Cell-Surface GRP78-Targeted Chimeric Antigen Receptor T Cells Eliminate Lung Cancer Tumor Xenografts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
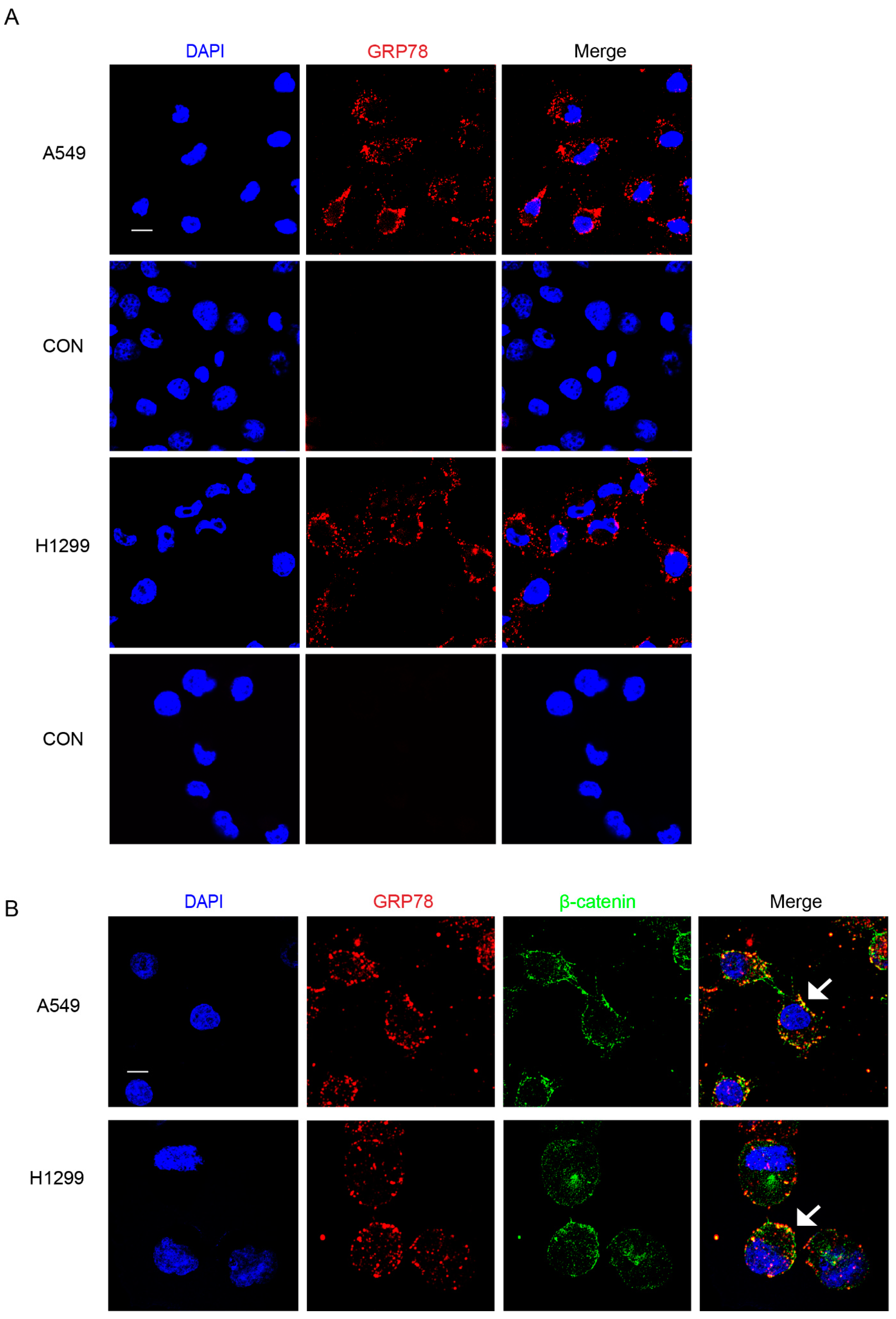
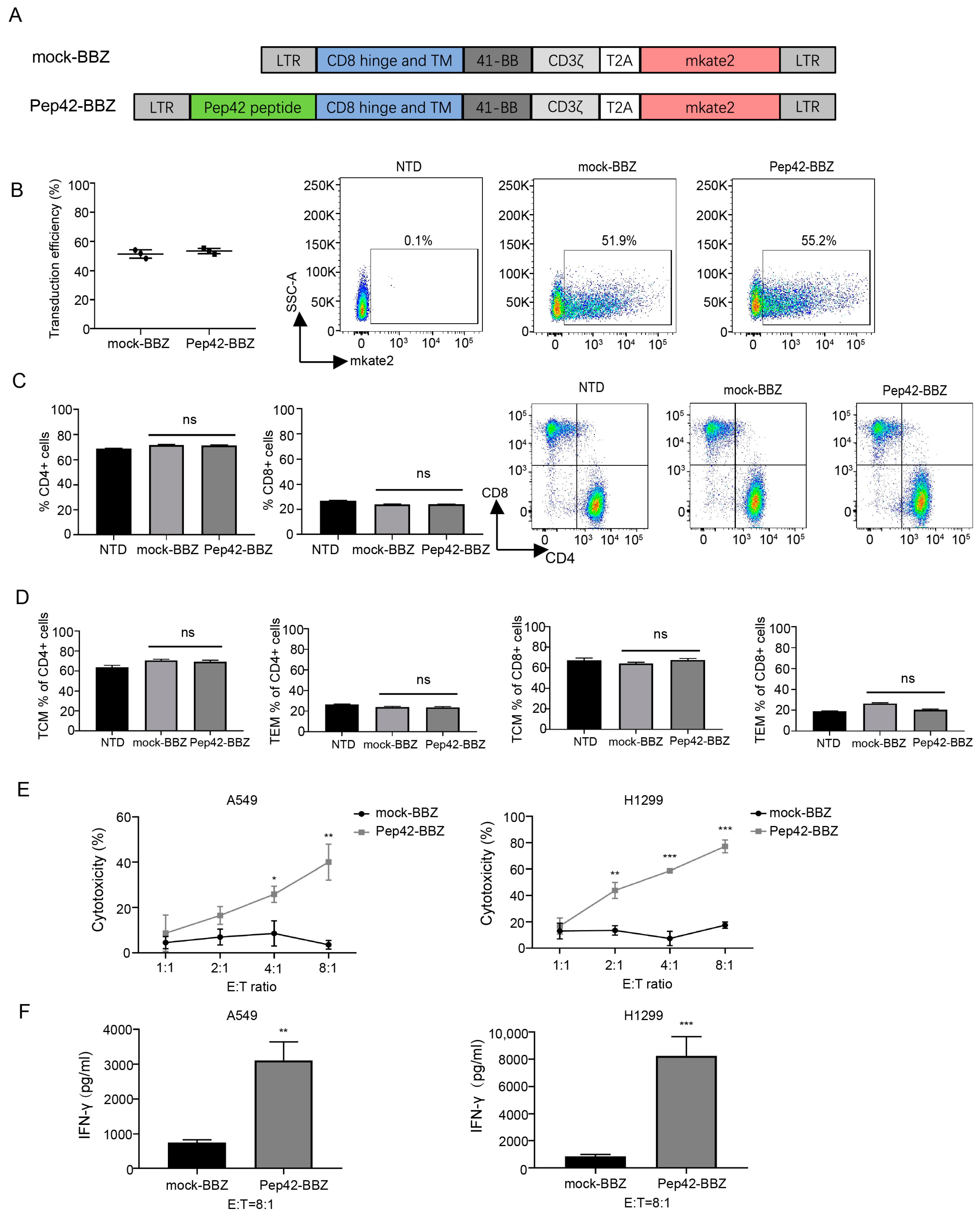
2.1. Pep42-BBZ CAR-T Cells Are Cytotoxic to Lung Cancer Cell Lines That Express csGRP78
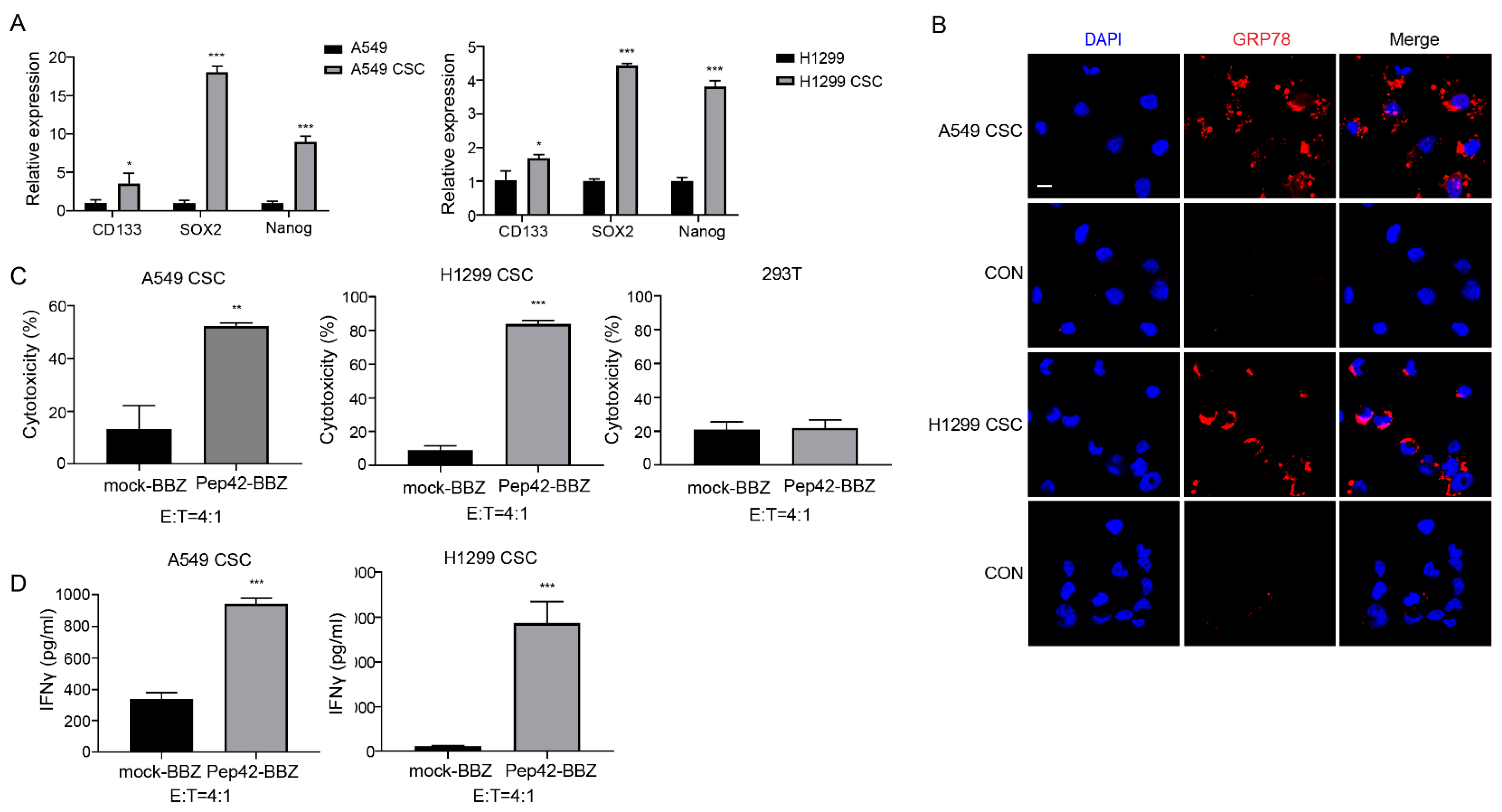
2.2. Pep42-BBZ CAR-T Cells Are Cytotoxic to CSCs In Vitro
2.3. Pep42-BBZ CAR-T Cells Eliminate A549 Tumor Xenografts
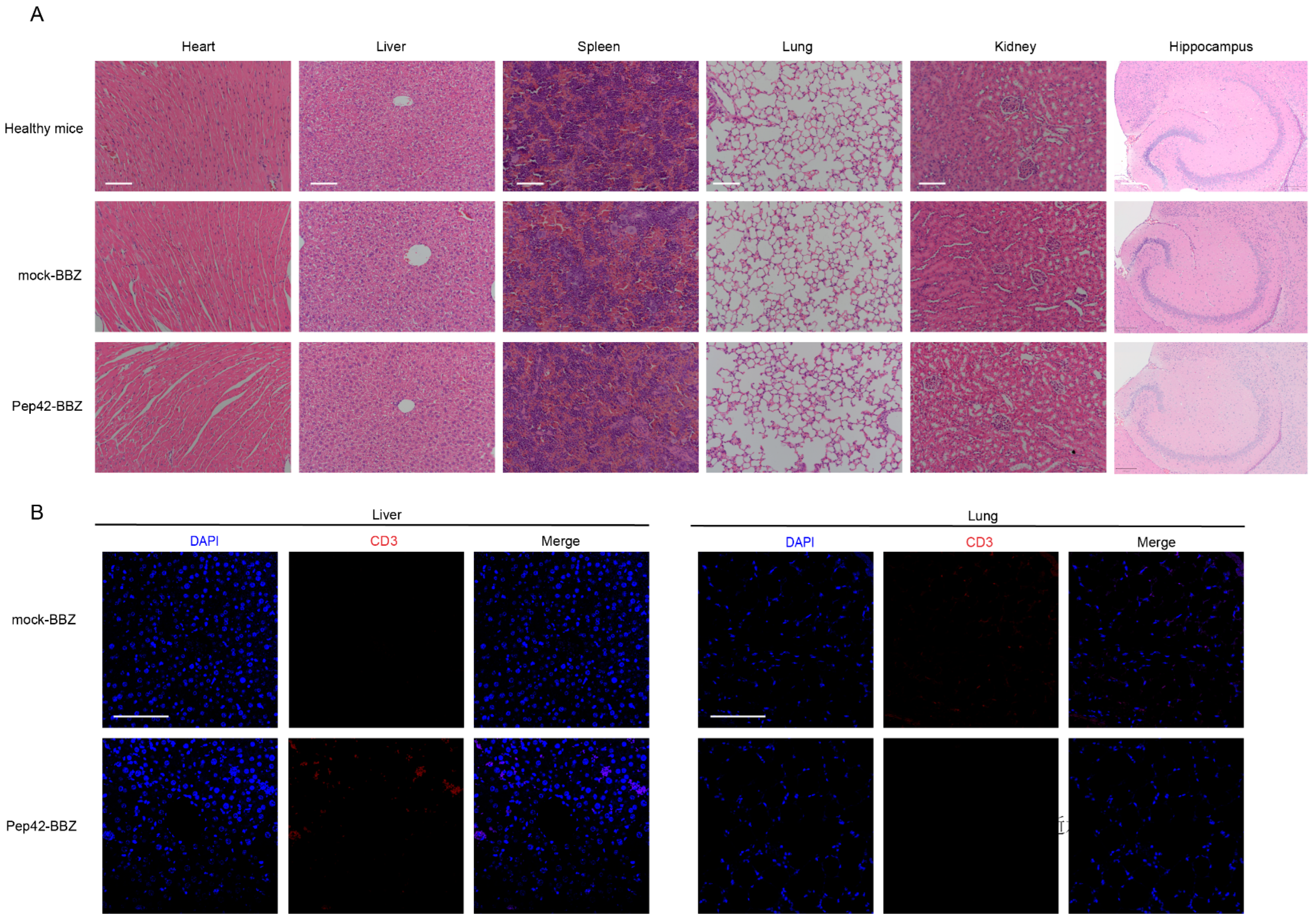
2.4. Pep42-BBZ CAR-T Cells Are Safe to Normal Organs
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Plasmid Design and Lentivirus Package
4.3. Production of CAR-T Cells
4.4. T Cell Phenotyping
4.5. Immunofluorescence
4.6. Cytotoxicity Assays In Vitro
4.7. ELISA
4.8. Xenograft Mouse Model
4.9. H&E Staining and Immunofluorescence Analysis
4.10. RNA Extraction and Quantitative Real-Time PCR
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chen, R.; Manochakian, R.; James, L.; Azzouqa, A.G.; Shi, H.; Zhang, Y.; Zhao, Y.; Zhou, K.; Lou, Y. Emerging therapeutic agents for advanced non-small cell lung cancer. J. Hematol. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Leong, T.L. Contemporary Concise Review 2021: Pulmonary nodules from detection to intervention. Respirology 2022, 27, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.L. Lung Cancer: Epidemiology and Screening. Surg. Clin. North Am. 2022, 102, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Zhang, C.Q.; Chen, F.F.; Lin, X.Y. Upregulation of Neural Precursor Cell Expressed Developmentally Downregulated 4-1 is Associated with Poor Prognosis and Chemoresistance in Lung Adenocarcinoma. Chin. Med. J. 2018, 131, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kumarakulasinghe, N.B.; van Zanwijk, N.; Soo, R.A. Molecular targeted therapy in the treatment of advanced stage non-small cell lung cancer (NSCLC). Respirology 2015, 20, 370–378. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Scharpenseel, H.; Hanssen, A.; Loges, S.; Mohme, M.; Bernreuther, C.; Peine, S.; Lamszus, K.; Goy, Y.; Petersen, C.; Westphal, M.; et al. EGFR and HER3 expression in circulating tumor cells and tumor tissue from non-small cell lung cancer patients. Sci. Rep. 2019, 9, 7406. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, J.; Wu, X.; Zhang, M.; Luo, D.; Zhang, R.; Li, S.; He, Y.; Bian, H.; Chen, Z. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Front. Med. 2019, 13, 57–68. [Google Scholar] [CrossRef]
- Sotoudeh, M.; Shirvani, S.I.; Merat, S.; Ahmadbeigi, N.; Naderi, M. MSLN (Mesothelin), ANTXR1 (TEM8), and MUC3A are the potent antigenic targets for CAR T cell therapy of gastric adenocarcinoma. J. Cell Biochem. 2019, 120, 5010–5017. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lou, Y.; Lu, L.; Fan, X. Mesothelin-targeted second generation CAR-T cells inhibit growth of mesothelin-expressing tumors in vivo. Exp. Ther. Med. 2019, 17, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Li, W.; Yu, X.; Flores-Villanueva, P.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; Young, K.H.; Ma, X.; et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 2020, 9, 72. [Google Scholar] [CrossRef]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- Chen, L.; Chen, F.; Li, J.; Pu, Y.; Yang, C.; Wang, Y.; Lei, Y.; Huang, Y. CAR-T cell therapy for lung cancer: Potential and perspective. Thorac. Cancer 2022, 13, 889–899. [Google Scholar] [CrossRef]
- Xu, C.; Ju, D.; Zhang, X. Chimeric antigen receptor T-cell therapy: Challenges and opportunities in lung cancer. Antib. Ther. 2022, 5, 73–83. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Farshbaf, M.; Khosroushahi, A.Y.; Mojarad-Jabali, S.; Zarebkohan, A.; Valizadeh, H.; Walker, P.R. Cell surface GRP78: An emerging imaging marker and therapeutic target for cancer. J. Control. Release 2020, 328, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhang, Y.; Lee Amy, S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Duan, W.; Liu, W.; Zhang, X.; Wang, Q. GRP78 in lung cancer. J. Transl. Med. 2021, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J. Biol. Chem. 2006, 281, 13694–13707. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Wang, F.; Pizzo, S.V. A novel receptor function for the heat shock protein Grp78: Silencing of Grp78 gene expression attenuates alpha2M*-induced signalling. Cell Signal. 2004, 16, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Zhou, J.; Chai, Z.; Li, J.; Xie, C.; Mao, J.; Lu, L.; Zhang, Y.; Wu, S.; Zhan, C.; et al. All-stage precisional glioma targeted therapy enabled by a well-designed D-peptide. Theranostics 2020, 10, 4073–4087. [Google Scholar] [CrossRef]
- Kapoor, V.; Dadey, D.Y.; Nguyen, K.; Wildman, S.A.; Hoye, K.; Khudanyan, A.; Bandara, N.; Rogers, B.E.; Thotala, D.; Hallahan, D.E. Tumor-Specific Binding of Radiolabeled PEGylated GIRLRG Peptide: A Novel Agent for Targeting Cancers. J. Nucl. Med. 2016, 57, 1991–1997. [Google Scholar] [CrossRef]
- Hebbar, N.; Epperly, R.; Vaidya, A.; Thanekar, U.; Moore, S.E.; Umeda, M.; Ma, J.; Patil, S.L.; Langfitt, D.; Huang, S.; et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022, 13, 587. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, H.; Yuan, Y.; Tang, J.; Chen, X.; Liu, T.; Zhao, X. Chimeric Antigen Receptor T Cells Targeting Cell Surface GRP78 to Eradicate Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2022, 10, 928140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, Z.; Zhang, J.; Wang, Y.; Wang, T.; Tong, S.; Wang, L.; Wang, S.; Chen, Y. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect. Prev. 2005, 29, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer. 2012, 131, E362–E370. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.K.; Wang, H.; Yim, A.M.; Le Naour, F.; Brichory, F.; Jang, J.H.; Zhao, R.; Puravs, E.; Tra, J.; Michael, C.W.; et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 2003, 278, 7607–7616. [Google Scholar] [CrossRef]
- Paolillo, M.; Colombo, R.; Serra, M.; Belvisi, L.; Papetti, A.; Ciusani, E.; Comincini, S.; Schinelli, S. Stem-Like Cancer Cells in a Dynamic 3D Culture System: A Model to Study Metastatic Cell Adhesion and Anti-Cancer Drugs. Cells 2019, 8, 1434. [Google Scholar] [CrossRef]
- Wang, L.; He, J.; Hu, H.; Tu, L.; Sun, Z.; Liu, Y.; Luo, F. Lung CSC-derived exosomal miR-210-3p contributes to a pro-metastatic phenotype in lung cancer by targeting FGFRL1. J. Cell Mol. Med. 2020, 24, 6324–6339. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Yuan, Y.; Sun, B.; Yang, D.; Liu, N.; Zhao, X. Chimeric antigen receptor T cells targeting cell surface GRP78 efficiently kill glioblastoma and cancer stem cells. J. Transl. Med. 2023, 21, 493. [Google Scholar] [CrossRef]
- Yuan, Y.; Fan, J.; Liang, D.; Wang, S.; Luo, X.; Zhu, Y.; Liu, N.; Xiang, T.; Zhao, X. Cell surface GRP78-directed CAR-T cells are effective at treating human pancreatic cancer in preclinical models. Transl. Oncol. 2023, 39, 101803. [Google Scholar] [CrossRef]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, Z.; Fan, H.; Song, J.; Liu, Y.; Gao, Z.; Wang, Q. Cancer-associated fibroblasts promote non-small cell lung cancer cell invasion by upregulation of glucose-regulated protein 78 (GRP78) expression in an integrated bionic microfluidic device. Oncotarget 2016, 7, 25593–25603. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015, 6, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Tsukahara, T.; Murata, K.; Hamada, S.; Kubo, T.; Kanaseki, T.; Hirohashi, Y.; Emori, M.; Teramoto, A.; Nakatsugawa, M.; et al. Development of CAR-T cells specifically targeting cancer stem cell antigen DNAJB8 against solid tumours. Br. J. Cancer 2023, 128, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining "stemness". Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, H.; Zhang, Z.; Xu, J.; Qi, Y.; Xue, H.; Gao, Z.; Zhao, R.; Wang, S.; Zhang, S.; et al. Cell surface GRP78 regulates BACE2 via lysosome-dependent manner to maintain mesenchymal phenotype of glioma stem cells. J. Exp. Clin. Cancer Res. 2021, 40, 20. [Google Scholar] [CrossRef]
- Conner, C.; Lager, T.W.; Guldner, I.H.; Wu, M.Z.; Hishida, Y.; Hishida, T.; Ruiz, S.; Yamasaki, A.E.; Gilson, R.C.; Belmonte, J.C.I.; et al. Cell surface GRP78 promotes stemness in normal and neoplastic cells. Sci. Rep. 2020, 10, 3474. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Xiao, B.F.; Zhang, J.T.; Zhu, Y.G.; Cui, X.R.; Lu, Z.M.; Yu, B.T.; Wu, N. Chimeric Antigen Receptor T-Cell Therapy in Lung Cancer: Potential and Challenges. Front. Immunol. 2021, 12, 782775. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Wei, W.; Yuan, Y.; Guo, J.; Liang, D.; Zhao, X. Cell-Surface GRP78-Targeted Chimeric Antigen Receptor T Cells Eliminate Lung Cancer Tumor Xenografts. Int. J. Mol. Sci. 2024, 25, 564. https://doi.org/10.3390/ijms25010564
Wang S, Wei W, Yuan Y, Guo J, Liang D, Zhao X. Cell-Surface GRP78-Targeted Chimeric Antigen Receptor T Cells Eliminate Lung Cancer Tumor Xenografts. International Journal of Molecular Sciences. 2024; 25(1):564. https://doi.org/10.3390/ijms25010564
Chicago/Turabian StyleWang, Shijie, Wenwen Wei, Yuncang Yuan, Jing Guo, Dandan Liang, and Xudong Zhao. 2024. "Cell-Surface GRP78-Targeted Chimeric Antigen Receptor T Cells Eliminate Lung Cancer Tumor Xenografts" International Journal of Molecular Sciences 25, no. 1: 564. https://doi.org/10.3390/ijms25010564
APA StyleWang, S., Wei, W., Yuan, Y., Guo, J., Liang, D., & Zhao, X. (2024). Cell-Surface GRP78-Targeted Chimeric Antigen Receptor T Cells Eliminate Lung Cancer Tumor Xenografts. International Journal of Molecular Sciences, 25(1), 564. https://doi.org/10.3390/ijms25010564