
Systemic Treatment with Fas-Blocking Peptide Attenuates Apoptosis in Brain Ischemia

 ,
,  , and
, and
Abstract
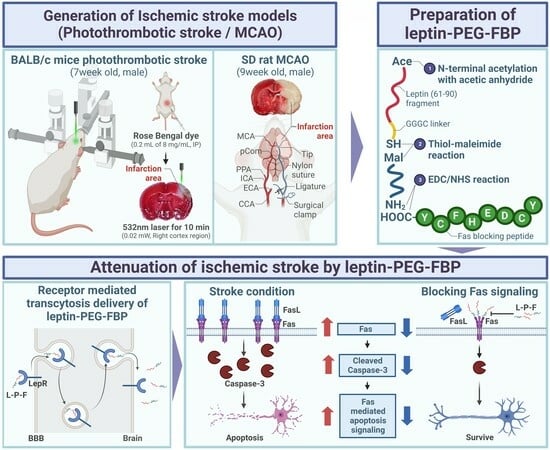
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Leptin-PEG-FBP Conjugate Inhibits Apoptosis in Hypoxic-N2a Cells
2.2. Intravenous Delivery of Leptin-PEG-FBP Localizes to Fas-Expressing Brain Regions in Photothrombotic Stroke Mouse Models of Ischemia
2.3. Leptin-Mediated Transcytosis in the Systemic Delivery of FBP to the Brain
2.4. Localization of Intravenously Delivered Leptin-PEG-FBP in Infarcted Regions of MCAO Rat Models with Ischemia
2.5. The Systemic Delivery of Leptin-PEG-FBP Alleviates Brain Cell Death in Rat Ischemic Brain Models
2.6. Reduction of Ischemic Infarction in the Rat MCAO with Systemic Leptin-PEG-FBP Treatment
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Cell Culture Studies
4.3. Animals
4.4. Experimental Stroke Models
4.5. Bio-Distribution
4.6. Real-Time RT-PCR
- Fas (CD95) forward 5′-GGAGGTGGTGATAGCCGGTAT-3′ reverse 5′-TGGGTAATCCATAGAGCCCAG -3′
- GAPDH forward 5′-AGGTCGGTGTGAACGGATTTG-3′, reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′
4.7. TTC Staining
4.8. Nissl Staining
4.9. Histology
4.10. TUNEL Assay
4.11. Immunohistochemistry
4.12. Western Blot
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Amki, M.; Wegener, S. Improving Cerebral Blood Flow after Arterial Recanalization: A Novel Therapeutic Strategy in Stroke. Int. J. Mol. Sci. 2017, 18, 669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, T.; Gong, S.X.; Huang, W.Q.; Zhou, Q.Y.; Wang, A.P.; Tian, Y. Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural. Regen. Res. 2021, 16, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Vecchie, A.; Casula, M.; Carbone, F.; Dallegri, F.; Montecucco, F. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int. J. Mol. Sci. 2016, 17, 1967. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Tadi, P.; Patti, L. Ischemic stroke; StatPearls Publishing: Orlando, FL, USA, 2018. [Google Scholar]
- Feigin, V.L.; Brainin, M.; Norrving, B.; Martins, S.; Sacco, R.L.; Hacke, W.; Fisher, M.; Pandian, J.; Lindsay, P. World Stroke Organization (WSO): Global Stroke Fact Sheet 2022. Int. J. Stroke 2022, 17, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Davalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, O.; Hornung, R.; Khatri, P.; Kleindorfer, D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: A doubling of treatment rates over the course of 5 years. Stroke 2011, 42, 1952–1955. [Google Scholar] [CrossRef] [PubMed]
- Knecht, T.; Borlongan, C.; Dela Pena, I. Combination therapy for ischemic stroke: Novel approaches to lengthen therapeutic window of tissue plasminogen activator. Brain Circ. 2018, 4, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 2022, 7, 215. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K. The fate of medications evaluated for ischemic stroke pharmacotherapy over the period 1995–2015. Acta Pharm. Sin. B 2016, 6, 522–530. [Google Scholar] [CrossRef]
- Maida, C.D.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef]
- Choi, C.; Benveniste, E.N. Fas ligand/Fas system in the brain: Regulator of immune and apoptotic responses. Brain Res. Brain Res. Rev. 2004, 44, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, N.; Lee, J.; Kim, S.; Hong, J.; Son, S.; Heo, W. Dynamic Fas signaling network regulates neural stem cell proliferation and memory enhancement. Sci. Adv. 2020, 6, eaaz9691. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wu, Z.; Xu, J.; Xu, Y. Calreticulin Binds to Fas Ligand and Inhibits Neuronal Cell Apoptosis Induced by Ischemia-Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 895284. [Google Scholar] [CrossRef]
- Dzietko, M.; Boos, V.; Sifringer, M.; Polley, O.; Gerstner, B.; Genz, K.; Endesfelder, S.; Borner, C.; Jacotot, E.; Chauvier, D.; et al. A critical role for Fas/CD-95 dependent signaling pathways in the pathogenesis of hyperoxia-induced brain injury. Ann. Neurol. 2008, 64, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Corsini, N.S.; Sancho-Martinez, I.; Laudenklos, S.; Glagow, D.; Kumar, S.; Letellier, E.; Koch, P.; Teodorczyk, M.; Kleber, S.; Klussmann, S.; et al. The death receptor CD95 activates adult neural stem cells for working memory formation and brain repair. Cell Stem Cell 2009, 5, 178–190. [Google Scholar] [CrossRef]
- Beier, C.P.; Kolbl, M.; Beier, D.; Woertgen, C.; Bogdahn, U.; Brawanski, A. CD95/Fas mediates cognitive improvement after traumatic brain injury. Cell Res. 2007, 17, 732–734. [Google Scholar] [CrossRef][Green Version]
- Khuman, J.; Meehan, W.P., 3rd; Zhu, X.; Qiu, J.; Hoffmann, U.; Zhang, J.; Giovannone, E.; Lo, E.H.; Whalen, M.J. Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J. Cereb. Blood Flow. Metab. 2011, 31, 778–789. [Google Scholar] [CrossRef]
- Bersano, A.; Gatti, L. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 14848. [Google Scholar] [CrossRef]
- Lembach, A.; Stahr, A.; Ali, A.A.H.; Ingenwerth, M.; von Gall, C. Sex-Dependent Effects of Bmal1-Deficiency on Mouse Cerebral Cortex Infarction in Response to Photothrombotic Stroke. Int. J. Mol. Sci. 2018, 19, 3124. [Google Scholar] [CrossRef]
- Krivoshein, G.; Bakreen, A.; van den Maagdenberg, A.; Malm, T.; Giniatullin, R.; Jolkkonen, J. Activation of Meningeal Afferents Relevant to Trigeminal Headache Pain after Photothrombotic Stroke Lesion: A Pilot Study in Mice. Int. J. Mol. Sci. 2022, 23, 12590. [Google Scholar] [CrossRef]
- Sharifulina, S.; Dzreyan, V.; Guzenko, V.; Demyanenko, S. Histone Methyltransferases SUV39H1 and G9a and DNA Methyltransferase DNMT1 in Penumbra Neurons and Astrocytes after Photothrombotic Stroke. Int. J. Mol. Sci. 2021, 22, 12483. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, G.; Tian, Y.; Zhang, Y.; Hong, Y.; Hao, Y.; Chen, C.; Wang, P.; Lei, H. A Novel Ligustrazine Derivative T-VA Prevents Neurotoxicity in Differentiated PC12 Cells and Protects the Brain against Ischemia Injury in MCAO Rats. Int. J. Mol. Sci. 2015, 16, 21759–21774. [Google Scholar] [CrossRef] [PubMed]
- Chelluboina, B.; Klopfenstein, J.D.; Gujrati, M.; Rao, J.S.; Veeravalli, K.K. Temporal regulation of apoptotic and anti-apoptotic molecules after middle cerebral artery occlusion followed by reperfusion. Mol. Neurobiol. 2014, 49, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Gupta, G.; D’Amore, J.; Singh, M.; Weidenheim, K.; Zhang, H.; Kessler, J.A. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J. Neurosci. Res. 2000, 61, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Gao, J.; Su, Y.; Wang, Z. Nanomedicine for Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 7600. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, T.; Karjalainen-Lindsberg, M.L.; Paetau, A.; Ijas, P.; Lindsberg, P.J. Apoptosis dominant in the periinfarct area of human ischaemic stroke--a possible target of antiapoptotic treatments. Brain 2006, 129 Pt 1, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.N.; Zhang, X.; Hu, X.M.; Chen, J.; Chang, L.L.; Li, J.W.; Liu, Z.; Cao, W.; Xu, Y. Targeted mutation of Fas ligand gene attenuates brain inflammation in experimental stroke. Brain Behav. Immun. 2012, 26, 61–71. [Google Scholar] [CrossRef]
- Yu, W.R.; Fehlings, M.G. Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: Implications for translational, clinical application. Acta Neuropathol. 2011, 122, 747–761. [Google Scholar] [CrossRef]
- Ullah, I.; Chung, K.; Oh, J.; Beloor, J.; Bae, S.; Lee, S.C.; Lee, M.; Kumar, P.; Lee, S.K. Intranasal delivery of a Fas-blocking peptide attenuates Fas-mediated apoptosis in brain ischemia. Sci. Rep. 2018, 8, 15041. [Google Scholar] [CrossRef]
- Frank, D.; Zlotnik, A.; Boyko, M.; Gruenbaum, B.F. The Development of Novel Drug Treatments for Stroke Patients: A Review. Int. J. Mol. Sci. 2022, 23, 5796. [Google Scholar] [CrossRef]
- Ji, W.; Ren, Y.; Wei, X.; Ding, X.; Dong, Y.; Yuan, B. Ischemic stroke protected by ISO-1 inhibition of apoptosis via mitochondrial pathway. Sci. Rep. 2023, 13, 2788. [Google Scholar] [CrossRef]
- Feng, J.; Feng, L.; Zhang, G. Mitochondrial damage in hippocampal neurons of rats with epileptic protein expression of Fas and caspase-3. Exp. Ther. Med. 2018, 16, 2483–2489. [Google Scholar] [CrossRef]
- Ethell, D.W.; Buhler, L.A. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J. Clin. Immunol. 2003, 23, 439–446. [Google Scholar] [CrossRef]
- Pan, J.; Zhao, Y.X.; Wang, Z.Q.; Jin, L.; Sun, Z.K.; Chen, S.D. Expression of FasL and its interaction with Fas are mediated by c-Jun N-terminal kinase (JNK) pathway in 6-OHDA-induced rat model of Parkinson disease. Neurosci. Lett. 2007, 428, 82–87. [Google Scholar] [CrossRef]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas-Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Cutillas, B.; Ambrosio, S. Fas and Fas-L expression in Huntington’s disease and Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2000, 26, 424–433. [Google Scholar] [CrossRef]
- Su, J.H.; Anderson, A.J.; Cribbs, D.H.; Tu, C.; Tong, L.; Kesslack, P.; Cotman, C.W. Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in beta-amyloid-induced neuronal death. Neurobiol. Dis. 2003, 12, 182–193. [Google Scholar] [CrossRef]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. Biomed. Res. Int. 2014, 2014, 869269. [Google Scholar] [CrossRef]
- Partridge, B.; Eardley, A.; Morales, B.E.; Campelo, S.N.; Lorenzo, M.F.; Mehta, J.N.; Kani, Y.; Mora, J.K.G.; Campbell, E.Y.; Arena, C.B.; et al. Advancements in drug delivery methods for the treatment of brain disease. Front. Vet. Sci. 2022, 9, 1039745. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow. Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of Novel Therapeutics Targeting the Blood-Brain Barrier: From Barrier to Carrier. Adv. Sci. 2021, 8, e2101090. [Google Scholar] [CrossRef]
- Morrison, J.I.; Petrovic, A.; Metzendorf, N.G.; Rofo, F.; Yilmaz, C.U.; Stenler, S.; Laudon, H.; Hultqvist, G. Standardized Preclinical In Vitro Blood–Brain Barrier Mouse Assay Validates Endocytosis-Dependent Antibody Transcytosis Using Transferrin-Receptor-Mediated Pathways. Mol. Pharmaceutics. 2023, 20, 1564–1576. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Beloor, J.; Maes, N.; Ullah, I.; Uchil, P.; Jackson, A.; Fikrig, E.; Lee, S.K.; Kumar, P. Small Interfering RNA-Mediated Control of Virus Replication in the CNS Is Therapeutic and Enables Natural Immunity to West Nile Virus. Cell Host Microbe 2018, 23, 549–556.e3. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Shao, K.; Huang, R.; Ye, L.; Lou, J.; Jiang, C. A leptin derived 30-amino-acid peptide modified pegylated poly-L-lysine dendrigraft for brain targeted gene delivery. Biomaterials 2010, 31, 5246–5257. [Google Scholar] [CrossRef]
- Tosi, G.; Badiali, L.; Ruozi, B.; Vergoni, A.V.; Bondioli, L.; Ferrari, A.; Rivasi, F.; Forni, F.; Vandelli, M.A. Can leptin-derived sequence-modified nanoparticles be suitable tools for brain delivery? Nanomedicine 2012, 7, 365–382. [Google Scholar] [CrossRef]
- Chhajed, S.; Sangale, S.; Barhate, S. Advantageous nasal drug delivery system: A review. Int. J. Pharm. Sci. Res. 2011, 2, 1322. [Google Scholar]
- Hasegawa, A.; Cheng, X.; Kajino, K.; Berezov, A.; Murata, K.; Nakayama, T.; Yagita, H.; Murali, R.; Greene, M.I. Fas-disabling small exocyclic peptide mimetics limit apoptosis by an unexpected mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 6599–6604. [Google Scholar] [CrossRef]
- Liu, G.; Li, Y.; Yang, L.; Wei, Y.; Wang, X.; Wang, Z.; Tao, L. Cytotoxicity study of polyethylene glycol derivatives. RSC Adv. 2017, 7, 18252–18259. [Google Scholar] [CrossRef]
- Neves, V.; Aires-da-Silva, F.; Morais, M.; Gano, L.; Ribeiro, E.; Pinto, A.N.; Aguiar, S.; Gaspar, D.; Fernandes, C.L.; Correia, J.O.D. Novel peptides derived from Dengue virus capsid protein translocate reversibly the blood–brain barrier through a receptor-free mechanism. ACS Chem. Biol. 2017, 12, 1257–1268. [Google Scholar] [CrossRef]
- Tolomeo, A.M.; Zuccolotto, G.; Malvicini, R.; De Lazzari, G.; Penna, A.; Franco, C.; Caicci, F.; Magarotto, F.; Quarta, S.; Pozzobon, M.; et al. Biodistribution of Intratracheal, Intranasal, and Intravenous Injections of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles in a Mouse Model for Drug Delivery Studies. Pharmaceutics 2023, 15, 548. [Google Scholar] [CrossRef]
- Xiao, G.; Gan, L.S. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int. J. Cell Biol. 2013, 2013, 703545. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Ma, Z.; Wu, B. Effects of pharmaceutical PEGylation on drug metabolism and its clinical concerns. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1691–1702. [Google Scholar] [CrossRef]
- Herold, D.A.; Keil, K.; Bruns, D.E. Oxidation of polyethylene glycols by alcohol dehydrogenase. Biochem. Pharmacol. 1989, 38, 73–76. [Google Scholar] [CrossRef]
- Friman, S.; Egestad, B.; Sjovall, J.; Svanvik, J. Hepatic excretion and metabolism of polyethylene glycols and mannitol in the cat. J. Hepatol. 1993, 17, 48–55. [Google Scholar] [CrossRef]
- Jevsevar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Alconcel, S.N.; Baas, A.S.; Maynard, H.D. FDA-approved poly (ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar]
- Onufriev, M.V.; Moiseeva, Y.V.; Zhanina, M.Y.; Lazareva, N.A.; Gulyaeva, N.V. A Comparative Study of Koizumi and Longa Methods of Intraluminal Filament Middle Cerebral Artery Occlusion in Rats: Early Corticosterone and Inflammatory Response in the Hippocampus and Frontal Cortex. Int. J. Mol. Sci. 2021, 22, 13544. [Google Scholar] [CrossRef]
- Rudy, R.F.; Charoenvimolphan, N.; Qian, B.; Berndt, A.; Friedlander, R.M.; Weiss, S.T.; Du, R. A Genome-Wide Analysis of the Penumbral Volume in Inbred Mice following Middle Cerebral Artery Occlusion. Sci. Rep. 2019, 9, 5070. [Google Scholar] [CrossRef]
- Won, J.; Khan, Z.A.; Hong, Y. Effects of isoflurane and xylazine on inducing cerebral ischemia by the model of middle cerebral artery occlusion in mice. Lab. Anim. Res. 2023, 39, 11. [Google Scholar] [CrossRef]
- Shin, T.H.; Lee, D.Y.; Basith, S.; Manavalan, B.; Paik, M.J.; Rybinnik, I.; Mouradian, M.M.; Ahn, J.H.; Lee, G. Metabolome Changes in Cerebral Ischemia. Cells 2020, 9, 1630. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, M.; Duan, X.; Zhai, Y.; Ma, B.; Duan, Z.; Xu, J.; Liu, H. Optimisation of a Mouse Model of Cerebral Ischemia-Reperfusion to Address Issues of Survival and Model Reproducibility and Consistency. Comput. Intell. Neurosci. 2022, 2022, 7594969. [Google Scholar] [CrossRef]
- Labat-gest, V.; Tomasi, S. Photothrombotic ischemia: A minimally invasive and reproducible photochemical cortical lesion model for mouse stroke studies. J. Vis. Exp. 2013, 76, e50370. [Google Scholar] [CrossRef]
- Zhang, W.F.; Jin, Y.C.; Li, X.M.; Yang, Z.; Wang, D.; Cui, J.J. Protective effects of leptin against cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2019, 17, 3282–3290. [Google Scholar] [CrossRef]
- Mikami, T.; Takao, T.; Yanagi, K.; Nakazawa, H. N (alpha) Selective Acetylation of Peptides. Mass Spectrom. 2012, 1, A0010. [Google Scholar] [CrossRef]
- Zheng, C.; Ma, C.; Bai, E.; Yang, K.; Xu, R. Transferrin and cell-penetrating peptide dual-functioned liposome for targeted drug delivery to glioma. Int. J. Clin. Exp. Med. 2015, 8, 1658–1668. [Google Scholar]
- Pinheiro, R.G.R.; Granja, A.; Loureiro, J.A.; Pereira, M.C.; Pinheiro, M.; Neves, A.R.; Reis, S. RVG29-Functionalized Lipid Nanoparticles for Quercetin Brain Delivery and Alzheimer’s Disease. Pharm. Res. 2020, 37, 139. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: New York, NY, USA, 2013. [Google Scholar]
- Staros, J.V.; Wright, R.W.; Swingle, D.M. Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal. Biochem. 1986, 156, 220–222. [Google Scholar] [CrossRef]
- Kim, M.; Oh, J.; Lee, Y.; Lee, E.-H.; Ko, S.H.; Jeong, J.H.; Park, C.H.; Lee, M. Delivery of self-replicating messenger RNA into the brain for the treatment of ischemic stroke. J. Control. Release 2022, 350, 471–485. [Google Scholar] [CrossRef]
- Ullah, I.; Chung, K.; Bae, S.; Li, Y.; Kim, C.; Choi, B.; Nam, H.Y.; Kim, S.H.; Yun, C.O.; Lee, K.Y.; et al. Nose-to-Brain Delivery of Cancer-Targeting Paclitaxel-Loaded Nanoparticles Potentiates Antitumor Effects in Malignant Glioblastoma. Mol. Pharm. 2020, 17, 1193–1204. [Google Scholar] [CrossRef]
- Akeret, K.; Hugelshofer, M.; Schaer, D.J.; Buzzi, R.M. Spatial transcriptome data from coronal mouse brain sections after striatal injection of heme and heme-hemopexin. Data Brief 2022, 41, 107866. [Google Scholar] [CrossRef]
- Davoli, M.A.; Fourtounis, J.; Tam, J.; Xanthoudakis, S.; Nicholson, D.; Robertson, G.S.; Ng, G.Y.; Xu, D. Immunohistochemical and biochemical assessment of caspase-3 activation and DNA fragmentation following transient focal ischemia in the rat. Neuroscience 2002, 115, 125–136. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, S.; Yi, Y.; Ullah, I.; Chung, K.; Park, S.; Lim, J.; Kim, C.; Pyun, S.-H.; Kim, M.; Kim, D.; et al. Systemic Treatment with Fas-Blocking Peptide Attenuates Apoptosis in Brain Ischemia. Int. J. Mol. Sci. 2024, 25, 661. https://doi.org/10.3390/ijms25010661
Chung S, Yi Y, Ullah I, Chung K, Park S, Lim J, Kim C, Pyun S-H, Kim M, Kim D, et al. Systemic Treatment with Fas-Blocking Peptide Attenuates Apoptosis in Brain Ischemia. International Journal of Molecular Sciences. 2024; 25(1):661. https://doi.org/10.3390/ijms25010661
Chicago/Turabian StyleChung, Sungeun, Yujong Yi, Irfan Ullah, Kunho Chung, Seongjun Park, Jaeyeoung Lim, Chaeyeon Kim, Seon-Hong Pyun, Minkyung Kim, Dokyoung Kim, and et al. 2024. "Systemic Treatment with Fas-Blocking Peptide Attenuates Apoptosis in Brain Ischemia" International Journal of Molecular Sciences 25, no. 1: 661. https://doi.org/10.3390/ijms25010661
APA StyleChung, S., Yi, Y., Ullah, I., Chung, K., Park, S., Lim, J., Kim, C., Pyun, S.-H., Kim, M., Kim, D., Lee, M., Rhim, T., & Lee, S.-K. (2024). Systemic Treatment with Fas-Blocking Peptide Attenuates Apoptosis in Brain Ischemia. International Journal of Molecular Sciences, 25(1), 661. https://doi.org/10.3390/ijms25010661