022, 156, 113919. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gong, J.; Qi, Q.; Liu, C.; Su, H.; Xing, Y.; Zhao, J. 131I-Labeled Anti-HER2 Nanobody for Targeted Radionuclide Therapy of HER2-Positive Breast Cancer. Int. J. Nanomed. 2023, 18, 1915–1925. [Google Scholar] [CrossRef]
- Yan, Y.; Cheng, X.; Li, L.; Zhang, R.; Zhu, Y.; Wu, Z.; Ding, K. A Novel Small Molecular Antibody, HER2-Nanobody, Inhibits Tumor Proliferation in HER2-Positive Breast Cancer Cells In Vitro and In Vivo. Front. Oncol. 2021, 11, 669393. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; Blykers, A.; Vaneycken, I.; D’Huyvetter, M.; Heemskerk, J.; Lahoutte, T.; Devoogdt, N.; Caveliers, V. 18F-nanobody for PET imaging of HER2 overexpressing tumors. Nucl. Med. Biol. 2016, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luan, L.; Liu, X.; Jiang, D.; Deng, J.; Xu, J.; Yuan, Y.; Xing, J.; Chen, B.; Xing, D.; et al. A novel nanobody-based HER2-targeting antibody exhibits potent synergistic antitumor efficacy in trastuzumab-resistant cancer cells. Front. Immunol. 2023, 14, 1292839. [Google Scholar] [CrossRef]
- Yaman, S.; Ramachandramoorthy, H.; Iyer, P.; Chintapula, U.; Nguyen, T.; Sabnani, M.; Kotadia, T.; Ghaffari, S.; Pop, L.M.; Hannan, R.; et al. Targeted chemotherapy via HER2-based chimeric antigen receptor (CAR) engineered T-cell membrane coated polymeric nanoparticles. Bioact. Mater. 2024, 34, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Atik, A.F.; Suryadevara, C.M.; Schweller, R.M.; West, J.L.; Healy, P.; Ii, J.E.H.; Congdon, K.L.; Sanchez-Perez, L.; McLendon, R.E.; Archer, G.E.; et al. Hyaluronic acid based low viscosity hydrogel as a novel carrier for Convection Enhanced Delivery of CAR T cells. J. Clin. Neurosci. 2018, 56, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, L.; Li, H.; Miao, F.; Zhang, Z.; Hu, C.; Yu, W.; Tang, Q.; Shao, G. Application of lipid nanovesicle drug delivery system in cancer immunotherapy. J. Nanobiotechnol. 2022, 20, 214. [Google Scholar] [CrossRef]
- Karimi, M.; Solati, N.; Ghasemi, A.; Estiar, M.A.; Hashemkhani, M.; Kiani, P.; Mohamed, E.; Saeidi, A.; Taheri, M.; Avci, P.; et al. Carbon nanotubes part II: A remarkable carrier for drug and gene delivery. Expert Opin. Drug Deliv. 2015, 12, 1089–1105. [Google Scholar] [CrossRef]
- Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanomedicine. N. Engl. J. Med. 2010, 363, 2434–2443. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Challenges in Development of Nanoparticle-Based Therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Bawa, R. Regulating Nanomedicine—Can the FDA Handle It? Curr. Drug Deliv. 2011, 8, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Duvall, M.N.; Knight, K. FDA Regulation of Nanotechnology; Beveridge and Diamond, PG: Washington, DC, USA, 2012. [Google Scholar]
- Clément, O.; Siauve, N.; Cuénod, C.A.; Frija, G. Liver imaging with ferumoxides (Feridex): Fundamentals, controversies, and practical aspects. Top. Magn. Reson. Imaging 1998, 9, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wei, Y. For Better or Worse, Iron Overload by Superparamagnetic Iron Oxide Nanoparticles as a MRI Contrast Agent for Chronic Liver Diseases. Chem. Res. Toxicol. 2016, 30, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.J. Current status of superparamagnetic iron oxide contrast agents for liver magnetic resonance imaging. World J. Gastroenterol. 2015, 21, 13400–13402. [Google Scholar] [CrossRef] [PubMed]
- Patel, J. Liposomal doxorubicin: Doxil®. J. Oncol. Pharm. Pr. 1996, 2, 201–210. [Google Scholar] [CrossRef]
- Ning, X.; Peng, C.; Li, E.S.; Xu, J.; Vinluan, R.D.; Yu, M.; Zheng, J. Physiological stability and renal clearance of ultrasmall zwitterionic gold nanoparticles: Ligand length matters. APL Mater. 2017, 5, 053406. [Google Scholar] [CrossRef] [PubMed]
- Notarianni, M.; Vernon, K.; Chou, A.; Aljada, M.; Liu, J.; Motta, N. Plasmonic effect of gold nanoparticles in organic solar cells. Sol. Energy 2014, 106, 23–37. [Google Scholar] [CrossRef]
- Qian, H.; Pretzer, L.A.; Velazquez, J.C.; Zhao, Z.; Wong, M.S. Gold nanoparticles for cleaning contaminated water. J. Chem. Technol. Biotechnol. 2013, 88, 735–741. [Google Scholar] [CrossRef]
- Ray, P.C.; Yu, H.; Fu, P.P. Toxicity and Environmental Risks of Nanomaterials: Challenges and Future Needs. J. Environ. Sci. Health Part C 2009, 27, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Sani, A.; Cao, C.; Cui, D. Toxicity of gold nanoparticles (AuNPs): A review. Biochem. Biophys. Rep. 2021, 26, 100991. [Google Scholar] [CrossRef] [PubMed]
- Sonavane, G.; Tomoda, K.; Makino, K. Biodistribution of colloidal gold nanoparticles after intravenous administration: Effect of particle size. Colloids Surf. B Biointerfaces 2008, 66, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.K.; Jittiwat, J.; Manikandan, J.; Ong, C.-N.; Yu, L.E.; Ong, W.-Y. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials 2010, 31, 2034–2042. [Google Scholar] [CrossRef]
- Lipka, J.; Semmler-Behnke, M.; Sperling, R.A.; Wenk, A.; Takenaka, S.; Schleh, C.; Kissel, T.; Parak, W.J.; Kreyling, W.G. Biodistribution of PEG-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials 2010, 31, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xu, K.; Ji, L.; Tang, B. Effect of gold nanoparticles on glutathione depletion-induced hydrogen peroxide generation and apoptosis in HL7702 cells. Toxicol. Lett. 2011, 205, 86–95. [Google Scholar] [CrossRef]
- Li, J.J.; Lo, S.-L.; Ng, C.-T.; Gurung, R.L.; Hartono, D.; Hande, M.P.; Ong, C.-N.; Bay, B.-H.; Yung, L.-Y.L. Genomic instability of gold nanoparticle treated human lung fibroblast cells. Biomaterials 2011, 32, 5515–5523. [Google Scholar] [CrossRef]

| Types | Examples | Application to (CAR)-T Cell Therapy |
|---|---|---|
| Polymeric | Poly(lactic-co-glycolic acid) (PLGA) | Carrier for T cells to enhance survival, increase uptake, and improve sustained drug release |
| Hydrogel | Hyaluronic acid | Carrier for enabling the continuous infusion of (CAR)-T cells, reducing sedimentation of the cells, and enhancing distribution |
| Lipid-based | Liposomes | Carrier for T cells to enhance survival |
| Lipid Nanoparticles (LNPs) | Surface functionalization for targeting | |
| Carbon-based | Carbon nanotubes (CNTs) | Provide mechanical strength High surface area for cargo loading The surface can be functionalized |
| Organism | Particle | Effects |
|---|---|---|
| Mice | AuNPs | AuNPs accumulate in spleen, liver, and lungs [109] |
| Rats | AuNPs | AuNPs accumulate in spleen and liver [110] |
| Mice | PEG-coated AuNPs | Apoptosis and acute inflammation [111] |
| Rats | PEG-coated AuNPs | Accumulation in spleen and liver [111] |
| Human liver cells | AuNPs | Depolarization of mitochondrial transmembrane potential and apoptosis [112] |
| Airway epithelial cells | AuNPs | Elevated lipid peroxidase and DNA damage [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ku, K.S.; Tang, J.; Chen, Y.; Shi, Y. Current Advancements in Anti-Cancer Chimeric Antigen Receptor T Cell Immunotherapy and How Nanotechnology May Change the Game. Int. J. Mol. Sci. 2024, 25, 5361. https://doi.org/10.3390/ijms25105361
Ku KS, Tang J, Chen Y, Shi Y. Current Advancements in Anti-Cancer Chimeric Antigen Receptor T Cell Immunotherapy and How Nanotechnology May Change the Game. International Journal of Molecular Sciences. 2024; 25(10):5361. https://doi.org/10.3390/ijms25105361
Chicago/Turabian StyleKu, Kimberly S., Jie Tang, Yuan Chen, and Yihui Shi. 2024. "Current Advancements in Anti-Cancer Chimeric Antigen Receptor T Cell Immunotherapy and How Nanotechnology May Change the Game" International Journal of Molecular Sciences 25, no. 10: 5361. https://doi.org/10.3390/ijms25105361
APA StyleKu, K. S., Tang, J., Chen, Y., & Shi, Y. (2024). Current Advancements in Anti-Cancer Chimeric Antigen Receptor T Cell Immunotherapy and How Nanotechnology May Change the Game. International Journal of Molecular Sciences, 25(10), 5361. https://doi.org/10.3390/ijms25105361