Abstract
UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) is a zinc amidase that catalyzes the second step of the biosynthesis of lipid A, which is an outer membrane essential structural component of Gram-negative bacteria. Inhibitors of this enzyme can be attributed to two main categories, non-hydroxamate and hydroxamate inhibitors, with the latter being the most effective given the chelation of Zn2+ in the active site. Compounds containing diacetylene or acetylene tails and the sulfonic head, as well as oxazoline derivatives of hydroxamic acids, are among the LpxC inhibitors with the most profound antibacterial activity. The present article describes the synthesis of novel functional derivatives of hydroxamic acids—bioisosteric to oxazoline inhibitors—containing 1,2,4- and 1,3,4-oxadiazole cores and studies of their cytotoxicity, antibacterial activity, and antibiotic potentiation. Some of the hydroxamic acids we obtained (9c, 9d, 23a, 23c, 30b, 36) showed significant potentiation in nalidixic acid, rifampicin, and kanamycin against the growth of laboratory-strain Escherichia coli MG1655. Two lead compounds (9c, 9d) significantly reduced Pseudomonas aeruginosa ATCC 27853 growth in the presence of nalidixic acid and rifampicin.
1. Introduction
Human infections caused by pathogenic bacteria kill millions of people each year, many of which are caused by diarrheal diseases, bacteria-driven tuberculosis, and lower respiratory infections [1,2,3]. Severe sepsis caused by bacterial infections affects up to 18 million people worldwide each year, with mortality rates ranging from 28% to 50% [1,4]. Bacterial infections are a major health problem in the world, and the treatment of these infectious diseases is becoming increasingly difficult given the development of antibiotic resistance [5,6,7,8,9,10]. Back in 1945, Alexander Fleming, who discovered penicillin, warned that bacteria could become resistant to antibiotics in the future. The development of resistance to antibiotics is a natural evolutionary process for microorganisms. The development of antibiotic resistance is aggravated by the uncontrolled use of antibiotics in medicine and agriculture [6]. Resistant strains can multiply and spread where infection prevention and control measures are not followed. They are also found in the environment (water, soil, and air).
Policies recommended by the World Health Organization (WHO) include research on new antibiotics, but no new class of antibiotics has been discovered since daptomycin and linezolid in the 1980s, and only optimizations or combinations of already known compounds have been recently commercialized [7]. Widespread resistance to available antibiotics in clinically important pathogenic bacteria is currently a global challenge because of an ever-increasing number of strains that are resistant to multiple classes of antibiotics. This means that progress in modern medicine, which relies on the availability of effective antibacterial drugs, is at risk. Thus, the need for the development of new antibiotics, delivery systems, and bacterial diagnostics is urgent, as well as the use of “antibiotic adjuvants/potentiators” in combination with antibiotics [11].
The majority of pathogenic microorganisms that can cause various serious diseases are Gram-negative bacteria; in particular, multidrug-resistant Gram-negative bacteria (MDRGNs) have become a major threat to hospitalized patients, with mortality rates ranging from 30% to 70% [12]. For example, E. coli is the leading cause of urinary tract infections, P. aeruginosa causes hospital-acquired pneumonia and bloodstream infections, N. gonorrhoeae is the source of gonorrhea, and K. pneumoniae causes urinary tract infections and pneumonia [1,13]. In addition, it is alarming that multidrug resistance is also common among the deadliest and most human-pathogenic bacterial species, such as the plague pathogen, Y. pestis, which have been isolated to different parts of the world (for example, Madagascar and Mongolia) [14].
Lipopolysaccharides (LPSs) are bacterial surface glycolipids produced by Gram-negative bacteria that are present in their outer membranes. An LPS is composed of three domains: lipid A, the core oligosaccharide, and the O antigen. The main function of LPSs is structural, as they act as barriers against agents toxic to the bacteria, for instance, antibiotics [15]. Lipid A, which is the hydrophobic anchor of lipopolysaccharides (LPSs), is an attractive target for the development of both new antibiotics and potentiators of action of already known antibiotics since bacteria lacking lipid A are generally unviable, while mutants with reduced biosynthesis of lipid A have slow growth and high sensitivity to a wide range of antibiotics [1].
A Zn-dependent metalloamidase, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC), is considered to be a promising target for the commitment step involved in catalyzing lipid A biosynthesis [16]. Deacetylase LpxC is one of the most studied enzymes involved in the biosynthesis of lipid A. Inhibitors of this enzyme can potentially be both antibiotics and potentiators of action of already known antibiotics given the key role of LpxC in the biosynthesis of lipid A and its lack of homology with mammalian proteins. Also, it is worth noting that, at the present time, inhibitors of enzyme LpxC in a variety of Gram-negative microorganisms have been described in the literature and have shown promise in the treatment of diseases caused by corresponding bacterial infections (P. aeruginosa, E. coli, N. gonorrhoeae, K. pneumoniae, B. pseudomallei, B. pertussis).
Most currently known LpxC deacetylase inhibitors fall into two main categories, namely, hydroxamate inhibitors and non-hydroxamate inhibitors. The mechanism of pharmacological action of hydroxamate inhibitors is the chelation of Zn2+ ions in the active site of the enzyme. Existing hydroxamate inhibitors can mainly be subdivided into those containing a butadiyne tail, those containing an acetylene tail, those containing a sulfone head, and other types of hydroxamate inhibitors [1]. The main difficulty encountered in the development of this class of LpxC deacetylase inhibitors is the toxicity of hydroxamic acids and their metabolic breakdown intermediates [17]. Studies devoted to the development of potential inhibitors of enzyme LpxC with a non-hydroxamate structure have also been published in the periodical literature [18,19,20,21]. As shown in some of them [22,23], the conversion of hydroxamic acid into carboxylic acid leads to a loss of enzyme LpxC inhibition and, as a consequence, antibacterial activity in the target compounds. When summarizing the data, the antibacterial activity of LpxC inhibitors with a non-hydroxamate structure is most often not as pronounced as compared with similar activity in hydroxamic acids [1].
Another class of LpxC hydroxamate inhibitor, oxazoline inhibitors (L-161,240, L-159,692, and L-159,463 [24]), can be noted, which are some of the first compounds with high antibacterial activity [16,24,25] (Figure 1). It was found that L-161,240 and its derivatives are potent inhibitors of E. coli LpxC but have no effect on the growth and in vitro activity of P. aeruginosa [16]. It was shown in investigations of the antibacterial activity of this chemotype that replacing the oxygen atom with a sulfur atom in the oxazoline ring, as well as increasing the size of the heterocycle, leads to a loss of in vitro antibacterial activity against P. aeruginosa in the target compounds. However, the introduction of a trifluoromethoxy substituent into the meta- or para-position of the benzene ring, as well as the introduction of halogens into the para-position, results in micromolar concentrations of half-maximal P. aeruginosa growth inhibition (0.5–6.0 μM). In the para-fluorinated analogs of L-161,240, the addition of an alkyl or alkoxy group to the meta-position also has a beneficial effect on effectiveness [25].
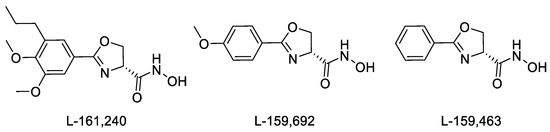
Figure 1.
Structure of inhibitors of E. coli LpxC.
The oxazoline LpxC inhibitors mentioned above (Figure 1) contain a hydroxamic acid functional group, which plays a key role in coordinating zinc ions in the active site of the enzyme, as well as a phenyl ring, which forms extensive interactions with hydrophobic substrate channels. The nitrogen atom of the oxazoline ring also forms interactions with the active site of the enzyme [26]. As shown previously [25], replacing the oxygen atom with another heteroatom in the oxazoline ring and increasing the size of the heterocycle lead to a loss of antibacterial activity in the inhibitors. However, in published studies devoted to oxazoline inhibitors, there are no examples of replacing the oxazoline heterocycle with another one of similar size containing nitrogen and oxygen atoms, the presence of which is considered to be mandatory. For example, oxazoline could be replaced with an oxadiazole ring, which has already established itself as a privileged heterocycle in medicinal chemistry with a wide range of pharmacological activity [27,28,29,30,31,32], including antibacterial activity [33,34,35]. We hypothesized that such a bioisosteric replacement of an oxazoline core with 1,2,4- and 1,3,4-oxadiazole would produce hydroxamic acids with a similar or improved antibacterial activity profile, particularly given the presence of an additional nitrogen atom as a hydrogen bond acceptor in the heterocyclic fragment, which may contribute to the better binding of target molecules at the enzyme site. On the other hand, replacing a non-aromatic heterocycle with an aromatic one leads to the formation of an additional planar fragment conjugated to the phenyl ring. This, in turn, greatly simplifies the synthesis and isolation of individual target hydroxamic acids, but it can also complicate the chelation of zinc ions in the active site of the enzyme because of the resulting conjugation of the heterocycle and the hydroxamate function. Therefore, we decided to synthesize hydroxamate derivatives linked to the heterocycle via an aliphatic linker in order to increase the mobility of the acid function and also obtain hydroxamic acids that are conjugated with the heterocycle directly or are linked to the heterocycle via an aromatic linker to establish further structure–activity relationships.
2. Results and Discussion
2.1. Synthesis of Hydroxamic Acids Containing a 1,2,4-Oxadiazole Nucleus
The synthesis of hydroxamic acids 4a–f was carried out in three stages from commercially available benzonitriles (1a–f) (Figure 2). The following benzonitriles were selected: 4-trifluoromethoxybenzonitrile, 1b; 3,4,5-trifluorobenzonitrile, 1c; 3-methylbenzonitrile, 1d; 4-iodobenzonitrile, 1f, all of which, according to data in the literature, make it possible to obtain target products containing an aromatic substituent that has a positive effect on potential antibacterial activity, which has been shown in the case of oxazolines—that is, inhibitors of LpxC [25]. Additionally, 2,6-difluorobenzonitrile, 1a, and 4-methoxybenzonitrile, 1e, were chosen as the starting benzonitriles. The nucleophilic addition of hydroxylamine to the starting benzonitriles, 1a–f, afforded amidoximes 2a–f, which were then converted into corresponding 1,2,4-oxadiazoles, 3a–f, via heterocyclization with ethyl oxalyl chloride in good yields. The subsequent interaction of esters 3a–f with excess hydroxylamine in the presence of diisopropylethylamine (DIPEA) led to the formation of the target hydroxamic acids, 4a–f (Figure 2).
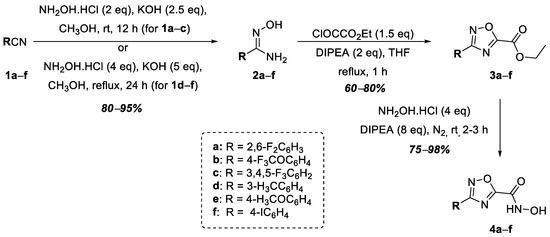
Figure 2.
Synthesis of hydroxamic acids 4a–f.
At the next stage, hydroxamic acids 9a–d were synthesized, in which the acid functional group was connected to the 1,2,4-oxadiazole heterocycle at position 5 through a methylene linker (Figure 3). At the first stage, malonic acid monomethyl ester, 6, was obtained from Meldrum’s acid, 5, according to a known procedure [36]. Then, compound 6 was activated with N,N′-carbonyldiimidazole (CDI), after which, amidoximes 2a–e were acylated to form O-acylamidoximes 7a–e in good yields. In this case, fluorine-containing 2a–c and 3-alkyl-substituted 2d were chosen as the starting amidoximes. The heterocyclization of compounds 7a–d into the target 1,2,4-oxadiazoles, 8a–d, was carried out by refluxing in THF in the presence of tetrabutylammonium fluoride (TBAF) according to a known procedure [37]. The subsequent interaction of esters 8a–d with excess hydroxylamine in the presence of DIPEA led to the formation of the target hydroxamic acids, 9a–d, in good yields (Figure 3).
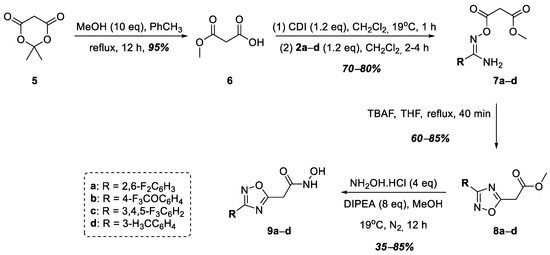
Figure 3.
Synthesis of hydroxamic acids 9a–d.
In order to study the effect of the substituent of the hydroxamic acid functional group on the antibacterial activity of the target compounds, we decided to synthesize a series of hydroxamic acids in which the hydroxamic acid functional group is directly conjugated to the benzene ring. Thus, at the next stage, a series of hydroxamic acids, 14a–e, was obtained, in which the hydroxamic acid functional group connected to the 1,2,4-oxadiazole heterocycle at position 5 through a phenylene linker (Figure 4). Monomethyl esters of terephthalic and isophthalic acids, 10a,b, were used as starting compounds. O-Acylamidoximes 11a–e were obtained in good yields from acids 10a,b similar to O-acylamidoximes 7a–d using amidoximes 2a–c and N-hydroxybenzimidamide. The cyclization of compounds 11a–e in the presence of TBAF led to the formation of 1,2,4-oxadiazoles 12a–e, which were further hydrolyzed to the corresponding 3- and 4-substituted benzoic acids—13a–c and 13d–e. Using this synthetic scheme, we decided to obtain the target hydroxamic acids from carboxylic acids given the lack of reaction between the corresponding esters, 12a–e, and a large excess of hydroxylamine. Thus, the target hydroxamic acids, 14a–e, were obtained from benzoic acids 13a–e in two stages: the formation of the corresponding acyl chlorides via the action of SOCl2 in CH2Cl2, followed by a reaction with hydroxylamine hydrochloride in the presence of Na2CO3, according to a modified known procedure [38] (Figure 4).
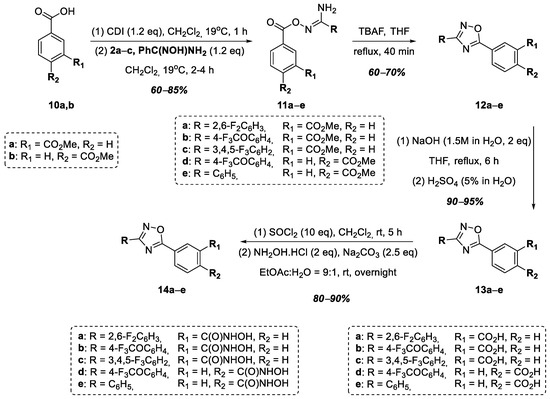
Figure 4.
Synthesis of hydroxamic acids 14a–e.
A series of hydroxamic acids containing a 1,2,4-oxadiazole ring linked at position 5 of the heterocycle through methylene and phenylene linkers was then completed with compounds 17a–d containing an ethylene linker linking the heterocycle nucleus to the hydroxamic acid functional group (Figure 5). Thus, at the first stage of synthesis, aromatic amidoximes 2a–d entered into a cyclocondensation reaction with succinic anhydride neatly, according to a known procedure [39]. The resulting carboxylic acids, 15a–d, were converted into methyl esters 16a–d in good yields. At the final stage of synthesis, esters 16a–d were refluxed in methanol for 8–10 h with a large excess of hydroxylamine in the presence of DIPEA to obtain hydroxamic acids 17a–d (Figure 5).
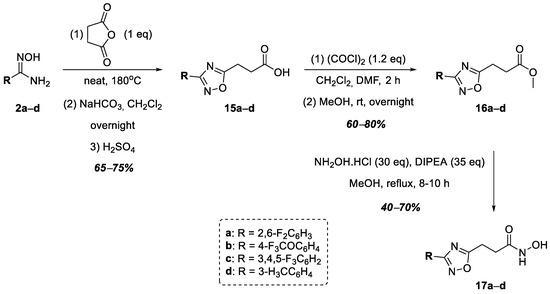
Figure 5.
Synthesis of hydroxamic acids 17a–d.
The next stage of this work was the synthesis of hydroxamic acids containing a 1,2,4-oxadiazole nucleus linked to the hydroxamic acid functional group by a methylene group at position 3 of the heterocycle (Figure 6). Thus, at the first stage, intermediate 19 was obtained via the nucleophilic addition of hydroxylamine to ethyl cyanoacetate, 18, which was then used in the synthesis without purification. Halogen-substituted benzoic acids 20a–d were activated by CDI followed by reactions with intermediate 19, which led to the formation of a series of O-acylamidoximes, 21a–d, in good yields. The target 1,2,4-oxadiazoles, 22a–e, were obtained via the heterocyclization of O-acylamidoximes 21a–e by refluxing their solution in toluene. Compounds 22a–d were converted into hydroxamic acids 23a–d in good yields under refluxing conditions using a solution of esters in methanol with an excess of hydroxylamine in the presence of DIPEA (Figure 6).
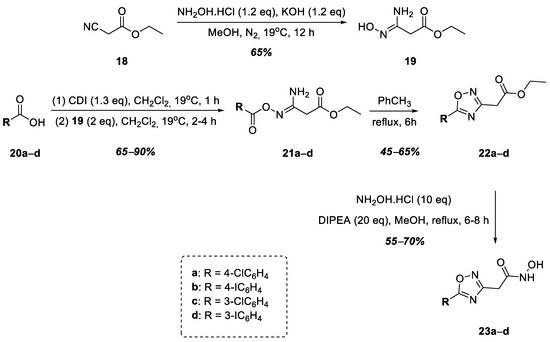
Figure 6.
Synthesis of hydroxamic acids 23a–d.
A commercially available ester, 24, was used as the starting compound for the synthesis of hydroxamic acid containing a 1,2,4-oxadiazole nucleus linked to the hydroxamic acid functional group by a phenylene group at position 3 of the heterocycle (Figure 7). Ester 24 was hydrolyzed to ataluren, 25, which was further converted to hydroxamic acid 26 in good yield via the sequential action of SOCl2 in CH2Cl2 and hydroxylamine in the presence of Na2CO3 (Figure 7).
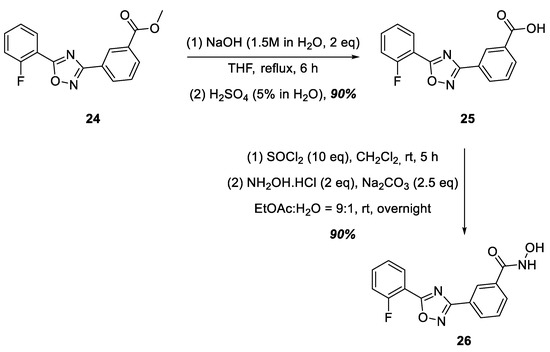
Figure 7.
Synthesis of hydroxamic acid 26.
2.2. Synthesis of Hydroxamic Acids Containing a 1,3,4-Oxadiazole Nucleus
The next stage of this work was the replacement of the 1,2,4-oxadiazole nucleus with the 1,3,4-oxadiazole nucleus in the target hydroxamic acids containing the functional group of hydroxamic acid at position 2 of the heterocycle (Figure 8). In this case, the choice fell on hydroxy- and halogen-substituted benzoic acids based on the structures of the known LpxC inhibitors mentioned earlier. The synthesis began with commercially available benzoic acids, 20b,d,e–h, which were converted into esters in the case of compounds 20b,d,e, and in the case of compounds 20f–h, the hydroxyl groups were at the same time methylated to form compounds 27a–f in good yields. Further, a series of hydrazides, 28a–f, without isolation or purification were obtained from esters 27a–f, after which, compounds 28a–f were converted into 1,3,4-oxadiazoles 29a–f, according to a modified known procedure [40]. The target hydroxamic acids, 30a–f, were obtained in good yields via the reaction of excess hydroxylamine hydrochloride with esters 29a–f in the presence of DIPEA (Figure 8).
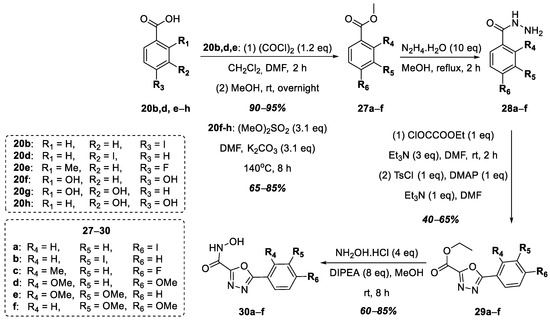
Figure 8.
Synthesis of hydroxamic acids 30a–f.
To increase the lipophilicity of the aromatic substituent at position 5 of the 1,3,4-oxadiazole ring, we decided to synthesize hydroxamic acid 36, structurally similar to acids 30a–f, containing an indole nucleus at position 5 of the heterocycle (Figure 9). Thus, 1H-indole-2-carboxylic acid, 31, was successively converted into its methyl ester, 32, and then into methyl 1-methyl-1H-indole-2-carboxylate, 33. Next, hydroxamic acid 36 was prepared similarly to the synthetic route shown in Figure 8: the formation of hydrazide 34 from methyl ester 33, followed by heterocyclization into 1,3,4-oxadiazole 35 and a reaction with hydroxylamine hydrochloride in the presence of DIPEA.
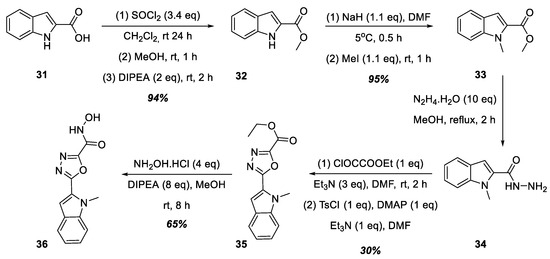
Figure 9.
Synthesis of hydroxamic acid 36.
2.3. In Vitro Studies of Potentiation of Antimicrobial Drugs via LpxC inhibitors
Preliminary screening of the synthesized substances was carried out on the laboratory strain of E. coli MG1655 since deacetylase LpxC has a high degree of homology in key amino acid residues of the active center of the enzyme among a wide range of Gram-negative pathogens [13]. The results of testing substances demonstrating inhibitory activity in the presence of nalidixic acid (NaI), rifampicin (Rif), and kanamycin (Km) are presented in Figure 10 and Figure 11. The greatest potentiating activity at low doses of nalidixic acid (17.22 μM) was demonstrated by substances 9d and 9c (Figure 10). Substances 23a, 23c, 36, and 30b inhibited bacterial growth less effectively (Figure 10).
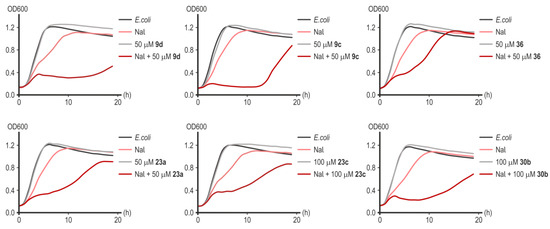
Figure 10.
Representative growth curves of E. coli MG1655 in the presence of nalidixic acid (17.22 μM), without or with inhibitors of LpxC in concentrations of 50–100 µM. Data points are averages of optical density at a wavelength of 600 nm (OD600) with a <5% margin of error.
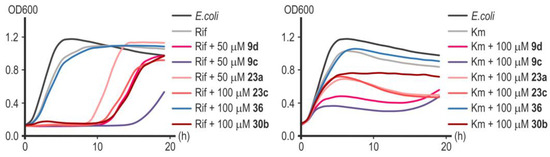
Figure 11.
Representative growth curves of E. coli MG1655 in the presence of rifampicin (6.08 μM) and kanamycin (0.62 μM) with or without inhibitors of LpxC in concentrations of 50 or 100 µM. Data points are averages of optical density at a wavelength of 600 nm (OD600), with a <5% margin of error.
Without antibiotics, all the target compounds synthesized were generally nontoxic to E. coli at all concentrations tested.
Substance 9c showed the greatest effectiveness in potentiating the action of rifampicin and kanamycin (Figure 11). Remarkably, substances 23a and 23c, which turned out to be less effective in combination with nalidixic acid (Figure 10), significantly increased the sensitivity of E. coli cells to rifampicin and kanamycin (Figure 11). Compound 30b significantly increased the sensitivity of cells to rifampicin but not to kanamycin (Figure 11). Rifampicin is not the antibiotic of choice for the treatment of infections caused by Gram-negative microflora, but increasing its effectiveness is potentially useful in preventing the development of opportunistic infections during anti-tuberculosis therapy [41]. Compound 36 was completely ineffective in combination with rifampicin or kanamycin (Figure 11). In the studied concentration range (50–100 µM), none of the substances d exhibited a toxic effect on cell growth in the absence of an antibiotic (Figure 10). However, despite the powerful bacteriostatic effect of compounds 9c and 9d in the first 10 h of incubation, we subsequently observed a resumption of culture growth, which can be explained by the metabolic transformation of the compound or by an occurrence of adaptive mutations.
Based on the results obtained for E. coli, two lead compounds were selected for their potentiation evaluation.
Substances 9c and 9d significantly reduced P. aeruginosa ATCC 27853 growth and lowered the MICs of the antibiotics tested when used together in combination with those products. The MIC value for nalidixic acid in the presence of 100 μM of 9c or 100 μM of 9d decreased approximately eight-fold from 17.22 μM to 2.15 μM (Figure 12). The combination of 50 μM of substance 9c to 2.15 μM of nalidixic acid completely inhibited the growth of P. aeruginosa within 14 h, while the addition of 50 μM of substance 9d to the same amount of nalidixic acid suppressed the growth of the culture only within 8 h (Figure 12). However, the addition of both substances at this concentration reduced the MIC of nalidixic acid by four times. Compound 9c at a concentration of 25 μM reduced the MIC of nalidixic acid by only two times. Compound 9d was ineffective at this concentration.
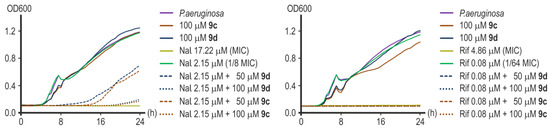
Figure 12.
Representative growth curves of P. aeruginosa ATCC 27853 in the presence of nalidixic acid (left) and rifampicin (right) with or without 9c or 9d.
Great results were obtained when investigating the effectiveness of rifampicin potentiation. Compounds 9c and 9d were equally effective in reducing the MIC of rifampicin by 64 times from 4.86 μM to 0.08 μM at both concentrations of 100 μM and 50 μM (Figure 12). The addition of 25 μM of substance 9c led to a twofold decrease in the MIC value of rifampicin, and 25 μM of compound 9d was inactive.
Without antibiotics, both compounds were generally nontoxic to P. aeruginosa at all concentrations tested.
When comparing the microbiological results obtained in this work and those obtained for the oxazoline inhibitors shown in Figure 1, several points should be noted. Firstly, oxazoline inhibitors had standalone antibacterial activity against the E. coli cell line [24], while the compounds we synthesized did not have such activity. Secondly, in a previous work, the LpxC inhibition of oxazoline derivatives was carried out against the purified P. aeruginosa enzyme [25], while in the present work, the data on the inhibition of the growth of P. aeruginosa are presented. There are no data about the potentiating activity of oxazoline inhibitors [24,25] in the presence of antibiotics; at the same time, our work shows that oxadiazole derivatives exhibit a synergistic effect, significantly enhancing the effectiveness of antibacterial drugs. Thus, our results show a new chemotype that can be effectively utilized for potentiating the action of known antibiotics against Gram-negative bacteria resistant to these antibiotics.
In general, the results obtained during our studies of the antibiotic-potentiating activities of the target hydroxamic acids correlate with the structure–activity relationships observed for oxazoline inhibitors [25]. The presence of an sp3-hybrid carbon atom linked with the hydroxamic acid functional group appears to be extremely important, and it likely facilitates the chelation of zinc ions at the intended target. The phenylene linker between the hydroxamate function and the heterocycle core not only appears to hinder the chelation of zinc ions in the active site of LpxC but also significantly increases the size of the target molecules, which may affect their binding to the enzyme in general. The preferred positions for introducing substituents into the phenyl moiety conjugated with the heterocycle are meta- and/or para-positions. Preference is given to small substituents that do not extend outside the plane of the aromatic ring—electron-withdrawing groups (-F, -Cl) or electron-donating groups (-CH3).
2.4. Cytotoxicity Assay
The results of the cytotoxicity assay of the target hydroxamic acids against the HEK293 cell line are presented in Table 1. From the presented data regarding the cytotoxicity of the target hydroxamic acids, the following patterns can be noted. In general, hydroxamic acids directly conjugated to a 1,2,4- or 1,3,4-oxadiazole nucleus (compounds 4a–f, 30a–f) have high CC50 values, except compound 30a (CC50 = 62.19 μM), which is noteworthy since its regioisomer, 4f, has a CC50 value of 367 μM. The replacement of the phenyl substituent in the 1,3,4-oxadiazole ring with a 1-methylindole nucleus leads to the increased toxicity of hydroxamic acid 36. The introduction of a methylene linker into hydroxamic acids containing a 1,2,4-oxadiazole ring at position 5 of the heterocycle leads to an increase in the toxicity of the target compounds by 2.5–3.5 times (compounds 4a–d and 9a–d, respectively). An increase in the number of methylene groups in the linker leads to a significant decrease in the CC50 values of hydroxamic acids 17a–d (CC50 values ranged from 21.30 to 59.22 μM) compared with compounds 9a–d (CC50 values ranged from 108.10 to 188.60 μM). At the same time, the introduction of a phenylene linker between the hydroxamic acid functional group and the 1,2,4-oxadiazole core leads to a significant increase in the toxicity of the target compounds (compounds 4a–c and 14a–c, respectively). It is worth noting that the position of the hydroxamic acid functional group in the phenyl substituent does not have a particular effect on the cytotoxicity of the target compounds (compounds 14b (CC50 = 6.39 μM) and 14d (CC50 = 5.28 μM)). Hydroxamic acids 23a–d containing a methylene group between the functional group of hydroxamic acid and position 3 of the 1,2,4-oxadiazole nucleus have similar cytotoxicity indices (CC50 values ranging from 112.50 to 171.10 μM) as acids 9a–d. Replacing the methylene linker with a phenylene one in this case leads to compound 26, which, like other examples of N-hydroxybenzamides in this work, is highly toxic (CC50 = 21.74 μM).

Table 1.
Cytotoxicity of target hydroxamic acids synthesized 1.
2.5. Computer Modeling of Active Compounds
2.5.1. Docking of Active Compounds to LpxC Structure
All six active chemical compounds were docked by the MOE 2019 program to the product UDP-(3-O-(R-3-hydroxymyristoyl))-glucosamine binding site of the LpxC structure (4mdt) (Figure 13). The docking experiments showed that the locations of the 9c, 9d, 23a, 23c, 30b, and 36 ligand binding sites are the same as for the 4mdt ligand UDP-(3-O-(R-3-hydroxymyristoyl))-glucosamine (Figure 13, blue surface) and that they include mostly hydrophobic residue: Leu18, Thr191, Phe192, Ile198, Gly210, and Ala215.
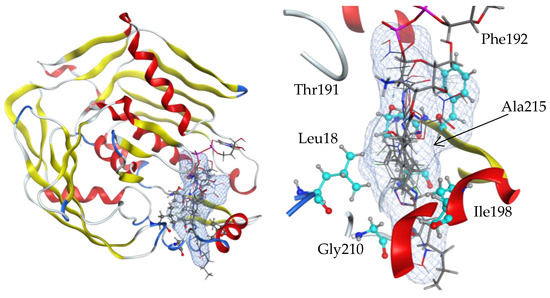
Figure 13.
Side views of ligand-binding pockets in the structure of an LpxC protein (4mdt). Locations of 9c, 9d, 23a, 23c, 30b, and 36 ligand binding sites are the same as for the 4mdt ligand UDP-(3-O-(R-3-hydroxymyristoyl))-glucosamine (blue surface), and they include mostly hydrophobic residues: Leu18, Thr191, Phe192, Ile198, Gly210, and Ala215. Ligand interaction figures with the amino acid residues of the binding sites are given in Supplementary Materials Figures S1–S6. Full lists of amino acids involved in the interaction are given in Supplementary Materials Table S1.
2.5.2. Pharmacophore Models of Active Compounds
Chemical compounds were pre-aligned with the flexible alignment function of the MOE 2019 program to compose the pharmacophore model. The pharmacophore centers were selected via the Consensus option according to the maximum density of atoms in the superposition. A simple model of four pharmacophore centers finds all compounds synthesized, both active and inactive (Figure 14). Since the structure of the active compounds found does not differ significantly from inactive compounds and the differences between the two active compounds are often greater than between the active and inactive compounds, a selective pharmacophore model could not be constructed.
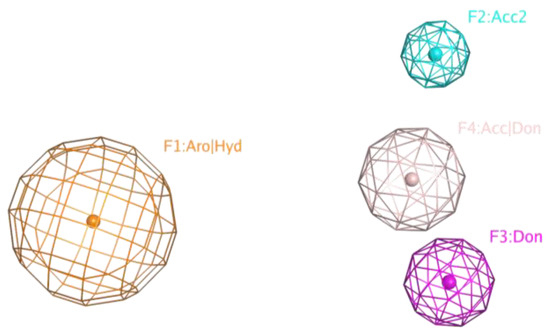
Figure 14.
A simple pharmacophore model of four pharmacophore centers finds all compounds synthesized, both active and inactive.
2.5.3. QSAR Models of Active Compounds
All compounds synthesized were marked in the MOE 2019 database as active (1) or inactive (0). In this case, the term activity was understood as the combined action together with the antibiotic. As mentioned above, the structure of the found active compounds differed from inactive compounds by one atom. We could not visually find a logical relationship between the active and inactive compounds, but this was possible using the QSAR method (Figure 15). Descriptors calculated from the structures of chemical compounds are often difficult to understand and logically analyze, but it is possible to build a highly efficient discriminative model from this application. It was found that the best-performing QSAR model is binary and includes 23 descriptors (Figure 15; Tables S2 and S3 in the Supplementary Materials).
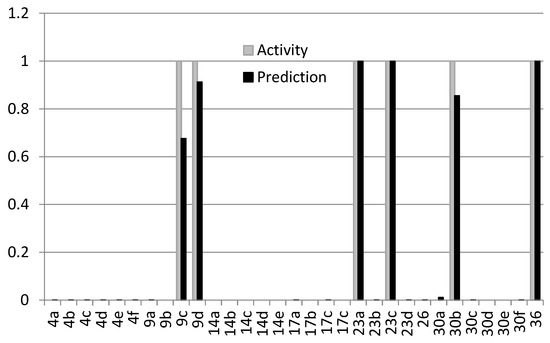
Figure 15.
Activity (gray) and prediction (black) comparison of the best-performing QSAR model.
The Binary Classification model includes, in particular, a number of aromatic atoms and a number of aromatic bonds; a number of double bonds; a number of rigid bonds; a number of rings; a number of chiral centers; a number of unconstrained chiral centers; relative negative partial charge; a polar surface area; and some less obvious descriptors. A full list of the descriptors used is in the Supplementary Materials (Table S2).
3. Materials and Methods
3.1. Materials
Mueller–Hinton agar (MHA) and Mueller–Hinton broth (MHB) were purchased from “HiMedia” (Mumbai, India); rifampicin (Rif) and nalidixic acid (Nal) were purchased from “Sigma-Aldrich” (St. Louis, MO, USA).
3.2. Cell Culture
The HEK293 cell line was obtained from the ECACC (European Collection of Authenticated Cell Cultures, cat. 85120602, Salisbury, UK). The cells were grown in monolayer culture in Dulbecco’s Modified Eagle Medium (DMEM) (PanEco, Moscow, Russia) supplemented with 2 mM of L-glutamine, 1% penicillin/streptomycin (Capricorn Scientific, Ebsdorfergrund, Germany), and 10% heat-inactivated fetal bovine serum (HyClone, Cytiva, Pasching, Austria) in a humidified atmosphere of 5% CO2 and at 37 °C (Binder, Tuttlingen, Germany).
Escherichia coli strain MG1655 and Pseudomonas aeruginosa strain ATCC 27853 were obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA). Subculturing and inoculum preparation were prepared according to the Clinical and Laboratory Standards Institute (CLSI) recommended method, M7-A9 [42], using MHA and MHB.
3.3. Generation of Growth Curves
Growth curves were obtained with a Bioscreen C automated growth analysis system. Cultures of E. coli MG1655 were grown overnight at 37 °C, diluted 1:100 in fresh medium, inoculated into honeycomb wells in triplicate, and grown at 37 °C with maximum shaking using the platform of the Bioscreen C instrument. When the cultures reached an optical density (OD600) of 0.2, cells were treated with nalidixic acid (17.22 μM), rifampicin (6.08 μM), or kanamycin (0.62 μM) in the presence or absence of LpxC inhibitors at a concentration of 0.01–0.1 mM and incubated at 37 °C for 19 h. OD600 values were recorded automatically at specified times, and the means of the triplicate cultures were plotted.
3.4. Antibiotic Activity Evaluation
Minimum inhibitory concentrations (MICs) for rifampicin and nalidixic acid were determined via the modified broth microdilution method in triplicate according to CLSI guidelines M7-A9 and M100-A30 [43]. The antibiotics were serially diluted twofold in 100 μL of MHB. The bacteria inoculum was 100 μL of a 1.0 × 106 CFU/mL dilution in MHB. MICs for antibiotics in the presence of 9c or 9d were determined with the same method. Rifampicin or nalidixic acid ranged from 4·MIC to 1/128·MIC and included zero concentration for each antibiotic in combination with 9c or 9d (100 μM, 50 μM, and 25 μM) in an equal volume of 50 μL. 9c or 9d stock solutions were prepared in DMSO; the total DMSO concentration in a well did not exceed 1%.
MIC endpoints were read after 24 h of incubation with an iMark microplate absorbance reader (Bio-Rad, Hercules, CA, USA). Growth curves of P. aeruginosa ATCC 27853 in the presence of rifampicin/nalidixic acid and 9c/9d were obtained using a multifunction microplate reader, SuperMax 3100 (ShanghaiFlash, Shanghai, China). Cells were grown in triplicate at 37 °C with aeration.
3.5. Cytotoxicity Assay
For analysis, the HEK293 cells were seeded into a 96-well plate (Corning Incorporated, Corning, NY, USA) in 100 μL of complete culture medium at a final concentration of 1 × 104 cells/well and cultured for 24 h for proper attachment. Subsequently, the medium was removed, and cells were exposed to the test inhibitor with different concentrations for 72 h in 100 μL of fresh complete culture medium.
The sensitivity of the cells to test inhibitor molecules was detected using the vital dye Alamar Blue (Invitrogen, Carlsbad, CA, USA). The Alamar Blue assay was performed according to the procedure described by O’Brien et al. [44]. After incubation with test inhibitor molecules, 10 μL of Alamar Blue reagent was added to each well, cell culture plates were returned to a humidified incubator, and the fluorescence was read after 3 h. The plates were exposed to an excitation wavelength of 545 nm, and the emission at 600 nm was measured with the CLARIOstar Plus (BMG Labtech, Ortenberg, Germany, Software Version 5.70 R2). To determine corresponding 50% cellular cytotoxicity (CC50) values, a four-parameter sigmoidal fit curve model (GraphPad Prism 8 software) was used to measure the data.
4. Conclusions
In sum, a series of compounds were synthesized containing a hydroxamic acid functional group conjugated directly to 1,2,4-oxadiazole and 1,3,4-oxadiazole, as well as through a methylene, ethylene, or phenylene linker, at positions 3 and 5 of the heterocycle (in the case of 1,2,4-oxadiazole). It was shown that the target hydroxamic acids containing a phenylene linker and an ethylene linker demonstrated low CC50 values (<59.22 μM), while acids directly conjugated to the heterocycle had low cytotoxicity. Among the above-mentioned acids, only two had weak potentiating activity in the presence of nalidixic acid against laboratory-strain E. coli MG1655—compounds 30b and 36—containing a hydroxamic acid functional group directly conjugated with 1,3,4-oxadiazole. Additionally, compound 30b significantly increased the sensitivity of E. coli cells to rifampicin but not to kanamycin, while compound 36 was completely ineffective in combination with these antibiotics. Among the compounds containing a hydroxamic acid functional group linked by a methylene linker to a 1,2,4-oxadiazole core, the greatest potentiating activity in the presence of nalidixic acid, rifampicin, and kanamycin against E. coli cells was demonstrated by substances 9c and 9d. At the same time, substances 23a and 23c, which turned out to be less effective in combination with nalidixic acid, significantly increased the sensitivity of E. coli cells to rifampicin and kanamycin. Lead compounds 9c and 9d showed the best results in nalidixic acid and rifampicin action potentiation against P. aeruginosa ATCC 27853 growth. All six active chemical compounds may have two partially overlapping binding sites; both are involved in interactions with the reaction product, UDP-(3-O-(R-3-hydroxymyristoyl))-glucosamine. In the foreseeable future, expanding the library of target hydroxamic acids and a more detailed study of their potentiating effect in combination with antibiotics against the growth of Gram-negative bacterial cells are expected.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms25010096/s1, Experimental procedures for synthesis and characterization data of all target hydroxamic acids, as well as intermediates of their synthesis, copies of their 1H- and 13C-NMR spectra, and HRMS; Tables S1–S3.
Author Contributions
Conceptualization, A.A.Z., V.V.C., V.A.M. and R.A.I.; methodology, A.A.Z., V.V.C., A.Z.A., P.N.S. and V.A.M.; software, A.A.A. (docking, QSAR); supervision, V.V.C., A.Z.A., M.A.K., P.N.S. and V.A.M.; data curation, R.A.I.; investigation, A.A.Z., V.V.C. and A.I.C. (1,2,4-oxadiazoles), A.Z.A. and T.A.M. (1,3,4-oxadiazoles), T.A.S., S.V.R., E.A.M. and V.V.K. (bacterial assay), M.A.K., Y.E.I. and A.I.C. (cytotoxicity); writing—original draft preparation, A.A.Z., V.V.C., A.Z.A., T.A.S., S.V.R., M.A.K., P.N.S. and V.A.M.; writing—review and editing, A.A.Z., V.V.C., A.A.A., P.N.S. and V.A.M.; project administration, R.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement № 075-10-2021-113, unique project ID RF----193021X0001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The authors would like to express their gratitude to the Resource Centers for Analytical Methods and Medicinal Chemistry of Sirius University for the obtained spectra and analytical data and R.A. Novikov (Engelhardt Institute of Molecular Biology RAS) for conducting NMR experiments, S.A. Silonov (Institute of Cytology RAS, Sirius University) for assistance in calculations of CC50, and M.B. Nawrozkij (Sirius University) for motivation and valuable advice. We thank E.S. Mozhaytsev (NIOCH SB RAS) and S.A. Kuleshova (Sirius University) for fruitful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Niu, Z.; Lei, P.; Wang, Y.; Wang, J.; Yang, J.; Zhang, J. Small Molecule LpxC Inhibitors against Gram-Negative Bacteria: Advances and Future Perspectives. Eur. J. Med. Chem. 2023, 253, 115326. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, D.V.; Holl, R. LpxC Inhibitors: A Patent Review (2010–2016). Expert Opin. Ther. Pat. 2017, 27, 1227–1250. [Google Scholar] [CrossRef] [PubMed]
- Frackenpohl, J.; Schneider, L.; Decker, L.J.B.; Dittgen, J.; Fenkl, F.; Fischer, C.; Franke, J.; Freigang, J.; Getachew, R.; Gonzalez Fernandez-Nino, S.M.; et al. Identifying New Lead Structures to Enhance Tolerance towards Drought Stress via High-Throughput Screening Giving Crops a Quantum of Solace. Bioorg. Med. Chem. 2019, 27, 115142. [Google Scholar] [CrossRef] [PubMed]
- Perman, S.M.; Goyal, M.; Gaieski, D.F. Initial Emergency Department Diagnosis and Management of Adult Patients with Severe Sepsis and Septic Shock. Scand. J. Trauma Resusc. Emerg. Med. 2012, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Negash, K.H.; Norris, J.K.S.; Hodgkinson, J.T. Siderophore–Antibiotic Conjugate Design: New Drugs for Bad Bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef]
- Chernyshov, V.V.; Kuzovlev, A.S.; Cherepanova, N.D.; Kasatkina, M.A.; Ivanov, R.A. Siderophore-antibiotic Conjugates: Structural Diversity and Antibacterial Activity. Clin. Microbiol. Antimicrob. Chemother. 2022, 24, 314–344. [Google Scholar] [CrossRef]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic Discovery: History, Methods and Perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef]
- Baym, M.; Stone, L.K.; Kishony, R. Multidrug Evolutionary Strategies to Reverse Antibiotic Resistance. Science 2016, 351, aad3292. [Google Scholar] [CrossRef]
- Cal, P.M.S.D.; Matos, M.J.; Bernardes, G.J.L. Trends in Therapeutic Drug Conjugates for Bacterial Diseases: A Patent Review. Expert Opin. Ther. Pat. 2017, 27, 179–189. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Bashiri, S.; Yuan, Y.; Ziora, Z.M.; Nabil, O.; Masuda, K.; Khongkow, M.; Rimsueb, N.; Cabral, H.; Ruktanonchai, U.; et al. Antimicrobial Activity Enhancers: Towards Smart Delivery of Antimicrobial Agents. Antibiotics 2022, 11, 412. [Google Scholar] [CrossRef]
- Chawla, M.; Verma, J.; Gupta, R.; Das, B. Antibiotic Potentiators Against Multidrug-Resistant Bacteria: Discovery, Development, and Clinical Relevance. Front. Microbiol. 2022, 13, 887251. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef] [PubMed]
- Barb, A.W.; Jiang, L.; Raetz, C.R.H.; Zhou, P. Structure of the Deacetylase LpxC Bound to the Antibiotic CHIR-090: Time-Dependent Inhibition and Specificity in Ligand Binding. Proc. Natl. Acad. Sci. USA 2007, 104, 18433–18438. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, N.; Liang, X.; Najeeb, J.; Lee, C.-J.; Titecat, M.; Leteurtre, E.; Simonet, M.; Toone, E.J.; Zhou, P.; Sebbane, F. Curative Treatment of Severe Gram-Negative Bacterial Infections by a New Class of Antibiotics Targeting LpxC. MBio 2017, 8, e00674-17. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Tumey, L.N.; McClerren, A.L.; Raetz, C.R.H. High-Throughput Catch-and-Release Synthesis of Oxazoline Hydroxamates. Structure-Activity Relationships in Novel Inhibitors of Escherichia coli LpxC: In Vitro Enzyme Inhibition and Antibacterial Properties. J. Am. Chem. Soc. 2003, 125, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Jackman, J.E.; Fierke, C.A.; Tumey, L.N.; Pirrung, M.; Uchiyama, T.; Tahir, S.H.; Hindsgaul, O.; Raetz, C.R.H. Antibacterial Agents That Target Lipid A Biosynthesis in Gram-Negative Bacteria. Inhibition of Diverse UDP-3-O-(R-3-Hydroxymyristoyl)-N-Acetylglucosamine Deacetylases by Substrate Analogs Containing Zinc Binding Motifs. J. Biol. Chem. 2000, 275, 11002–11009. [Google Scholar] [CrossRef]
- Barb, A.W.; Leavy, T.M.; Robins, L.I.; Guan, Z.; Six, D.A.; Zhou, P.; Bertozzi, C.R.; Raetz, C.R.H. Uridine-Based Inhibitors as New Leads for Antibiotics Targeting Escherichia coli LpxC. Biochemistry 2009, 48, 3068–3077. [Google Scholar] [CrossRef]
- Furuya, T.; Shapiro, A.B.; Comita-Prevoir, J.; Kuenstner, E.J.; Zhang, J.; Ribe, S.D.; Chen, A.; Hines, D.; Moussa, S.H.; Carter, N.M.; et al. N-Hydroxyformamide LpxC Inhibitors, Their In Vivo Efficacy in a Mouse Escherichia coli Infection Model, and Their Safety in a Rat Hemodynamic Assay. Bioorg. Med. Chem. 2020, 28, 115826. [Google Scholar] [CrossRef]
- Shin, H.; Gennadios, H.A.; Whittington, D.A.; Christianson, D.W. Amphipathic Benzoic Acid Derivatives: Synthesis and Binding in the Hydrophobic Tunnel of the Zinc Deacetylase LpxC. Bioorg. Med. Chem. 2007, 15, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Tumey, L.N.; Raetz, C.R.H.; Jackman, J.E.; Snehalatha, K.; McClerren, A.L.; Fierke, C.A.; Gantt, S.L.; Rusche, K.M. Inhibition of the Antibacterial Target UDP-(3-O-Acyl)-N-Acetylglucosamine Deacetylase (LpxC): Isoxazoline Zinc Amidase Inhibitors Bearing Diverse Metal Binding Groups. J. Med. Chem. 2002, 45, 4359–4370. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Steiner, M.G.; de Laszlo, S.E.; Patchett, A.A.; Anderson, M.S.; Hyland, S.A.; Russell Onishi, H.; Silyer, L.L.; Raetz, C.R.H. Carbohydroxamido-Oxazolidines: Antibacterial Agents That Target Lipid A Biosynthesis. Bioorg. Med. Chem. Lett. 1999, 9, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.R.; Pelak, B.A.; Gerckens, L.S.; Silver, L.L.; Kahan, F.M.; Chen, M.-H.; Patchett, A.A.; Galloway, S.M.; Hyland, S.A.; Anderson, M.S.; et al. Antibacterial Agents That Inhibit Lipid A Biosynthesis. Science 1996, 274, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Kline, T.; Andersen, N.H.; Harwood, E.A.; Bowman, J.; Malanda, A.; Endsley, S.; Erwin, A.L.; Doyle, M.; Fong, S.; Harris, A.L.; et al. Potent, Novel in Vitro Inhibitors of the Pseudomonas aeruginosa Deacetylase LpxC. J. Med. Chem. 2002, 45, 3112–3129. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-J.; Liang, X.; Gopalaswamy, R.; Najeeb, J.; Ark, E.D.; Toone, E.J.; Zhou, P. Structural Basis of the Promiscuous Inhibitor Susceptibility of Escherichia coli LpxC. ACS Chem. Biol. 2014, 9, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Parikh, P.H.; Timaniya, J.B.; Patel, M.J.; Patel, K.P. Design, Synthesis, and Characterization of Novel Substituted 1,2,4-Oxadiazole and Their Biological Broadcast. Med. Chem. Res. 2020, 29, 538–548. [Google Scholar] [CrossRef]
- Karabanovich, G.; Zemanová, J.; Smutný, T.; Székely, R.; Šarkan, M.; Centárová, I.; Vocat, A.; Pávková, I.; Čonka, P.; Němeček, J.; et al. Development of 3,5-Dinitrobenzylsulfanyl-1,3,4-Oxadiazoles and Thiadiazoles as Selective Antitubercular Agents Active Against Replicating and Nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 2362–2380. [Google Scholar] [CrossRef]
- Chernyshov, V.V.; Yarovaya, O.I.; Esaulkova, I.L.; Sinegubova, E.; Borisevich, S.S.; Popadyuk, I.I.; Zarubaev, V.V.; Salakhutdinov, N.F. Novel O-Acylated Amidoximes and Substituted 1,2,4-Oxadiazoles Synthesised from (+)-Ketopinic Acid Possessing Potent Virus-Inhibiting Activity against Phylogenetically Distinct Influenza A Viruses. Bioorg. Med. Chem. Lett. 2022, 55, 128465. [Google Scholar] [CrossRef]
- Gonda, T.; Bérdi, P.; Zupkó, I.; Fülöp, F.; Szakonyi, Z. Stereoselective Synthesis, Synthetic and Pharmacological Application of Monoterpene-Based 1,2,4- and 1,3,4-Oxadiazoles. Int. J. Mol. Sci. 2017, 19, 81. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Jain, P.; Jain, P.; Tiwari, N.; Jain, R.; Jain, R.; Jain, K.A.; Agrawal, K.R. Synthesis and Biological Activities of Oxadiazole Derivatives: A Review. Mini-Rev. Med. Chem. 2016, 16, 825–845. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.V.; Sen’kova, A.V.; Popadyuk, I.I.; Salomatina, O.V.; Logashenko, E.B.; Komarova, N.I.; Ilyina, A.A.; Salakhutdinov, N.F.; Zenkova, M.A. Novel 3′-Substituted-1′,2′,4′-Oxadiazole Derivatives of 18βh-Glycyrrhetinic Acid and Their O-Acylated Amidoximes: Synthesis and Evaluation of Antitumor and Anti-Inflammatory Potential In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 3511. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin; Mazumder, A.; Yar, M.S.; Mazumder, R.; Chakraborthy, G.S.; Ahsan, M.J.; Rahman, M.U. Updates on Synthesis and Biological Activities of 1,3,4-Oxadiazole: A Review. Synth. Commun. 2017, 47, 1805–1847. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Chudasma, S.J.; Patel, T.M.; Dave, B.P. A Review on Synthetic Account of 1,2,4-Oxadiazoles as Anti-Infective Agents. Mol. Divers. 2022, 26, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Hendawy, O.M. A Comprehensive Review of Recent Advances in the Biological Activities of 1,2,4-Oxadiazoles. Arch. Pharm. 2022, 355, 2200045. [Google Scholar] [CrossRef]
- Kumar, N.; Ghosh, S.; Bhunia, S.; Bisai, A. Synthesis of 2-Oxindoles via “transition-Metal-Free” Intramolecular Dehydrogenative Coupling (IDC) of sp2 C-H and sp3 C-H Bonds. Beilstein J. Org. Chem. 2016, 12, 1153–1169. [Google Scholar] [CrossRef]
- Gangloff, A.R.; Litvak, J.; Shelton, E.J.; Sperandio, D.; Wang, V.R.; Rice, K.D. Synthesis of 3,5-Disubstituted-1,2,4-Oxadiazoles Using Tetrabutylammonium Fluoride as a Mild and Efficient Catalyst. Tetrahedron Lett. 2001, 42, 1441–1443. [Google Scholar] [CrossRef]
- Yamada, T.; Shibata, Y.; Kawauchi, S.; Yoshizaki, S.; Tanaka, K. Formal Lossen Rearrangement/[3 + 2] Annulation Cascade Catalyzed by a Modified Cyclopentadienyl RhIII Complex. Chem. Eur. J. 2018, 24, 5723–5727. [Google Scholar] [CrossRef]
- Maciel, L.G.; Barbosa, A.D.S.; de Alencar-Filho, E.B.; Soares, T.A.; Dos Anjos, J.V. A Second Generation of 1,2,4-Oxadiazole Derivatives with Enhanced Solubility for Inhibition of 3-Hydroxykynurenine Transaminase (HKT) FromAedes Aegypti. RSC Med. Chem. 2021, 12, 222–236. [Google Scholar] [CrossRef]
- Brahmayya, M.; Liu, F.S.; Suen, S.Y.; Huang, C.C.; Dai, S.A.; Guo, Y.S.; Chen, Y.F. A New and Efficient Method for the Synthesis of 2-N-(Aryl)-1,3,4-Oxadiazole-2,5-Diamine Derivatives. Res. Chem. Intermed. 2016, 42, 1965–1975. [Google Scholar] [CrossRef]
- Fenner, L.; Reid, S.E.; Fox, M.P.; Garone, D.; Wellington, M.; Prozesky, H.; Zwahlen, M.; Schomaker, M.; Wandeler, G.; Kancheya, N.; et al. Tuberculosis and the Risk of Opportunistic Infections and Cancers in HIV-Infected Patients Starting ART in Southern Africa. Trop. Med. Int. Health 2013, 18, 194–198. [Google Scholar] [CrossRef] [PubMed]
- CLSI Document M7-A9; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically (9th ed.). Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012.
- CLSI Document M100-A30; Performance Standards for Antimicrobial Susceptibility Testing (30th ed.). Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020.
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).