

Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis?


Abstract
1. Introduction
2. Selected Cells Involved in the Liver Fibrosis Process
2.1. Hepatocytes and Hepatic Stellate Cells (HSC)
2.2. Liver Macrophages
2.3. Lymphocytes
3. Regression of Liver Fibrosis
Mechanisms of Regression
4. The Role of Selected Nutraceuticals in Liver Fibrosis
4.1. Vitamin C
4.2. Omega-3 Polyunsaturated Fatty Acids (PUFAs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Intervention Duration | Methods | Results | Reference |
|---|---|---|---|---|
| Patients diagnosed with MASLD n = 24 Randomly assigned to 2 groups: Study group n = 13 Control group n = 11 | 6 months | Patients received 503 mg DHA and 103 mg EPA 3 times daily or placebo. | Docosahexaenoic acid (DHA) and omega index increased significantly in RBC in addition to a significant reduction in alkaline phosphatase (ALP) and liver fibrosis. There has been a reduction in ALP and liver stiffness measure (LSM) as measured by FibroScan® Reductions in waist circumference (WC), gamma-glutamyl transferase (GGT), total cholesterol (CT), triglycerides (TG), and controlled attenuation parameter (CAP) were noted, although statistical significance was not attained. No significant changes in the assessed parameters were observed in the placebo group. There was no statistically significant difference in the relative expression of miR-122 when comparing the baseline period to the six-month intervention, observed in both the placebo group and the n-3 PUFA group. | Cansanção et al. 2020 [122] |
| Patients diagnosed with MASH n = 78 Randomly assigned to 2 groups: Study group n = 39 Control group n = 39 | 6 months | Patients received 50 mL of PUFA in a 1:1 ratio of EHA and DHA in their daily diet). Additionally, both groups were recommended to engage in moderate physical exercise lasting 30 min at least 5 days a week. A low-fat, low-cholesterol, and low-carbohydrate diet was also recommended. | There was an improvement in parameters such as AST (aspartate aminotransferase), ALT (alanine aminotransferase), GGTP (gamma-glutamyl transpeptidase), reactive C protein (CRP), malondialdehyde (MDA) as well as collagen type IV and P-III-P. Positive results were obtained in terms of steatosis grade, necrotic-inflammatory grade, fibrosis grade, and ballooning results compared to the control group. Histological characteristics were comparable between the control group and the PUFA-treated group. | Li et al. 2015 [123] |
| Patients diagnosed with MASH n = 34 Randomly assigned to 2 groups: Study group n = 17 Control group n = 17 | 12 months | Patients received n-3 at a dose of 3000 mg/day with advice on 150 min of physical activity per week and a hypocaloric diet (the reduction of 30% of daily kcal intake). | There was a greater decrease in liver fat content. Supplementation did not lead to improvement in the primary histological activity score in patients with NASH (≥2-point reduction in NAS). No independent effects on the markers of hepatocyte damage or the indicators of insulin sensitivity were observed. There was no improvement in cell injury biomarkers (M30 and M65) that did not occur uniformly with N-3 PUFA and only occurred with concomitant weight loss. There were no consistent beneficial effects on blood lipid composition (aside from a trend in triglyceride reduction) or insulin sensitivity. | Argo et al. 2015 [125] |
| Patients diagnosed with MASLD n = 103 Randomly assigned to 2 groups: Study group n = 51 Control group n = 52 | 15–18 months | Patients received 4 g/day of DHA + EPA (460 mg EPA and 380 mg DHA) or placebo. | There was a trend toward improvement in liver fat percentage. DHA erythrocyte enrichment was independently associated with a decrease in liver fat percentage. No improvement in fibrosis scores occurred. | Scorletii et al. 2014 [126] |
4.3. Carotenoids
4.4. Curcumin
- ○
- Curcumin;
- ○
- Demethoxycurcumin;
- ○
- Bisdemethoxycurcumin;
- ○
- Cyclocurcumin.
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Delgado, M.E.; Cárdenas, B.I.; Farran, N.; Fernandez, M. Metabolic Reprogramming of Liver Fibrosis. Cells 2021, 10, 3604. [Google Scholar] [CrossRef] [PubMed]
- Aydın, M.M.; Akçalı, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed]
- Dokmak, A.; Lizaola-Mayo, B.; Trivedi, H.D. The Impact of Nonalcoholic Fatty Liver Disease in Primary Care: A Population Health Perspective. Am. J. Med. 2021, 134, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Salomone, F.; Webb, M.; Shibolet, O.; Kariv, R.; Zelber-Sagi, S. Dietary vitamin E and C intake is inversely associated with the severity of nonalcoholic fatty liver disease. Dig. Liver Dis. 2019, 51, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Liu, S.; Yang, M. Treatment of liver fibrosis: Past, current, and future. World J. Hepatol. 2023, 15, 755–774. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Electronic address: Easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J. Hepatol. 2019, 70, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Colletti, A.; Penson, P.E.; Katsiki, N.; Mikhailidis, D.P.; Toth, P.P.; Gouni-Berthold, I.; Mancini, J.; Marais, D.; International Lipid Expert Panel (ILEP); et al. Nutraceutical approaches to non-alcoholic fatty liver disease (NAFLD): A position paper from the International Lipid Expert Panel (ILEP). Pharmacol Res. 2023, 189, 106679. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Colletti, A.; Bellentani, S. Nutraceutical Approach to Non-Alcoholic Fatty Liver Disease (NAFLD): The Available Clinical Evidence. Nutrients 2018, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Hoti, G.; Matencio, A.; Rubin Pedrazzo, A.; Cecone, C.; Appleton, S.L.; Khazaei Monfared, Y.; Caldera, F.; Trotta, F. Nutraceutical Concepts and Dextrin-Based Delivery Systems. Int. J. Mol. Sci. 2022, 23, 4102. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Baratta, F.; Pastori, D.; Angelico, F. The role of nutraceuticals for the treatment of non-alcoholic fatty liver disease. Br. J. Clin. Pharmacol. 2017, 83, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Casari, M.; Siegl, D.; Deppermann, C.; Schuppan, D. Macrophages and platelets in liver fibrosis and hepatocellular carcinoma. Front. Immunol. 2023, 14, 1277808. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 40875. [Google Scholar] [CrossRef] [PubMed]
- Loft, A.; Alfaro, A.J.; Schmidt, S.F.; Pedersen, F.B.; Terkelsen, M.K.; Puglia, M.; Chow, K.K.; Feuchtinger, A.; Troullinaki, M.; Maida, A.; et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab. 2021, 33, 1685–1700. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Gan, C.; Cai, Q.; Tang, C.; Gao, J. Inflammasomes and Pyroptosis of Liver Cells in Liver Fibrosis. Front. Immunol. 2022, 30, 896473. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Postigo-Fernandez, J.; Kim, K.; Zhu, C.; Yu, J.; Meroni, M.; Mayfield, B.; Bartolomé, A.; Dapito, D.H.; Ferrante, A.W., Jr.; et al. Notch-mediated hepatocyte MCP-1 secretion causes liver fibrosis. J. Clin. Insight. 2023, 8, e165369. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kuo, L.M.; Wu, Y.H.; Chang, Y.C.; Lai, K.H.; Hwang, T.L. BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells. Biomedicines 2020, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef]
- Kozłowska, J.; Jabłońska, J.; Wiercińska-Drapało, A. Toll-like receptors in viral hepatitis. Postepy Hig Med. Dosw. 2009, 63, 351–354. [Google Scholar]
- Cheng, D.; Chai, J.; Wang, H.; Fu, L.; Peng, S.; Ni, X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. [Google Scholar] [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease—Novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef] [PubMed]
- Daemen, S.; Gainullina, A.; Kalugotla., G.; He., L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, Y.; Orito, E.; Sugauchi, F.; Hasegawa, I.; Sakurai, M.; Fujiwara, K.; Ohno, T.; Ueda, R.; Mizokami, M. Transforming growth factor-beta-1 genetic polymorphism in Japanese patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2003, 18, 1139–1143. [Google Scholar] [CrossRef]
- Pan, P.; Chen, C.; Hong, J.; Gu, Y. Autoimmune pathogenesis, immunosuppressive therapy and pharmacological mechanism in aplastic anemia. Int. Immunopharmacol. 2023, 117, 110036. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 803037. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Wang, H.; Ni, M.; Wang, Z.; Wang, Z.; Wei, S.; Liu, M.; Wang, P.; Qiu, J.; Zhang, L.; et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut 2022, 71, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, B.; Xue, M.; Zhu, J.; Tong, G.; Fan, J.; Zhu, K.; Hu, Z.; Chen, R.; Dong, Y.; et al. Macrophage-specific FGF12 promotes liver fibrosis progression in mice. Hepatology 2023, 77, 816–833. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Y.; Liu, K.; Liu, L.; Shan, S.; Liu, H.; Ren, J.; Sun, S.; Wang, M.; Jia, J.; et al. GITRL impairs hepatocyte repopulation by liver progenitor cells to aggravate inflammation and fibrosis by GITR+CD8+ T lymphocytes in CDE Mice. Cell Death Dis. 2024, 15, 114. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, N.; Barget, N.; Andrieu, M.; Roulot, D.; Letoumelin, P.; Grando, V.; Trinchet, J.C.; Ganne-Carrié, N.; Beaugrand, M.; Deny, P.; et al. Interferon gamma-secreting HCV-specific CD8+ T cells in the liver of patients with chronic C hepatitis: Relation to liver fibrosis--ANRS HC EP07 study. J. Viral Hepat. 2006, 13, 474–481. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, P.; Peng, B.; Ming, Y.Z. Cross-talk between hepatic stellate cells and T lymphocytes in liver fibrosis. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, H.; Yao, Y.; Zhang, X.; Guan, Y.; Zheng, F. CD4+ T cell activation and inflammation in NASH-related fibrosis. Front. Immunol. 2022, 10, 967410. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yin, S. Natural killer T cells in liver injury, inflammation and cancer. Expert. Rev. Gastroenterol. Hepatol. 2015, 9, 1077–1085. [Google Scholar] [CrossRef]
- Fasbender, F.; Widera, A.; Hengstler, J.G.; Watzl, C. Natural Killer Cells and Liver Fibrosis. Front. Immunol. 2016, 29, 19. [Google Scholar] [CrossRef]
- Baroni Pietto, M.C.; Lev, P.R.; Glembotsky, A.C.; Marín Oyarzún, C.P.; Gomez, G.; Collado, V.; Pisoni, C.; Gomez, R.A.; Grodzielski, M.; Gonzalez, J.; et al. Pathogenic mechanisms contributing to thrombocytopenia in patients with systemic lupus erythematosus. Platelets 2022, 33, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Lua, I.; Li, Y.; Pappoe, L.S.; Asahina, K. Myofibroblastic Conversion and Regeneration of Mesothelial Cells in Peritoneal and Liver Fibrosis. Am. J. Pathol. 2015, 185, 3258–3273. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.C.; Kim, H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin. Mol. Hepatol. 2017, 23, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. 2020, 245, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Kothari, A.; Sarma, P.; Medhi, B.; Omar, B.J.; Kaushal, K. Recent Advancements in Antifibrotic Therapies for Regression of Liver Fibrosis. Cells 2022, 11, 1500. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Puengel, T.; Loomba, R.; Friedman, S.L. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J. Hepatol. 2023, 79, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Sun, Y.; Zhou, J.; Wu, X.; Chen, Y.; Piao, H.; Lu, L.; Ding, H.; Nan, Y.; Jiang, W.; et al. Early steep decline of liver stiffness predicts histological reversal of fibrosis in chronic hepatitis B patients treated with entecavir. J. Viral Hepat. 2019, 26, 576–585. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, R.; Aghemo, A.; Rumi, M.G.; Ronchi, G.; Donato, M.F.; Paradis, V.; Colombo, M.; Bedossa, P. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 2012, 56, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.; Kleinman, L.; Barker, C.M.; Revicki, D.A.; Green, J. Relationship of health-related quality of life to treatment adherence and sustained response in chronic hepatitis C patients. Hepatology 2002, 35, 704–708. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. J. Am. Med. Assoc. 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; McHutchison, J.; Manns, M.; Trepo, C.; Lindsay, K.; Goodman, Z.; Ling, M.H.; Albrecht, J. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002, 122, 1303–1313. [Google Scholar] [CrossRef]
- Maylin, S.; Martinot-Peignoux, M.; Moucari, R.; Boyer, N.; Ripault, M.P.; Cazals-Hatem, D.; Giuily, N.; Castelnau, C.; Cardoso, A.C.; Asselah, T.; et al. Eradication of hepatitis C virus in patients successfully treated for chronic hepatitis C. Gastroenterology 2008, 135, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Moussalli, J.; Munteanu, M.; Thabut, D.; Lebray, P.; Rudler, M.; Ngo, Y.; Thibault, V.; Mkada, H.; Charlotte, F.; et al. Slow regression of liver fibrosis presumed by repeated biomarkers after virological cure in patients with chronic hepatitis C. J. Hepatol. 2013, 59, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Aleman, S.; Rahbin, N.; Weiland, O.; Davidsdottir, L.; Hedenstierna, M.; Rose, N.; Verbaan, H.; Stål, P.; Carlsson, T.; Norrgren, H.; et al. A risk for hepatocellular carcinoma persists long-term after sustained virologic response in patients with hepatitis C-associated liver cirrhosis. Clin. Infect. Dis. 2013, 57, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shigefuku, R.; Maeyama, S.; Suzuki, M. Cirrhosis improvement to alcoholic liver fibrosis after passive abstinence. BMJ Case Rep. 2014, 10, bcr2013201618. [Google Scholar] [CrossRef]
- Glass, L.M.; Dickson, R.C.; Anderson, J.C.; Suriawinata, A.A.; Putra, J.; Berk, B.S.; Toor, A. Total body weight loss of ≥10% is associated with improved hepatic fibrosis in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Anstee, Q.M.; Trauner, M.; Lawitz, E.J.; Abdelmalek, M.F.; Ding, D.; Han, L.; Jia, C.; Huss, R.S.; Chung, C.; et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology 2022, 75, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Chen, S.Y.; You, H. Regression of liver fibrosis: Evidence and challenges. Chin. Med. J. 2020, 133, 1696–1702. [Google Scholar] [CrossRef]
- Hartl, J.; Ehlken, H.; Sebode, M.; Peiseler, M.; Krech, T.; Zenouzi, R.; von Felden, J.; Weiler-Normann, C.; Schramm, C.; Lohse, A.W. Usefulness of biochemical remission and transient elastography in monitoring disease course in autoimmune hepatitis. J. Hepatol. 2018, 68, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Morandeau, E.; Anderson, G.J.; Ramm, G.A.; Ramm, L.E.; Morcet, J.; Bouzille, G.; Dixon, J.; Clouston, A.D.; Lainé, F.; et al. Regression of Fibrosis Stage with Treatment Reduces Long-Term Risk of Liver Cancer in Patients with Hemochromatosis Caused by Mutation in HFE. Clin. Gastroenterol. Hepatol. 2020, 18, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Sayaf, K.; Zanotto, I.; Gabbia, D.; Alberti, D.; Pasqual, G.; Zaramella, A.; Fantin, A.; De Martin, S.; Russo, F.P. Sex Drives Functional Changes in the Progression and Regression of Liver Fibrosis. Int. J. Mol. Sci. 2023, 24, 16452. [Google Scholar] [CrossRef] [PubMed]
- Calvente, C.J.; Tameda, M.; Johnson, C.D.; Del Pilar, H.; Lin, Y.C.; Adronikou, N.; De Mollerat Du Jeu, X.; Llorente, C.; Boyer, J.; Feldstein, A.E. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J. Clin. Investig. 2019, 129, 4091–4109. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Syed, S.; Kehar, S.I. Glial Fibrillary Acidic Protein (GFAP) as a Mesenchymal marker of Early Hepatic Stellate Cells Activation in Liver Fibrosis in Chronic Hepatitis C Infection. Pak. J. Med. Sci. 2014, 30, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Xie, X.L.; Wang, M.M.; Yin, J.; Tian, J.-M.; Jiang, X.-Y.; Zhang, D.; Han, J.; Bai, Y.; Cui, Z.-J.; et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Oben, J.A.; Vinciguerra, M. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: A putative synergy between retinoic acid and PPAR-gamma signalings. Clin. Exp. Med. 2017, 17, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ki-Hyun, K.; Chih-Chiun, C.; Monzon, R.; Lau, L.F. Matricellular Protein CCN1 Promotes Regression of Liver Fibrosis through Induction of Cellular Senescence in Hepatic Myofibroblasts. Mol. Cell. Biol. 2013, 33, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Cen, Y.; Lu, D.; Luo, W.; Jiang, H. IL-22 inactivates hepatic stellate cells via downregulation of the TGF-β1/Notch signaling pathway. Mol. Med. Rep. 2018, 17, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Pacher, M.; Balakrishnan, A.; Yuan, Q.; Tsay, H.C.; Yang, D.; Reetz, J.; Brandes, S.; Dai, Z.; Pützer, B.M.; et al. Direct Reprogramming of Hepatic Myofibroblasts into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell 2016, 18, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Kamiya, A.; Sumiyoshi, H.; Tsuruya, K.; Kagawa, T.; Inagaki, Y. A Deactivation Factor of Fibrogenic Hepatic Stellate Cells Induces Regression of Liver Fibrosis in Mice. Hepatology 2020, 71, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. Gastroenterol. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P.; Leibovich, S.; et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Hepatic Stellate Cell Behavior during Resolution of Liver Injury. Semin. Liver Dis. 2001, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Verma, V.K.; Morton, L.A.; Shah, V.H.; LaRusso, N.F.; Gores, G.J.; Malhi, H. Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology 2016, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Österreicher, C.; Kluwe, J.; Osawa, Y.; A Brenner, D.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jiang, Z.; Ruan, B.; Duan, J.; Song, P.; Liu, J.; Han, H.; Wang, L. Disruption of myofibroblastic Notch signaling attenuates liver fibrosis by modulating fibrosis progression and regression. Int. J. Biol. Sci. 2021, 17, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Mabire, M.; Hegde, P.; Hammoutene, A.; Wan, J.; Caër, C.; Al Sayegh, R.; Cadoux, M.; Allaire, M.; Weiss, E.; Thibault-Sogorb, T.; et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat. Commun. 2023, 14, 1830. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.A.M.; Masroor, A.; Khorochkov, A.; Prieto, J.; Singh, K.B.; Nnadozie, M.C.; Abdal, M.; Shrestha, N.; Mohammed, L. The Role of Vitamins in Non-Alcoholic Fatty Liver Disease: A Systematic Review. Cureus 2021, 13, e16855. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency promote fatty liver disease development? Nutrients 2014, 6, 5473–5499. [Google Scholar] [CrossRef] [PubMed]
- Smith-Cortinez, N.; Fagundes, R.R.; Gomez, V.; Kong, D.; de Waart, D.R.; Heegsma, J.; Sydor, S.; Olinga, P.; de Meijer, V.E.; Taylor, C.T.; et al. Collagen release by human hepatic stellate cells requires vitamin C and is efficiently blocked by hydroxylase inhibition. FASEB J. 2021, 35, e21219. [Google Scholar] [CrossRef] [PubMed]
- Licata, A.; Zerbo, M.; Como, S.; Cammilleri, M.; Soresi, M.; Montalto, G.; Giannitrapani, L. The Role of Vitamin Deficiency in Liver Disease: To Supplement or Not Supplement? Nutrients 2021, 13, 4014. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, Y.; Wang, D.; Huang, Z.; Xiao, X.; Zheng, Q.; Li, S.; Long, D.; Feng, L. Mitochondrial Dysfunction in Metabolic Dysfunction Fatty Liver Disease (MAFLD). Int. J. Mol. Sci. 2023, 24, 17514. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Q.; Li, H.X.; Tan, W.L.; Yang, L.; Ma, X.W.; Li, W.X.; Wang, Q.B.; Shang, C.Z.; Chen, Y.J. Association of Serum Vitamin C with NAFLD and MAFLD among Adults in the United States. Front. Nutr. 2022, 8, 795391. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Liu, J.; Liu, K. Association of nonalcoholic fatty liver disease and liver fibrosis detected by transient elastography with serum retinol in American adults. Front. Nutr. 2023, 10, 1094161. [Google Scholar] [CrossRef] [PubMed]
- Barbakadze, G.; Khachidze, T.; Sulaberidze, G.; Burnadze, K.; Jebashvili, M. Comparative Analysis of Efficiency of Ursodeoxycholic Acid and Combination of Vitamin E and Vitamin C in Treatment of Non-Diabetic Nonalcoholic Steatohepatitis. Georgian Med. News 2019, 288, 81–85. [Google Scholar]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, X.; Yang, H.; Wu, P.; Wang, S.; Cao, D.; Guo, X.; Xu, Z.; Gao, J.; Zhang, W.; et al. Effects of Oral Vitamin C Supplementation on Liver Health and Associated Parameters in Patients with Non-Alcoholic Fatty Liver Disease: A Randomized Clinical Trial. Front. Nutr. 2021, 8, 745609. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Vuppalanch, R.; Gawrich, S.; Gahbril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Vitamin E Improves Transplant-FreeSurvival and Hepatic Decompensation among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology 2020, 71, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Maboud, M.; Menshawy, A.; Menshawy, E.; Emara, A.; Alshandidy, M.; Eid, M. The efficacy of vitamin E in reducing non-alcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Therap. Adv. Gastroenterol. 2020, 13, 1756284820974917. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Braghini, M.R.; Crudele, A.; Smeriglio, A.; Bianchi, M.; Condorelli, A.G.; Nobili, R.; Conti, L.A.; De Stefanis, C.; Lioci, G.; et al. Combination Treatment with Hydroxytyrosol and Vitamin E Improves NAFLD-Related Fibrosis. Nutrients 2022, 14, 3791. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Fouladvand, F.; Falahi, E.; Asbaghi, O.; Abbasnezhad, A. Effect of Vitamins C and E Co-Supplementation on Serum C-Reactive Protein Level: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Prev. Nutr. Food Sci. 2020, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. Antioxidant supplementation enhances antioxidant capacity and mitigates oxidative damage following acute ischaemic stroke. Eur. J. Clin. Nutr. 2005, 59, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, W.; He, Z.; Yang, H.; Gao, J.; Wu, P.; Ma, Z.F. Dietary Vitamin C Intake Is Associated with Improved Liver Function and Glucose Metabolism in Chinese Adults. Front. Nutr. 2022, 31, 779912. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Sakai, Y.; Terashima, T.; Shimode, T.; Seki, A.; Orita, N.; Takeshita, Y.; Shimakami, T.; Takatori, H.; Arai, K.; et al. Decline in serum albumin concentration is a predictor of serious events in nonalcoholic fatty liver disease. Medicine 2021, 100, e26835. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.R.; Verdelho Machado, M. New Insights About Albumin and Liver Disease. Ann. Hepatol. 2018, 17, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Ionele, C.M.; Subtirelu, M.S.; Ungureanu, B.S.; Serbanescu, M.S.; Rogoveanu, I. Calcium and Phosphorus Deficiencies in Patients with Liver Cirrhosis. Curr. Health Sci. J. 2022, 48, 311–316. [Google Scholar] [CrossRef]
- Nayila, I. Effect of Ascorbic Acid Supplementation on Liver Function Tests in Hepatitis C Patients. Open J. Intern. Med. 2020, 10, 263–279. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 25, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Kousparou, C.; Fyrilla, M.; Stephanou, A.; Patrikios, I. DHA/EPA (Omega-3) and LA/GLA (Omega-6) as Bioactive Molecules in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 10717. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.; Rosqvist, F.; Ahlström, H.; Niessen, H.G.; Schultheis, C.; Hockings, P.; Hulthe, J.; Gummesson, A.; Wanders, A.; Rorsman, F.; et al. Hepatic Unsaturated Fatty Acids Are Linked to Lower Degree of Fibrosis in Non-alcoholic Fatty Liver Disease. Front. Med. 2022, 10, 814951. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Wu, X.L.; Chen, R.P.; Chen, C.; Liu, X.G.; Wu, B.J.; Huang, Z.M. Effectiveness of Omega-3 Polyunsaturated Fatty Acids in Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 6, e0162368. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shao, C.; Zhang, W.; Wang, G.; Lu, D.C.; Han, W.; Wu, Z.S.; Chen, C.B. Omega-3 polyunsaturated fatty acids prevent progression of liver fibrosis and promote liver regeneration after partial hepatectomy in cirrhotic rats. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10151–10160. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, Y.; Zhu, Y.; Gao, Y.; Chen, H.; Zhang, Y.; Yin, W.; Li, Y.; Wang, K.; Xiao, J. Protective effect of the ω-3 polyunsaturated fatty acids on the schistosomiasis liver fibrosis in mice. Int. J. Clin. Exp. Med. 2015, 15, 9470–9476. [Google Scholar]
- Zhang, K.; Chang, Y.; Shi, Z.; Han, X.; Han, Y.; Yao, Q.; Hu, Z.; Cui, H.; Zheng, L.; Han, T.; et al. ω-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci. Rep. 2016, 6, 30029. [Google Scholar] [CrossRef] [PubMed]
- Vell, M.S.; Creasy, K.T.; Scorletti, E.; Seeling, K.S.; Hehl, L.; Rendel, M.D.; Schneider, K.M.; Schneider, C.V. Omega-3 intake is associated with liver disease protection. Front. Public Health 2023, 11, 1192099. [Google Scholar] [CrossRef] [PubMed]
- Cansanção, K.; Citelli, M.; Carvalho Leite, N.; López de Las Hazas, M.C.; Dávalos, A.; Tavares do Carmo, M.D.G.; Peres, W.A.F. Impact of Long-Term Supplementation with Fish Oil in Individuals with Non-Alcoholic Fatty Liver Disease: A Double Blind Randomized Placebo Controlled Clinical Trial. Nutrients 2020, 12, 3372. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yang, L.H.; Sha, K.H.; Liu, T.G.; Zhang, L.G.; Liu, X.X. Efficacy of poly-unsaturated fatty acid therapy on patients with nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 7008–7013. [Google Scholar] [CrossRef] [PubMed]
- Padiadpu, J.; Garcia-Jaramillo, M.; Newman, N.K.; Pederson, J.W.; Rodrigues, R.; Li, Z.; Singh, S.; Monnier, P.; Trinchieri, G.; Brown, K.; et al. Multi-omic network analysis identified betacellulin as a novel target of omega-3 fatty acid attenuation of western diet-induced nonalcoholic steatohepatitis. EMBO Mol. Med. 2023, 15, e18367. [Google Scholar] [CrossRef] [PubMed]
- Argo, C.K.; Patrie, J.T.; Lackner, C.; Henry, T.D.; de Lange, E.E.; Weltman, A.L.; Shah, N.L.; Al-Osaimi, A.M.; Pramoonjago, P.; Jayakumar, S.; et al. Effects of n-3 fish oil on metabolic and histological parameters in NASH: A double-blind, randomized, placebo-controlled trial. J. Hepatol. 2015, 62, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D.; WELCOME Study. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: Results from the Welcome* study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M.; EPE-A Study Group. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 2014, 147, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Johra, F.T.; Bepari, A.K.; Bristy, A.T.; Reza, H.M. A Mechanistic Review of β-Carotene, Lutein, and Zeaxanthin in Eye Health and Disease. Antioxidants 2020, 9, 1046. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshani, A. Insights of hypercarotenaemia: A brief review. Clin. Nutr. ESPEN 2017, 23, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Sahin, K.; Bilen, H.; Bahcecioglu, I.H.; Bilir, B.; Ashraf, S.; Halazun, K.J.; Kucuk, O. Carotenoids and non-alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Vitaglione, P.; Morisco, F.; Caporaso, N.; Fogliano, V. Dietary antioxidant compounds and liver health. Crit. Rev. Food Sci. Nutr. 2004, 44, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Mohan, L.; Bharadvaja, N. Disease Prevention and Treatment Using β-Carotene: The Ultimate Provitamin A. Rev. Bras. Farmacogn. 2022, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Maiani, G.; Castón, M.J.; Catasta, G.; Toti, E.; Cambrodón, I.G.; Bysted, A.; Granado-Lorencio, F.; Olmedilla-Alonso, B.; Knuthsen, P.; Valoti, M.; et al. Carotenoids: Actual knowledge on food sources, intakes, stability and bioavailability and their protective role in humans. Mol. Nutr. Food Res. 2009, 53, S194–S218. [Google Scholar] [CrossRef] [PubMed]
- Villaça Chaves, G.; Pereira, S.E.; Saboya, C.J.; Ramalho, A. Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity. Obes. Surg. 2008, 18, 378–385. [Google Scholar] [CrossRef]
- Martin, K.R.; Failla, M.L.; Smith, J.C., Jr. Beta-carotene and lutein protect HepG2 human liver cells against oxidant-induced damage. J. Nutr. 1996, 126, 2098–20106. [Google Scholar] [CrossRef] [PubMed]
- Seifert, W.F.; Bosma, A.; Hendriks, H.F.; van Leeuwen, R.E.; van Thiel-de Ruiter, G.C.; Seifert-Bock, I.; Knook, D.L.; Brouwer, A. Beta-carotene (provitamin A) decreases the severity of CCl4-induced hepatic inflammation and fibrosis in rats. Liver 1995, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wardi, J.; Reifen, R.; Aeed, H.; Zadel, L.; Avni, Y.; Bruck, R. Beta-carotene attenuates experimentally induced liver cirrhosis in rats. Isr. Med. Assoc. J. 2001, 3, 151–154. [Google Scholar] [PubMed]
- El-Baz, F.K.; Salama, A.; Ali, S.I.; Elgohary, R. Haematococcus pluvialis carotenoids enrich fractions ameliorate liver fibrosis induced by thioacetamide in rats: Modulation of metalloproteinase and its inhibitor. BioMed Res. Int. 2021, 2021, 6631415. [Google Scholar] [CrossRef] [PubMed]
- El-Baz, F.K.; Salama, A.A.A.; Hussein, R.A. Dunaliella salina microalgae oppose thioacetamide-induced hepatic fibrosis in rats. Toxicol. Rep. 2019, 10, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, F.; Gul, M.; Ates, B.; Ozturk, I.C.; Cetin, A.; Vardi, N.; Otlu, A.; Yilmaz, I. Protective effect of apricot (Prunus armeniaca L.) on hepatic steatosis and damage induced by carbon tetrachloride in Wistar rats. Br. J. Nutr. 2009, 102, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bengmark, S.; Qu, S. Nutrigenomics therapy of hepatisis C virus induced-hepatosteatosis. BMC Gastroenterol. 2010, 10, 49. [Google Scholar] [CrossRef]
- Harari, A.; Harats, D.; Marko, D.; Cohen, H.; Barshack, I.; Kamari, Y.; Gonen, A.; Gerber, Y.; Ben-Amotz, A.; Shaish, A. A 9-cis beta-carotene-enriched diet inhibits atherogenesis and fatty liver formation in LDL receptor knockout mice. J. Nutr. 2008, 138, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Xing, F.; Huo, J.; Fung, M.; Liong, E.C.; Ching, Y.P.; Xu, A.; Chang, R.C.; So, K.F.; Tipoe, G.L. L. Lycium barbarum polysaccharides therapeutically improve hepatic functions in non-alcoholic steatohepatitis rats and cellular steatosis model. Sci Rep. 2014, 4, 5587. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, C.; Vásquez, B.; Souza-Mello, V.; Adeli, K.; Mandarim-de-Lacerda, C.; Del Sol, M. Morphoquantitative effects of oral β-carotene supplementation on liver of C57BL/6 mice exposed to ethanol consumption. Int. J. Clin. Exp. Pathol. 2019, 12, 1713–1722. [Google Scholar] [PubMed]
- Gopal, K.; Gowtham, M.; Sachin, S.; Ravishankar Ram, M.; Shankar, E.M.; Kamarul, T. Attrition of Hepatic Damage Inflicted by Angiotensin II with α-Tocopherol and β-Carotene in Experimental Apolipoprotein E Knock-out Mice. Sci. Rep. 2015, 5, 18300. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, H.; Chen, M.; Shao, J. Clinical Relevance of Vitamins and Carotenoids with Liver Steatosis and Fibrosis Detected by Transient Elastography in Adults. Front. Nutr. 2021, 8, 760985. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.M.; Cansanção, K.; Perez, R.M.; Leite, N.C.; Padilha, P.; Ramalho, A.; Peres, W. Association between serum and dietary antioxidant micronutrients and advanced liver fibrosis in non-alcoholic fatty liver disease: An observational study. PeerJ. 2020, 8, e9838. [Google Scholar] [CrossRef] [PubMed]
- Kataria, Y.; Deaton, R.J.; Enk, E.; Jin, M.; Petrauskaite, M.; Dong, L.; Goldenberg, J.R.; Cotler, S.J.; Jensen, D.M.; van Breemen, R.B.; et al. Retinoid and carotenoid status in serum and liver among patients at high-risk for liver cancer. BMC Gastroenterol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Khan, U.M.; Sevindik, M.; Zarrabi, A.; Nami, M.; Ozdemir, B.; Kaplan, D.N.; Selamoglu, Z.; Hasan, M.; Kumar, M.; Alshehri, M.M.; et al. Lycopene: Food Sources, Biological Activities, and Human Health Benefits. Oxid. Med. Cell Longev. 2021, 2021, 2713511. [Google Scholar] [CrossRef] [PubMed]
- Nishida, P.; Berg, B.; Shakersain, K.; Hecht, A.; Takikawa, R.; Tao, Y.; Kakuta, C.; Uragami, H.; Hashimoto, N.; Misawa, N.; et al. Astaxanthin: Past, Present, and Future. Mar. Drugs 2023, 21, 514. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, X.; Liu, M.; Zhao, H.; Sun, Y. Lycopene prevents non-alcoholic fatty liver disease through regulating hepatic NF-κB/NLRP3 inflammasome pathway and intestinal microbiota in mice fed with high-fat and high-fructose diet. Front. Nutr. 2023, 10, 1120254. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, Y.; Yu, T.T.; Hao, W.; Wang, G. Lycopene improves autophagy and attenuates carbon tetrachloride-induced hepatic fibrosis in rats. Croat. Med. J. 2023, 64, 243–255. [Google Scholar] [CrossRef]
- Ni, Y.; Zhuge, F.; Nagashimada, M.; Nagata, N.; Xu, L.; Yamamoto, S.; Fuke, N.; Ushida, Y.; Suganuma, H.; Kaneko, S.; et al. Lycopene prevents the progression of lipotoxicity-induced nonalcoholic steatohepatitis by decreasing oxidative stress in mice. Free Radic. Biol. Med. 2020, 152, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Albrahim, T.; Alonazi, M.A. A. Lycopene corrects metabolic syndrome and liver injury induced by high fat diet in obese rats through antioxidant, anti-inflammatory, antifibrotic pathways. Biomed. Pharmacother. 2021, 141, 111831. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Y. Astaxanthin prevents TGFβ1-induced pro-fibrogenic gene expression by inhibiting Smad3 activation in hepatic stellate cells. Biochim. Biophys. Acta 2015, 1850, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Lee, Y.; Pham, T.X.; Hu, S.; Park, Y.K.; Lee, J.Y. Y. Astaxanthin inhibits the reduction of glycolysis during the activation of hepatic stellate cells. Life Sci. 2020, 256, 117926. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Al Mamun, M.A.; Faruk, M.; Ul Islam, M.T.; Rahman, M.M.; Alam, M.N.; Rahman, A.F.M.T.; Reza, H.M.; Alam, M.A. Astaxanthin Ameliorates Hepatic Damage and Oxidative Stress in Carbon Tetrachloride-administered Rats. Pharmacogn. Res. 2017, 9 (Suppl. S1), S84–S91. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.A.; Gescher, A.J.; Steward, W.P. Curcumin: The story so far. Eur. J. Cancer 2005, 41, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar] [CrossRef] [PubMed]
- Li, S. Chemical Composition and Product Quality Control of Turmeric (Curcuma longa L.). Pharm. Crops 2011, 5, 28–54. [Google Scholar] [CrossRef]
- Mirzaei, H.; Shakeri, A.; Rashidi, B.; Jalili, A.; Banikazemi, Z.; Sahebkar, A. Phytosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed. Pharmacother. 2017, 85, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T. Curcumin as a functional food-derived factor: Degradation products, metabolites, bioactivity, and future perspectives. Food Funct. 2018, 9, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Shahab-Uddin, A.A.; Khan, U.; Abdul, H.; Mohiuddin, E.; Asif, M. Curcuma longa and curcumin: A review article. Rom. J. Biol. Plant Biol. 2010, 55, 65–70. [Google Scholar]
- Akter, J.; Hossain, M.A.; Takara, K.; Islam, M.Z.; Hou, D.X. Antioxidant activity of different species and varieties of turmeric (Curcuma spp.): Isolation of active compounds. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 215, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Dosoky, N.S.; Setzer, W.N. Chemical Composition and Biological Activities of Essential Oils of Curcuma Species. Nutrients 2018, 10, 1196. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600. [Google Scholar] [CrossRef] [PubMed]
- Farzaei, M.H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F.F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S.M.; Rahimi, R.; Abdollahi, M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients 2018, 10, 855. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Vaishnani, D.K.; Mansi; Zeng, J.; Xie, Z.; Jin, X.; Zhang, H.; Wut, Y.; Hla, K.; Ying, F. Novel Curcumin Analogue L6H4 in Treating Liver Fibrosis and Type 2 Diabetes. Diabetes Metab. Syndr. Obes. 2023, 16, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Park, Y.K.; Lee, J.Y. Food components with antifibrotic activity and implications in prevention of liver disease. J. Nutr. Biochem. 2018, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Qin, J.; Zhen, X.; Yang, Q.; Huang, L. Curcumin protects against hepatic stellate cells activation and migration by inhibiting the CXCL12/CXCR4 biological axis in liver fibrosis: A study in vitro and in vivo. Biomed. Pharmacother. 2018, 101, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Chen, L.; Kong, D.; Zhang, X.; Lu, C.; Lu, Y.; Zheng, S. Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J. Cell Mol. Med. 2014, 18, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; He, Y.; Ye, G.; Liu, X.; Huang, J.; Zhang, Q.; Tian, D.; Wang, T.; Shu, J. Curcumin inhibits the activity and induces apoptosis of activated hepatic stellate cell by suppressing autophagy. J. Cell Biochem. 2023, 124, 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Huang, R.; Xiong, Y.L.; Wu, C. Protective effects of curcumin against liver fibrosis through modulating DNA methylation. Chin. J. Nat. Med. 2016, 14, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-T.; Wang, N.; Tan, H.Y.; Li, S.; Feng, Y. Targeting Hepatic Stellate Cells for the Treatment of Liver Fibrosis by Natural Products: Is It the Dawning of a New Era? Front. Pharmacol. 2020, 11, 548. [Google Scholar] [CrossRef] [PubMed]

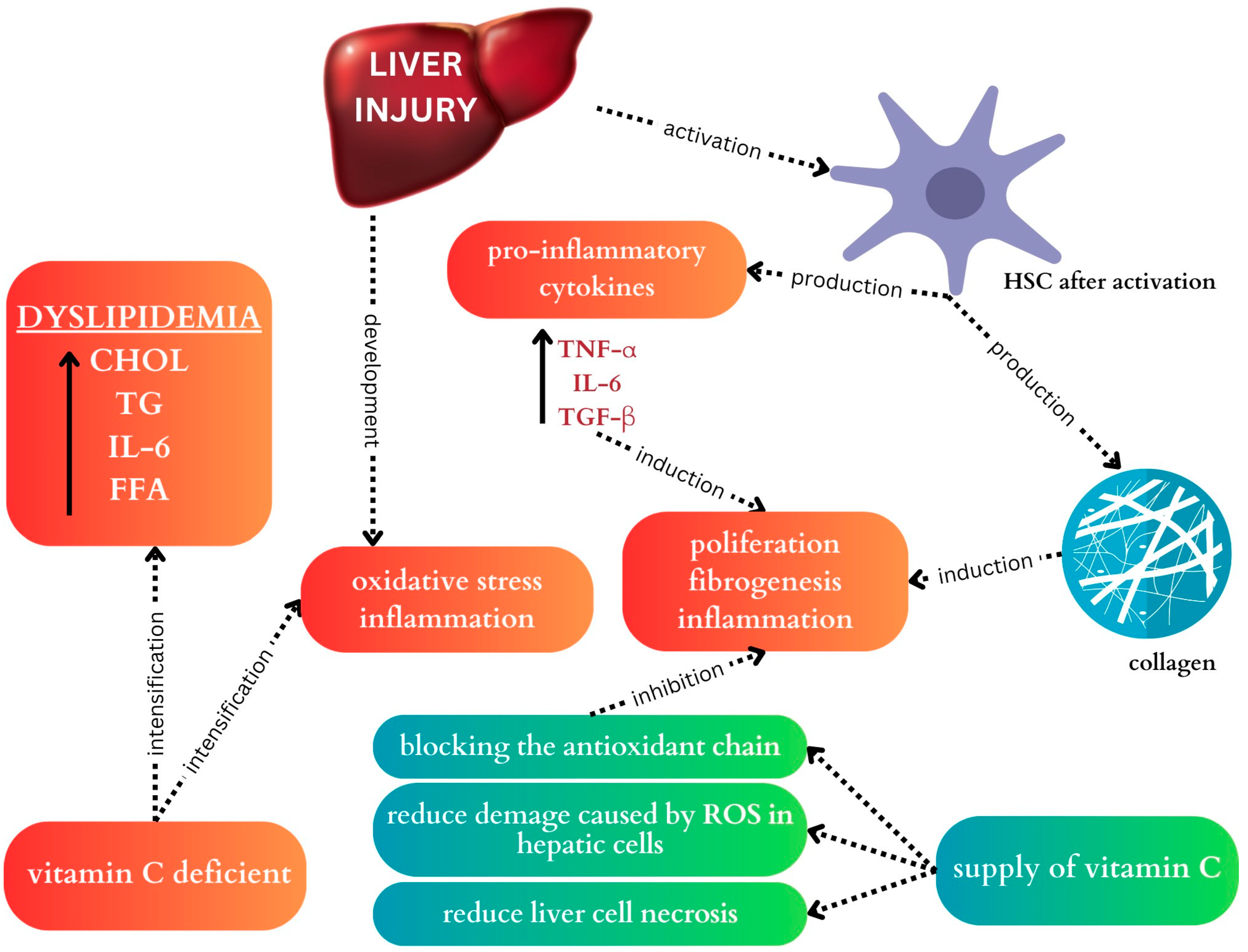

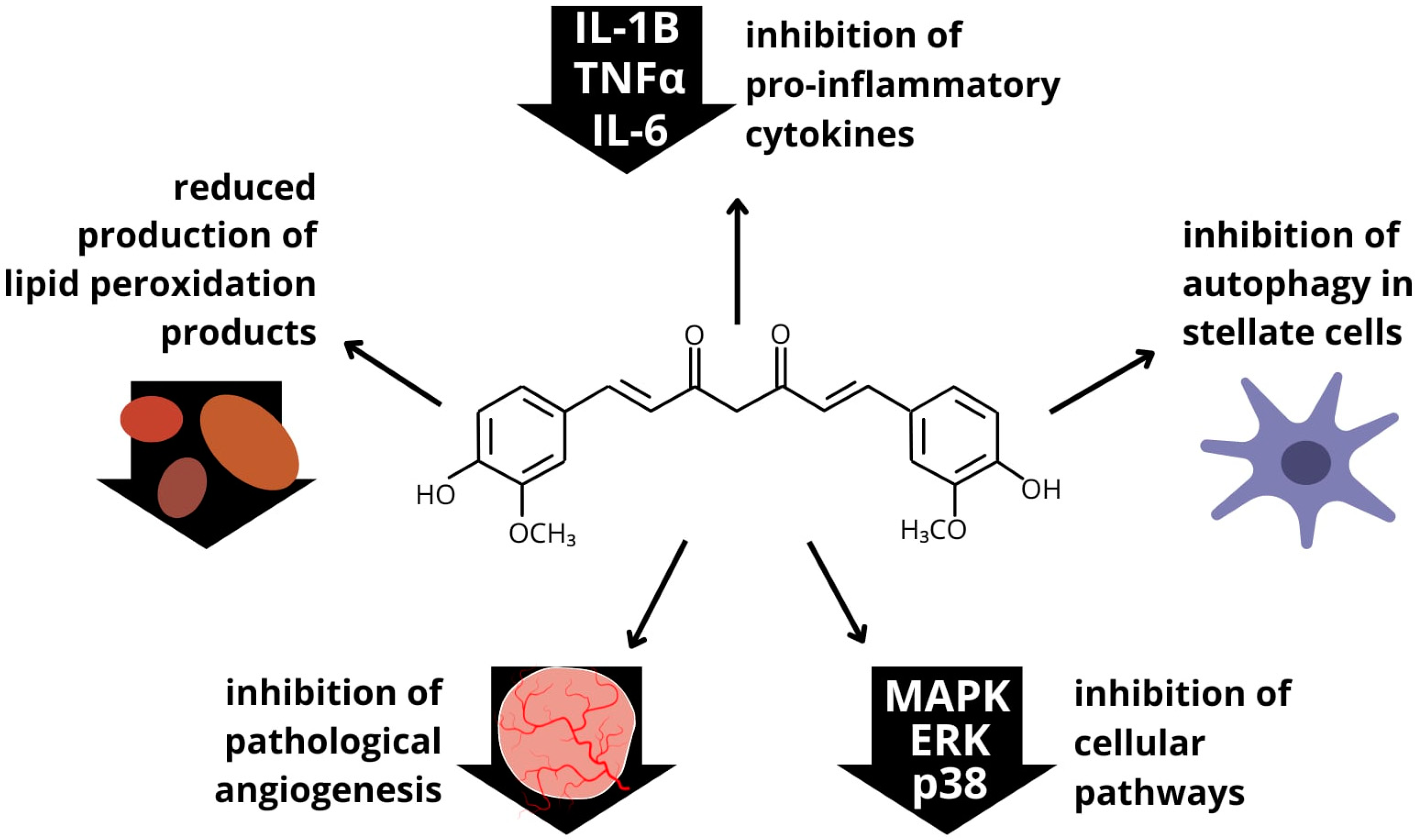
| Population | Intervention Duration | Methods | Results | Reference |
|---|---|---|---|---|
| Patients diagnosed with MASH n = 107 Randomly assigned to 3 groups: Study group n = 35 n = 52 Control group n = 20 | 12 months | Patients were randomly assigned to receive ursodeoxycholic acid (UDCA) 15 mg/kg/day (group A) or vitamin E 800 mg/day plus vitamin C 500 mg/day (group B) and a control group that did not receive any treatment. | After 12 months of treatment with vitamins E and C, compared to UDCA, a significant reduction in the average level of alanine aminotransferase (ALT) was observed. There was a reduction in both the mean steatosis score and fibrosis score. | Barbakadze et al. 2019 [100]. |
| Patients diagnosed with MASH n = 49 Randomly assigned to 2 groups: Study group n = 23 Control group n = 22 | 6 months | Patients randomized to receive vitamins E and C (1000 IU and 1000 mg, respectively) or placebo daily. | Significant improvement in fibrosis was noted within the group that received vitamins E and C but not in the placebo group. No statistically significant difference in fibrosis was noted between the vitamin and placebo groups. There were no changes in the ALT concentration in the study group and no differences in the AST value between the groups. The evaluation of the histologic data demonstrated no statistically significant differences in inflammation/necrosis score. | Harrisona et al. 2003 [101] |
| Patients diagnosed with MASLD n = 24 Randomly assigned to 3 groups with different vitamin C intakes: Study group n = 26 n = 30 n = 28 | 12 weeks | Patients treated with low (250 mg/day, n = 26) or medium (1000 mg/day, n = 30) or high (2000 mg/day, n = 28) doses of oral vitamin C supplements. | In the medium supply group, a more significant decrease in the concentration of AST and ALP was observed compared to the high supply group. There was no statistically significant difference in ALT or AST between the low- and high-dose vitamin C groups. Liver health indicators such as gamma-glutamyltransferase, alkaline phosphatase, total bilirubin, direct bilirubin, and glucose metabolism parameters such as fasting insulin, fasting glucose, and homeostasis model assessment for insulin resistance decreased after the intervention but were comparable in the three groups. | He et al. 2021 [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokal-Dembowska, A.; Jarmakiewicz-Czaja, S.; Ferenc, K.; Filip, R. Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis? Int. J. Mol. Sci. 2024, 25, 5238. https://doi.org/10.3390/ijms25105238
Sokal-Dembowska A, Jarmakiewicz-Czaja S, Ferenc K, Filip R. Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis? International Journal of Molecular Sciences. 2024; 25(10):5238. https://doi.org/10.3390/ijms25105238
Chicago/Turabian StyleSokal-Dembowska, Aneta, Sara Jarmakiewicz-Czaja, Katarzyna Ferenc, and Rafał Filip. 2024. "Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis?" International Journal of Molecular Sciences 25, no. 10: 5238. https://doi.org/10.3390/ijms25105238
APA StyleSokal-Dembowska, A., Jarmakiewicz-Czaja, S., Ferenc, K., & Filip, R. (2024). Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis? International Journal of Molecular Sciences, 25(10), 5238. https://doi.org/10.3390/ijms25105238