
Integrated Transcriptomic and Metabolomic Analyses Identify Critical Genes and Metabolites Associated with Seed Vigor of Common Wheat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Dynamic Transcriptional Variations in HT and LT Wheat Seeds by Artificial Aging
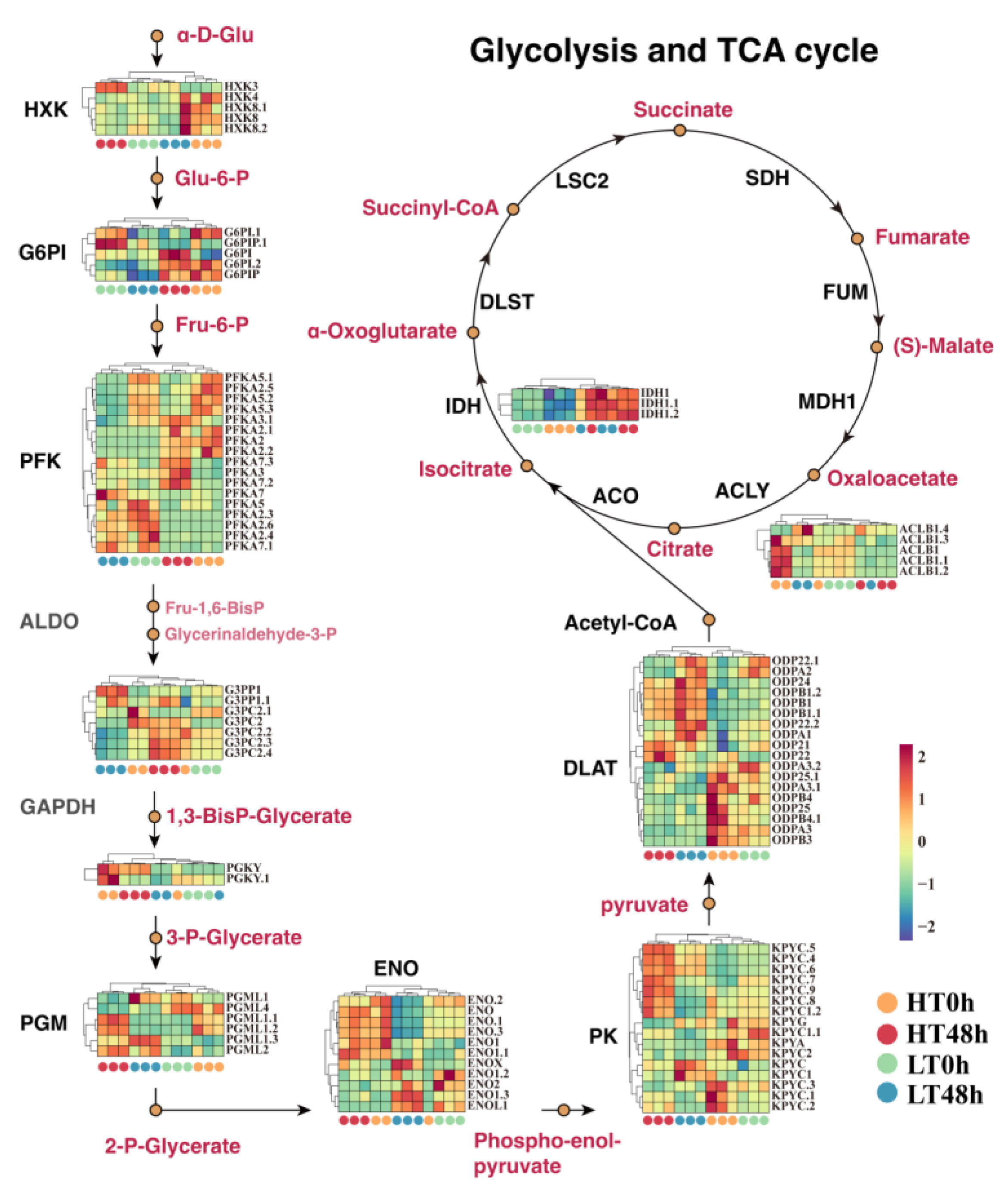
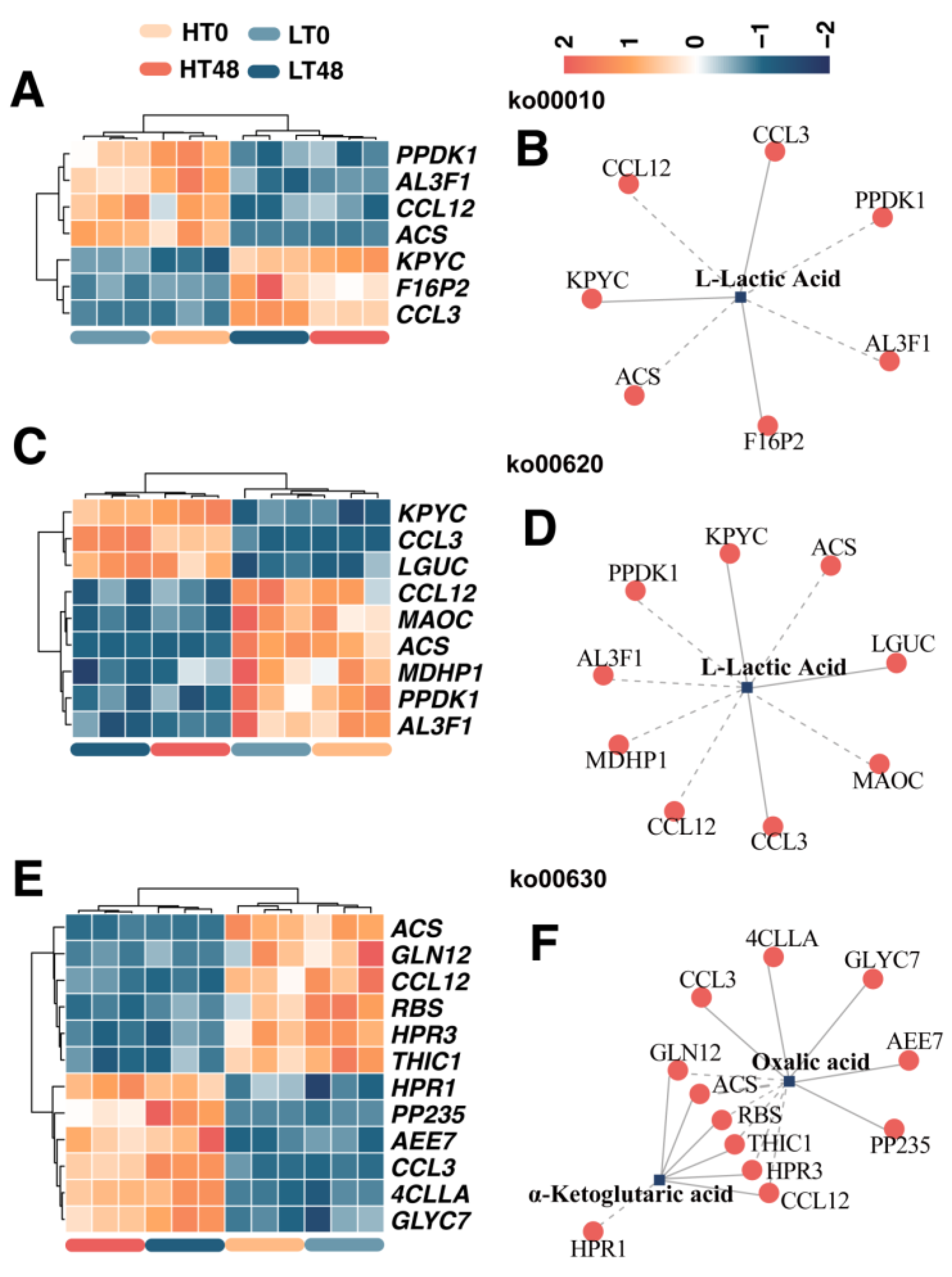
2.2. Differential Expression of Glycolysis and TCA-Cycle-Related Genes May Affect Wheat Seed Vigor
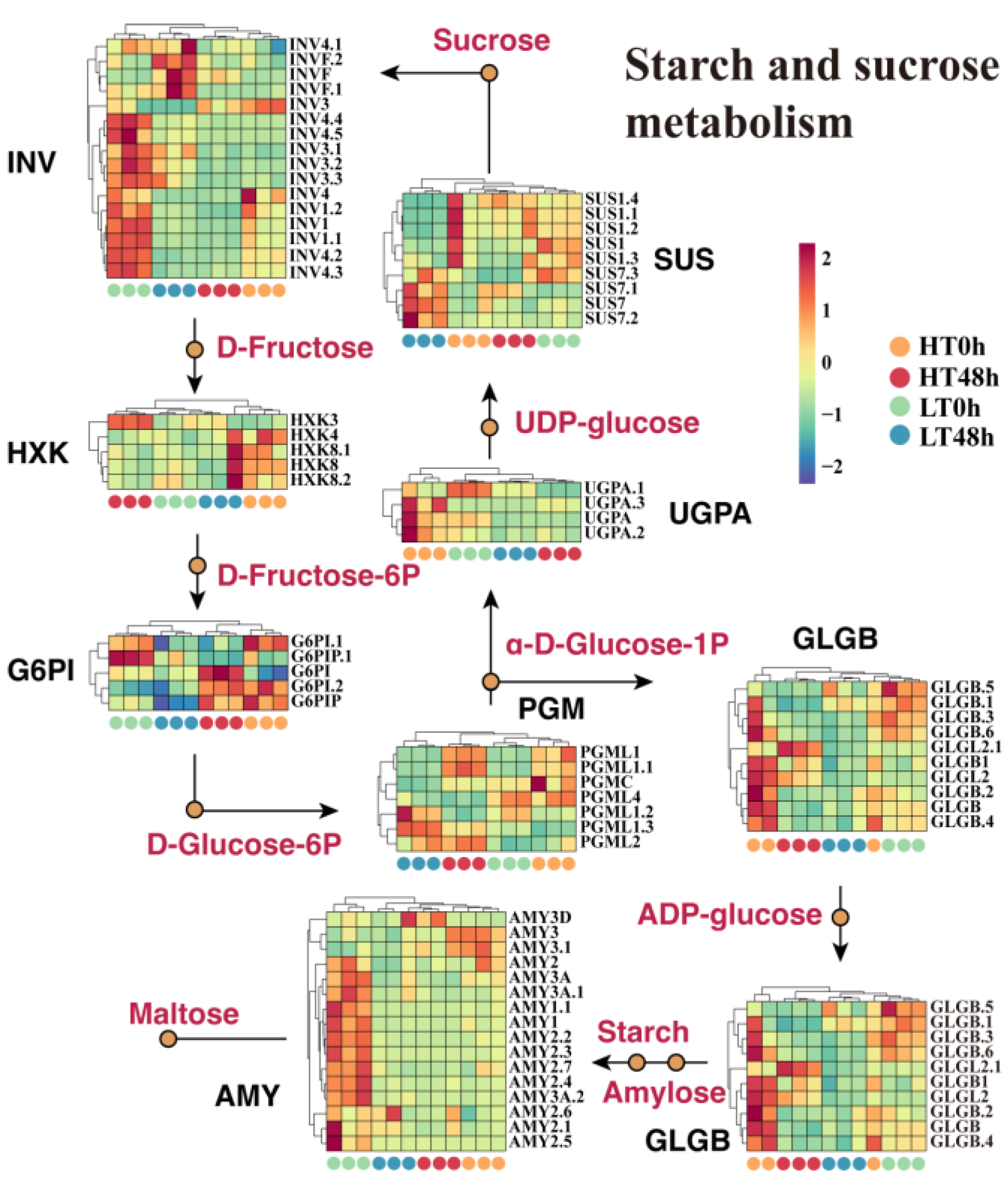
2.3. Reduced Expression of Starch Metabolic-Related Genes May Cause Decreased Activity in Wheat Seeds
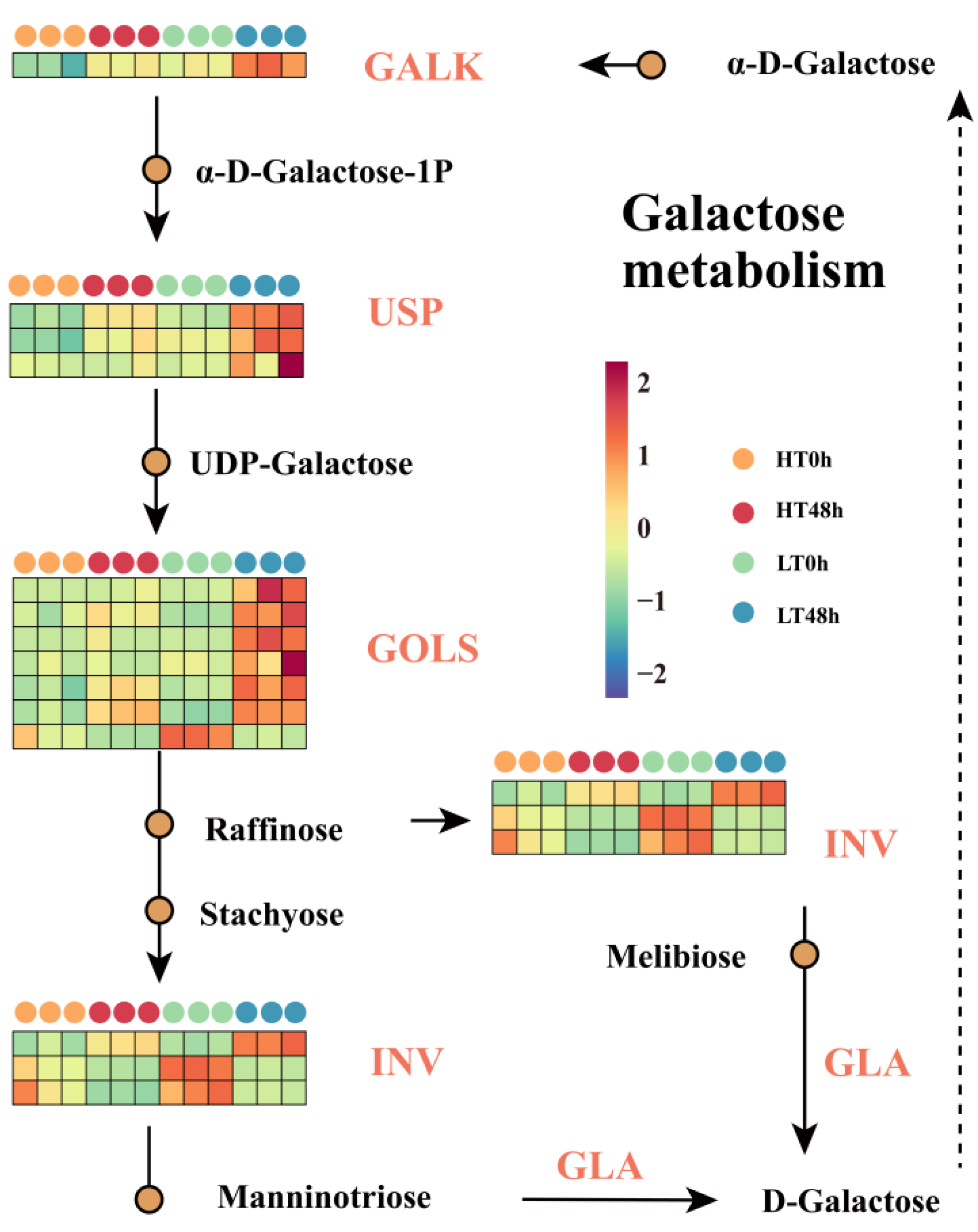
2.4. Alterations in the Expression of Galactose Metabolism-Related Genes Resulted in Variations in Wheat Seed Vigor
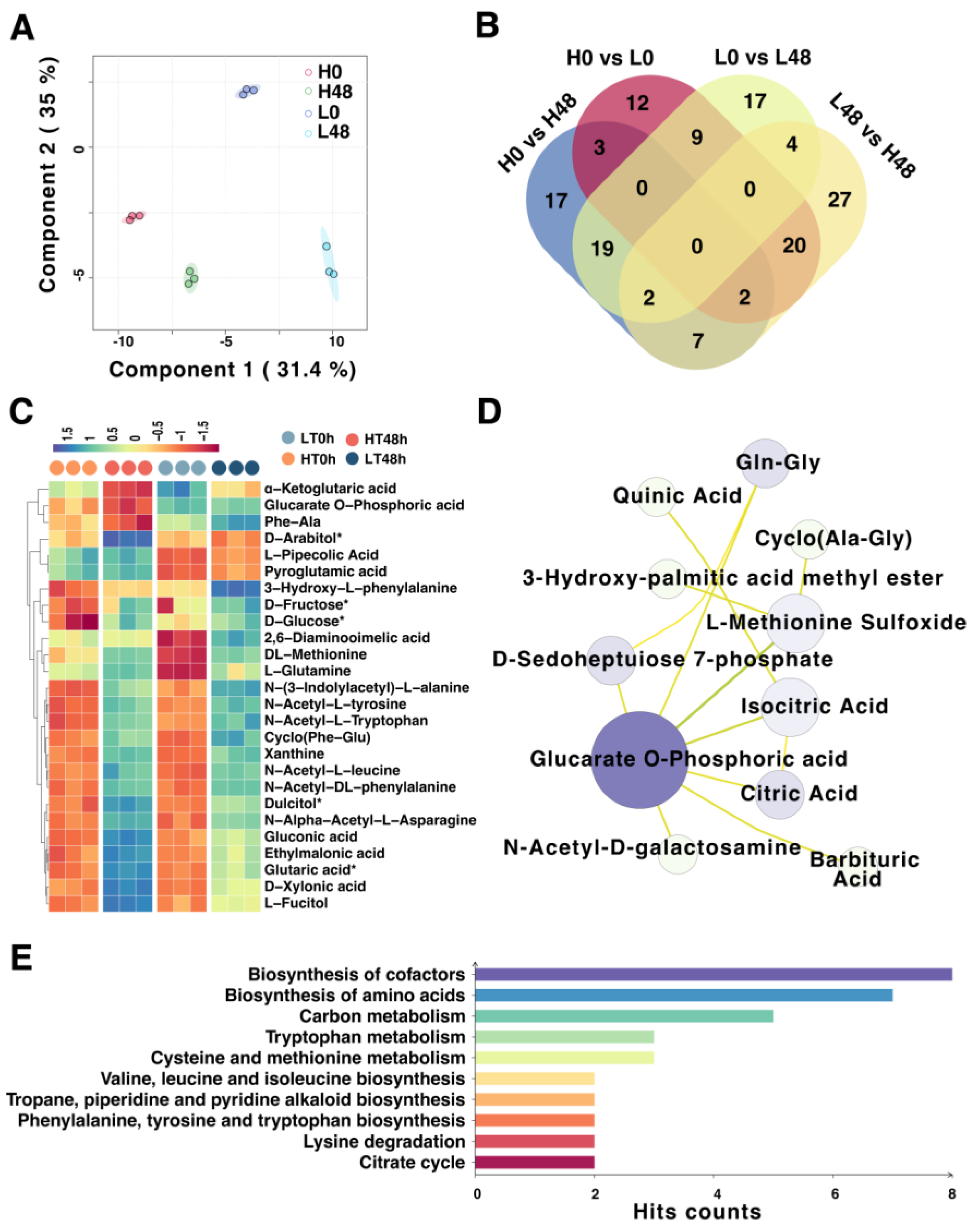
2.5. Dynamic Metabolomic Changes in HT and LT Wheat Seeds by Artificial Aging
2.6. Association Analysis of Metabolome and Transcriptome Data
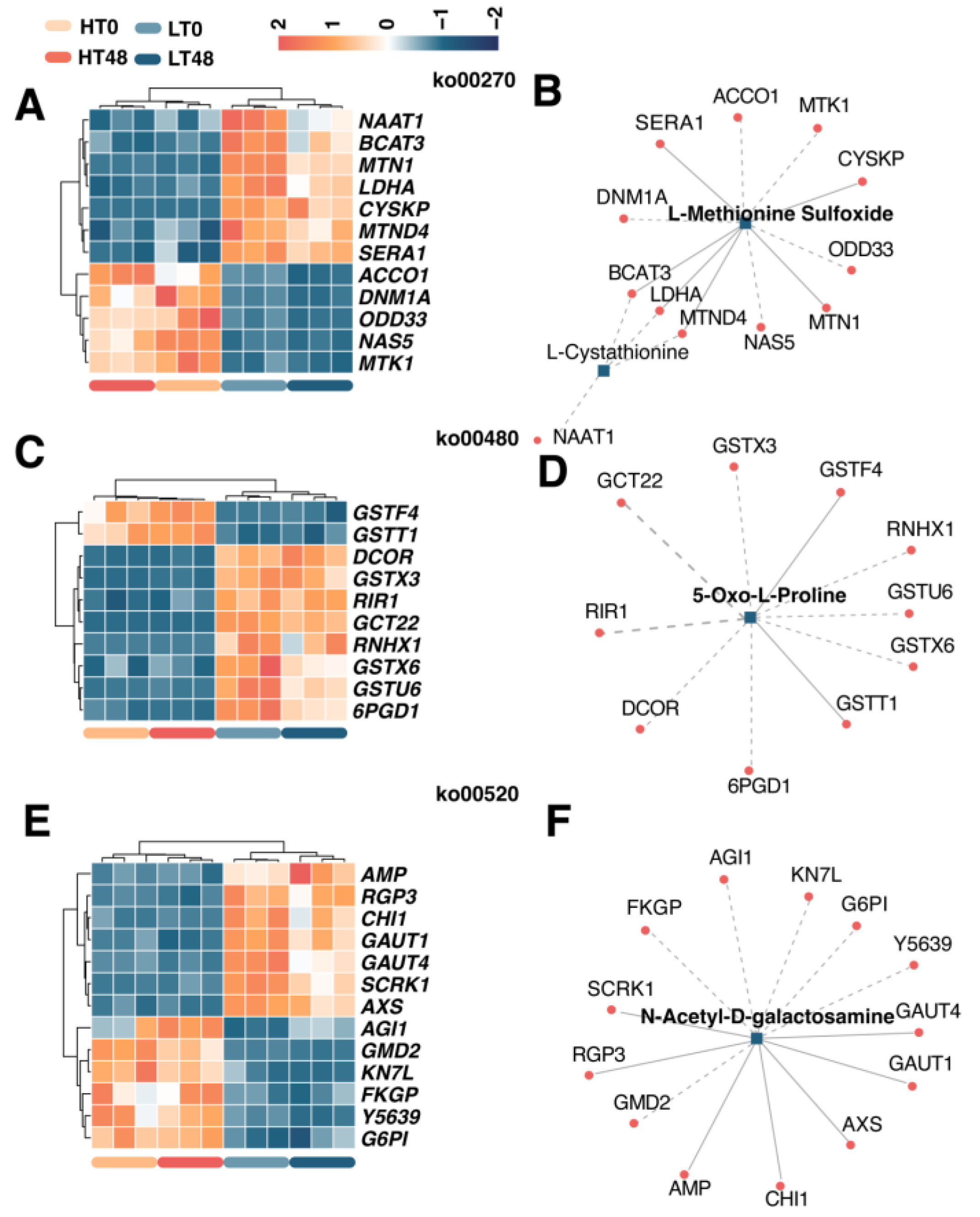
2.7. Transcript–Metabolite Correlation Network Demonstrating Correlations between DIMs and DEGs
2.8. Transcription Factors (TFs) Associated with Aging in Wheat Seeds
3. Discussion
4. Materials and Methods
4.1. Wheat Seed Materials and Treatment Conditions
4.2. Sample Preparation and Extraction for Widely Targeted Metabolic Profiling
4.3. UPLC-MS/MS Conditions
4.4. Transciptome Analysis
4.5. Statistical Analysis
4.6. Quantitative Real-Time Polymerase Chain Reaction (q-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, M.; Shen, J.; Yuan, L.; Jiang, R.; Chen, X.; Davies, W.J.; Zhang, F. Improving crop productivity and resource use efficiency to ensure food security and environmental quality in China. J. Exp. Bot. 2012, 63, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yun, L.; Song, B. Effects of storage time on seed vigor of Psathyrostachys juncea. Seed 2015, 34, 33–38. [Google Scholar]
- Dar, R.A.; Ahmad, M.; Kumar, S.; Reshi, M. Agriculture germplasm resources: A tool of conserving diversity. Sci. Res. Essays 2015, 10, 326–338. [Google Scholar]
- Liu, L.; Lai, Y.; Cheng, J.; Wang, L.; Du, W.; Wang, Z.; Zhang, H. Dynamic quantitative trait locus analysis of seed vigor at three maturity stages in rice. PLoS ONE 2014, 9, e115732. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, X.; Sun, D.; Liu, F.; Bao, Y.; He, Y. Rapid and nondestructive measurement of rice seed vitality of different years using near-infrared hyperspectral imaging. Molecules 2019, 24, 2227. [Google Scholar] [CrossRef]
- Reed, R.C.; Bradford, K.J.; Khanday, I. Seed germination and vigor: Ensuring crop sustainability in a changing climate. Heredity 2022, 128, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Chen, S.; Huang, Y.; Qin, Y.; Ruan, G. Effects of artificial aging on physiological characteristics of rice seeds with different dormancy characteristics. Agric. Biotechnol. 2019, 8, 52–56. [Google Scholar]
- Qin, P.; Kong, Z.; Liao, X.; Liu, Y. Effects of accelerated aging on physiological and biochemical characteristics of waxy and non-waxy wheat seeds. J. Northeast Agric. Univ. (Engl. Ed.) 2011, 18, 7–12. [Google Scholar] [CrossRef]
- Lin, Y.X.; Xu, H.J.; Yin, G.K.; Zhou, Y.C.; Lu, X.X.; Xin, X. Dynamic changes in membrane lipid metabolism and antioxidant defense during soybean (Glycine max L. Merr.) seed aging. Front. Plant Sci. 2022, 13, 908949. [Google Scholar] [CrossRef]
- Jiang, F.L.; Bo, L.P.; Xu, J.J.; Wu, Z. Changes in respiration and structure of non-heading Chinese cabbage seeds during gradual artificial aging. Sci. Hortic. 2018, 238, 14–22. [Google Scholar] [CrossRef]
- Kadavala, J.B.; Patel, M.B.; Parmar, P.K.; Patil, K. Seed ageing physiological, biochemical and molecular basis: A review. TPI 2023, 12, 1511–1517. [Google Scholar]
- Chen, H.; Osuna, D.; Colville, L.; Lorenzo, O.; Graeber, K.; Kuester, H.; Kranner, I. Transcriptome-wide mapping of pea seed ageing reveals a pivotal role for genes related to oxidative stress and programmed cell death. PLoS ONE 2013, 8, e78471. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Hay, F.R.; Dickie, J.B.; Trivedi, C.; Choi, J.; Fiegener, R. Seed storage: Maintaining seed viability and vigor for restoration use. Restor. Ecol. 2020, 28, S249–S255. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.; Sun, J.; Meng, J.; Tao, J. Deterioration of orthodox seeds during ageing: Influencing factors, physiological alterations and the role of reactive oxygen species. Plant Physiol. Biochem. 2021, 158, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xin, X.; Yin, G.; Zhou, J.; Zhou, Y.; Lu, X. Timing for antioxidant-priming against rice seed ageing: Optimal only in non-resistant stage. Sci. Rep. 2020, 10, 13294. [Google Scholar] [CrossRef] [PubMed]
- Katarzyna, K.; Plitta-Michalak, B.; Ratajczak, E. Reactive oxygen species as potential drivers of the seed aging process. Plants 2019, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Z.; Fu, Z.; Liu, Z.; Hu, Y.; Tang, J. Comparative QTL analysis of maize seed artificial aging between an immortalized F2 population and its corresponding RILs. Crop J. 2016, 4, 30–39. [Google Scholar] [CrossRef]
- Wang, B.; Wang, S.; Tang, Y.; Jiang, L.; He, W.; Lin, Q.; Wang, L. Transcriptome-wide characterization of seed aging in rice: Identification of specific long-lived mRNAs for seed longevity. Front. Plant Sci. 2022, 13, 857390. [Google Scholar] [CrossRef]
- Liu, M.; Wang, T.; Chen, S.; Wang, C.; Zhao, X. Physioloycial and seed vigour changes of maize seeds during artificial aging course. J. Nucl. Agric. Sci. 2008, 22, 510–513. [Google Scholar]
- Shi, H.; Guan, W.; Shi, Y.; Wang, S.; Fan, H.; Yang, J.; Chen, W.; Zhang, W.; Sun, D.; Jing, R. QTL mapping and candidate gene analysis of seed vigor-related traits during artificial aging in wheat (Triticum aestivum). Sci. Rep. 2020, 10, 22060. [Google Scholar] [CrossRef]
- Veselovsky, V.A.; Veselova, T.V. Lipid peroxidation, carbohydrate hydrolysis, and Amadori-Maillard reaction at early stages of dry seed aging. Russ. J. Plant Physiol. 2012, 59, 811–817. [Google Scholar] [CrossRef]
- Wen, W.; Li, D.; Li, X.; Gao, Y.; Li, W.; Li, H.; Liu, J.; Liu, H.; Chen, W.; Luo, J.; et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 3438. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Fang, M.; Din, G.; Gao, H.; Yang, Y.; Liu, T.; Chen, W.; Gao, L. Metabolomics analysis of grains of wheat infected and noninfected with Tilletia controversa Kühn. Sci. Rep. 2021, 11, 18876. [Google Scholar] [CrossRef] [PubMed]
- Datir, S.S.; Yousf, S.; Sharma, S.; Kochle, M.; Ravikumar, A.; Chugh, J. Cold storage reveals distinct metabolic perturbations in processing and non-processing cultivars of potato (Solanum tuberosum L.). Sci. Rep. 2020, 10, 6268. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Guo, H.; Zhang, L.; Tang, Z.; Yu, X.; Wu, J.; Zeng, F. Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 2019, 20, 2070. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Luo, S.; Jiao, J.; Yan, W.; Zeng, B.; He, H.; He, G. Integrated Transcriptomic and Metabolomic Analysis Reveals the Mechanism of Gibberillic Acid Regulates the Growth and Flavonoid Synthesis in Phellodendron chinense Schneid Seedlings. Int. J. Mol. Sci. 2023, 24, 16045. [Google Scholar] [CrossRef] [PubMed]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef]
- Ouzouline, M.; Tahani, N.; Demandre, C.; El Amrani, E.; Benhassaine-Kesri, G.; Caid, H. Effects of accelerated aging upon the lipid composition of seeds from two soft wheat varieties from. Grasasy Aceites 2009, 60, 367–374. [Google Scholar]
- Zhang, H.; Yang, G.; GaoMorocco, W.; Zhu, Y.; Huang, F.; Pei, H.; Li, Q. Study on the seed vigor of Toona sinensis under specific storage conditions. For. Sci. Res. 2019, 32, 152–159. [Google Scholar]
- Veselova, T.V.; Veselovsky, V.A.; Obroucheva, N.V. Deterioration mechanisms in air-dry pea seeds during early aging. Plant Physiol. Biochem. 2015, 87, 133–139. [Google Scholar] [CrossRef]
- Khan, M.M.; Abbas, M.; Awan, F.S.; Shahid, M.; Ali, M.; Ahmad, S. Physio-biochemic and genetic changes in stored pea (Pisum sativum) seeds. Int. J. Agric. Biol. 2013, 15, 951–956. [Google Scholar]
- Goel, A.; Sheoran, I.S. Lipid peroxidation and peroxide-scavenging enzymes in cotton seeds under natural ageing. Biol. Plant. 2003, 46, 429–434. [Google Scholar] [CrossRef]
- Chang, H.; Zhang, F.; Yang, Z.; Kong, D.; Zheng, Q.; Hao, L. Physiological and biochemical responses of Allium mongolicum seeds to storage aging. Plant Physiol. J. 2015, 51, 1075–1081. [Google Scholar]
- Yin, H.; Tang, Q.; Wang, Z. Changes of physiological and biochemical indexes of Welsh onion seeds during storage. South China Agric. 2007, 1, 66–67. [Google Scholar]
- Silva, D.D.; Stuepp, C.A.; Wendling, I.; Helm, C.V.; Angelo, A.C. Physiological and biochemical changes in Cedrela fissilis seeds during storage. Pesqui. Agropecuária Bras. 2020, 55. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, X.; Zhang, Z.; Zhou, Y.; Liu, M. Changes of protein components in wheat seed during aging. J. Yangzhou Univ. (Agric. Life Sci. Ed.) 2019, 40, 86–90. [Google Scholar]
- Hirohisa, T. Generation of poly-β-hydroxybutyrate from externally provided acetate in rice root. PPB 2012, 50, 35–43. [Google Scholar]
- Lin, M.; Oliver, D.J. The role of acetyl-coenzyme a synthetase in Arabidopsis. Plant Physiol. 2008, 147, 1822–1829. [Google Scholar] [CrossRef]
- Swartzberg, D.; Dai, N.; Gan, S.; Amasino, R.; Granot, D. Effects of cytokinin production under two SAG promoters on senescence and development of tomato plants. Plant Biol. (Stuttg.) 2006, 8, 579–586. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, L.; Lin, L.; Zhao, L.; Liu, Q.; Wei, C. A novel mutation of OsPPDKB, encoding pyruvate orthophosphate dikinase, affects metabolism and structure of starch in the rice endosperm. Int. J. Mol. Sci. 2018, 19, 2268. [Google Scholar] [CrossRef]
- Sterling, J.D.; Atmodjo, M.A.; Inwood, S.E.; Kumar Kolli, V.S.; Quigley, H.F.; Hahn, M.G.; Mohnen, D. Functional identification of an Arabidopsis pectin biosynthetic homogalacturonan galacturonosyltransferase. Proc. Natl. Acad. Sci. USA 2006, 103, 5236–5241. [Google Scholar] [CrossRef] [PubMed]
- Knill, T.; Schuster, J.; Reichelt, M.; Gershenzon, J.; Binder, S. Arabidopsis branched-chain aminotransferase 3 functions in both amino acid and glucosinolate biosynthesis. Plant Physiol. 2008, 146, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; McRoberts, J.; Shi, F.; Moreno, J.E.; Jones, A.D.; Howe, G.A. The flavonoid biosynthetic enzyme chalcone isomerase modulates terpenoid production in glandular trichomes of tomato. Plant Physiol. 2014, 164, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Kim, S.C.; Li, J. Transcriptional regulation of lipid catabolism during seedling establishmen. Mol. Plant 2020, 13, 984–1000. [Google Scholar] [CrossRef] [PubMed]
- Dolouch, J.C. Accelerated aging techniques for predicting the relative storability of seed lots. Seed Sci. Technol. 1973, 1, 427–452. [Google Scholar]
- Yu, Y.; Guo, G.; Lv, D.; Hu, Y.; Li, J.; Li, X.; Yan, Y. Transcriptome analysis during seed germination of elite Chinese bread wheat cultivar Jimai 20. BMC Plant Biol. 2014, 14, 20. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, R.; Song, Y.; He, J.; Sun, J.; Bai, J.; Abliz, Z. RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: Finding potential biomarkers for breast cancer. Analyst 2009, 134, 2003–2011. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yao, Y.; Chen, X.; Wu, J.; Wu, Q.; Liu, S.; Zhang, X. Metabolomic and transcriptomic analyses reveal the mechanism of sweet-acidic taste formation during pineapple fruit development. Front. Plant Sci. 2022, 13, 971506. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Chen, W.; Jia, T.; Shi, H.; Sun, D. Integrated Transcriptomic and Metabolomic Analyses Identify Critical Genes and Metabolites Associated with Seed Vigor of Common Wheat. Int. J. Mol. Sci. 2024, 25, 526. https://doi.org/10.3390/ijms25010526
Yang Z, Chen W, Jia T, Shi H, Sun D. Integrated Transcriptomic and Metabolomic Analyses Identify Critical Genes and Metabolites Associated with Seed Vigor of Common Wheat. International Journal of Molecular Sciences. 2024; 25(1):526. https://doi.org/10.3390/ijms25010526
Chicago/Turabian StyleYang, Zhenrong, Weiguo Chen, Tianxiang Jia, Huawei Shi, and Daizhen Sun. 2024. "Integrated Transcriptomic and Metabolomic Analyses Identify Critical Genes and Metabolites Associated with Seed Vigor of Common Wheat" International Journal of Molecular Sciences 25, no. 1: 526. https://doi.org/10.3390/ijms25010526
APA StyleYang, Z., Chen, W., Jia, T., Shi, H., & Sun, D. (2024). Integrated Transcriptomic and Metabolomic Analyses Identify Critical Genes and Metabolites Associated with Seed Vigor of Common Wheat. International Journal of Molecular Sciences, 25(1), 526. https://doi.org/10.3390/ijms25010526