
Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biology of Satellite Cell in Regulating Muscle Homeostasis and Regeneration
3. Skeletal Muscle Regeneration in Pathological Conditions
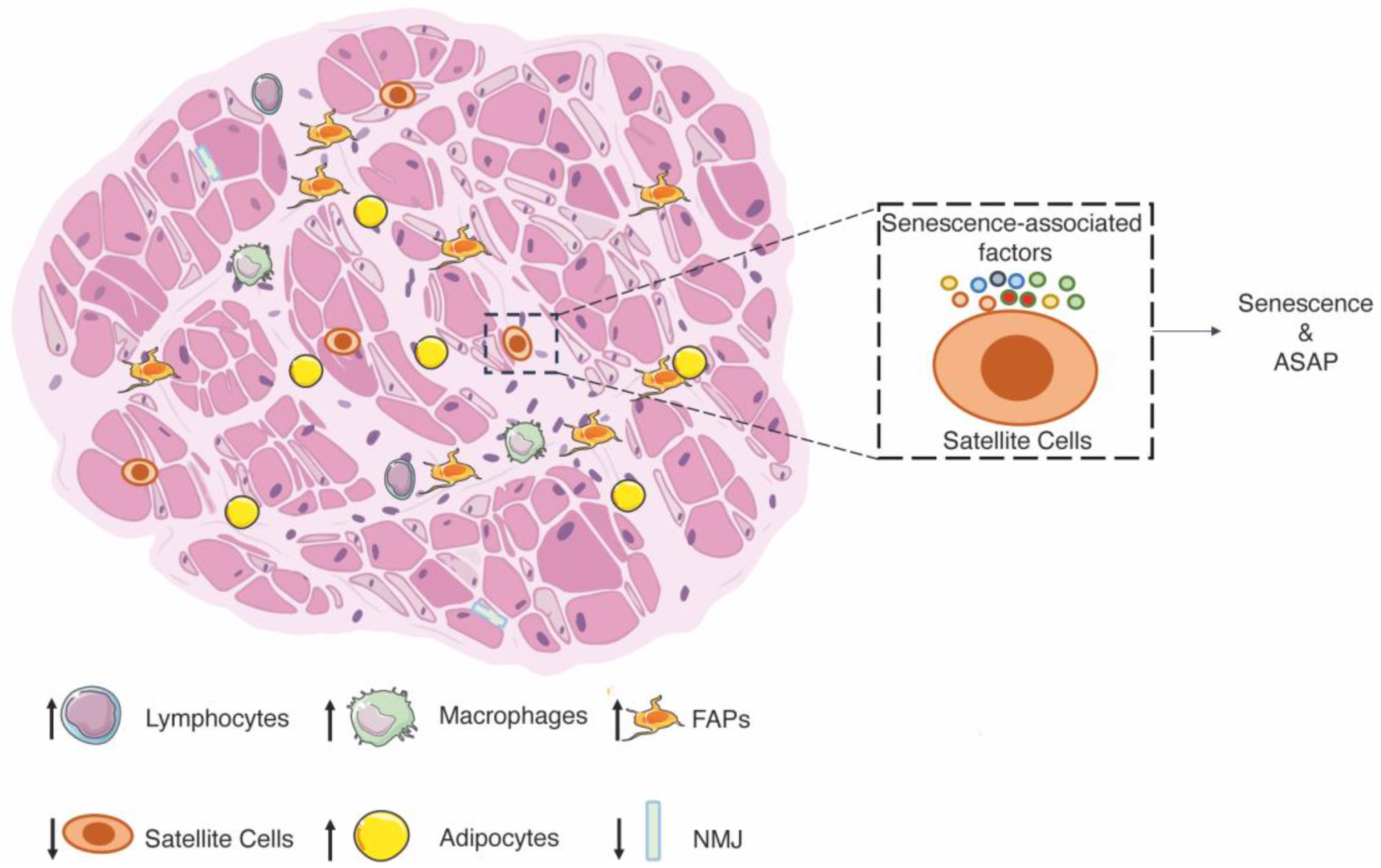
3.1. Age-Related Sarcopenia
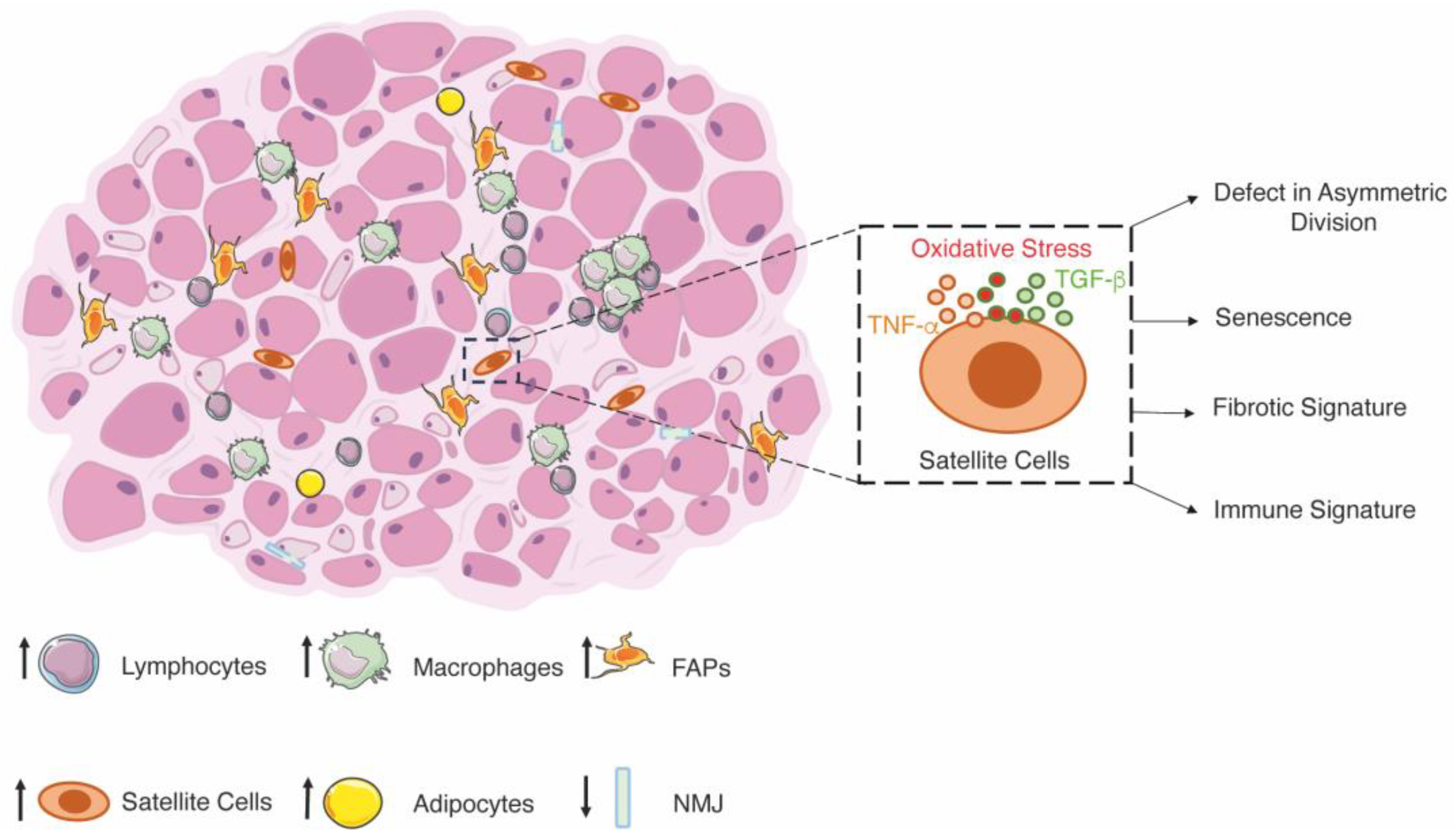
3.2. Duchenne Muscular Dystrophy
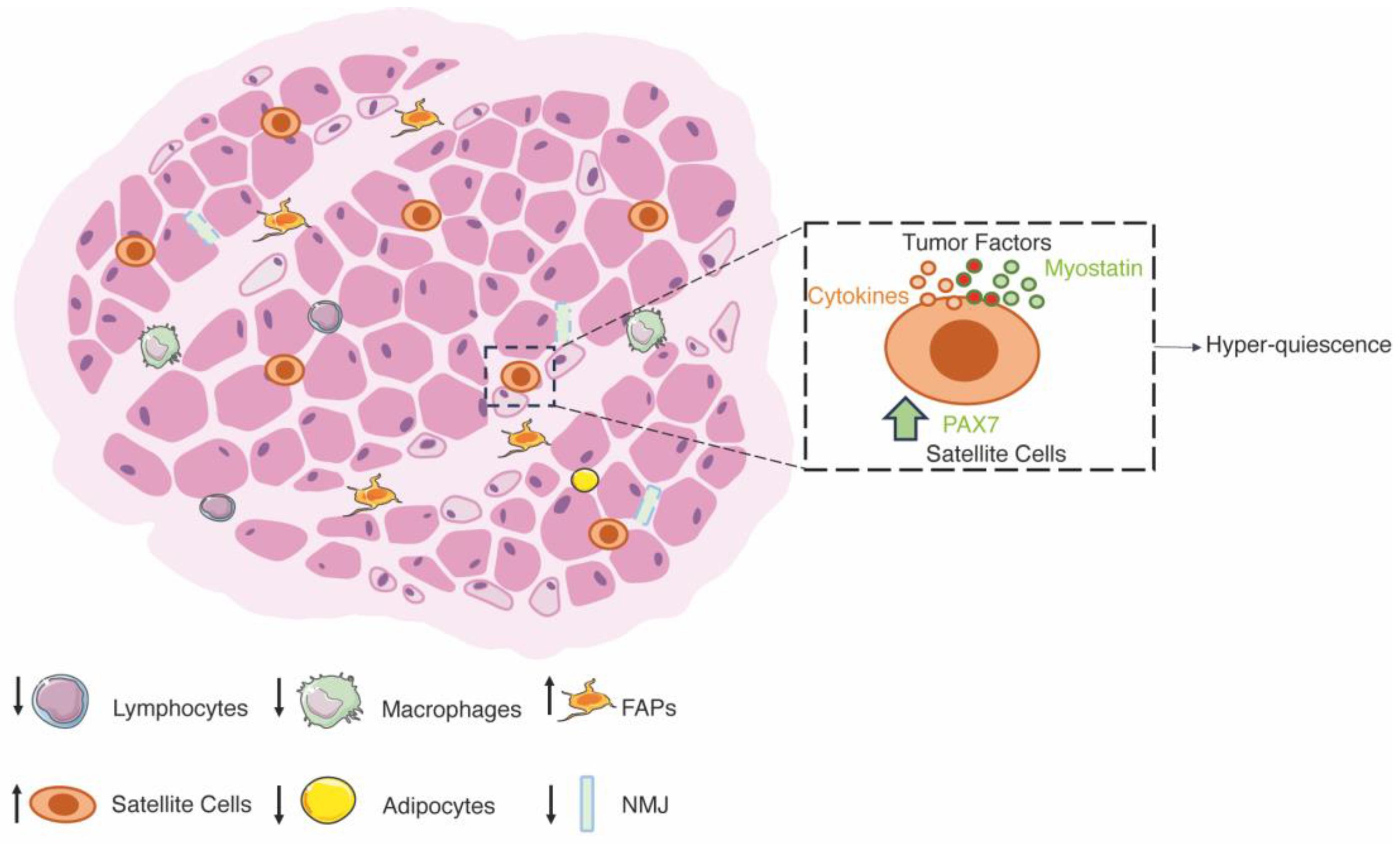
3.3. Cachexia
4. Summary and Future Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Bruusgaard, J.C.; Liestol, K.; Ekmark, M.; Kollstad, K.; Gundersen, K. Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J. Physiol. 2003, 551, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Roman, W.; Pinheiro, H.; Pimentel, M.R.; Segales, J.; Oliveira, L.M.; Garcia-Dominguez, E.; Gomez-Cabrera, M.C.; Serrano, A.L.; Gomes, E.R.; Munoz-Canoves, P. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science 2021, 374, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Kodippili, K.; Rudnicki, M.A. Satellite cell contribution to disease pathology in Duchenne muscular dystrophy. Front. Physiol. 2023, 14, 1180980. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Rudnicki, M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2011, 13, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Conboy, M.J.; Karasov, A.O.; Rando, T.A. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol. 2007, 5, e102. [Google Scholar] [CrossRef]
- Kuang, S.; Gillespie, M.A.; Rudnicki, M.A. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2008, 2, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Huo, F.; Liu, Q.; Liu, H. Contribution of muscle satellite cells to sarcopenia. Front. Physiol. 2022, 13, 892749. [Google Scholar] [CrossRef]
- Arneson, P.C.; Doles, J.D. Impaired Muscle Regeneration in Cancer-Associated Cachexia. Trends Cancer 2019, 5, 579–582. [Google Scholar] [CrossRef]
- Saleh, K.K.; Switzler, C.; Hicks, M.R.; Gane, L.; Gibbs, D.E.; Pyle, A.D. Duchenne muscular dystrophy disease severity impacts skeletal muscle progenitor cells systemic delivery. Front. Physiol. 2023, 14, 1190524. [Google Scholar] [CrossRef]
- Verdijk, L.B.; Koopman, R.; Schaart, G.; Meijer, K.; Savelberg, H.H.; van Loon, L.J. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E151–E157. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Webster, C.; Pavlath, G.K. Defective myoblasts identified in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 1983, 80, 4856–4860. [Google Scholar] [CrossRef] [PubMed]
- Decary, S.; Hamida, C.B.; Mouly, V.; Barbet, J.P.; Hentati, F.; Butler-Browne, G.S. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul. Disord. 2000, 10, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, S.; Wang, C.; Kuang, S. The hypoxia-inducible factors HIF1alpha and HIF2alpha are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J. Biol. Chem. 2017, 292, 5981–5991. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef]
- Cirillo, F.; Resmini, G.; Angelino, E.; Ferrara, M.; Tarantino, A.; Piccoli, M.; Rota, P.; Ghiroldi, A.; Monasky, M.M.; Ciconte, G.; et al. HIF-1alpha Directly Controls WNT7A Expression During Myogenesis. Front. Cell Dev. Biol. 2020, 8, 593508. [Google Scholar] [CrossRef]
- Cirillo, F.; Resmini, G.; Ghiroldi, A.; Piccoli, M.; Bergante, S.; Tettamanti, G.; Anastasia, L. Activation of the hypoxia-inducible factor 1alpha promotes myogenesis through the noncanonical Wnt pathway, leading to hypertrophic myotubes. FASEB J. 2017, 31, 2146–2156. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Bentzinger, C.F.; Rudnicki, M.A. Characteristics of Satellite Cells and Multipotent Adult Stem Cells in the Skeletal Muscle; Hayat, M.A., Ed.; Springer: Dordrecht, The Netherlands, 2013; Volume 12. [Google Scholar]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004, 18, 1088–1105. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, V.D.; Punch, V.G.; Kawabe, Y.; Jones, A.E.; Palidwor, G.A.; Porter, C.J.; Cross, J.W.; Carvajal, J.J.; Kockx, C.E.; van IJcken, W.F.; et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 2012, 22, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Sincennes, M.C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. STEM Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Sincennes, M.C.; Brun, C.E.; Lin, A.Y.T.; Rosembert, T.; Datzkiw, D.; Saber, J.; Ming, H.; Kawabe, Y.I.; Rudnicki, M.A. Acetylation of PAX7 controls muscle stem cell self-renewal and differentiation potential in mice. Nat. Commun. 2021, 12, 3253. [Google Scholar] [CrossRef]
- Rocheteau, P.; Gayraud-Morel, B.; Siegl-Cachedenier, I.; Blasco, M.A.; Tajbakhsh, S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 2012, 148, 112–125. [Google Scholar] [CrossRef]
- Shinin, V.; Gayraud-Morel, B.; Tajbakhsh, S. Template DNA-strand co-segregation and asymmetric cell division in skeletal muscle stem cells. Methods Mol. Biol. 2009, 482, 295–317. [Google Scholar] [CrossRef]
- Shinin, V.; Gayraud-Morel, B.; Gomes, D.; Tajbakhsh, S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat. Cell Biol. 2006, 8, 677–687. [Google Scholar] [CrossRef]
- Peng, J.; Han, L.; Liu, B.; Song, J.; Wang, Y.; Wang, K.; Guo, Q.; Liu, X.; Li, Y.; Zhang, J.; et al. Gli1 marks a sentinel muscle stem cell population for muscle regeneration. Nat. Commun. 2023, 14, 6993. [Google Scholar] [CrossRef]
- Palla, A.R.; Hilgendorf, K.I.; Yang, A.V.; Kerr, J.P.; Hinken, A.C.; Demeter, J.; Kraft, P.; Mooney, N.A.; Yucel, N.; Burns, D.M.; et al. Primary cilia on muscle stem cells are critical to maintain regenerative capacity and are lost during aging. Nat. Commun. 2022, 13, 1439. [Google Scholar] [CrossRef]
- Brun, C.E.; Sincennes, M.C.; Lin, A.Y.T.; Hall, D.; Jarassier, W.; Feige, P.; Le Grand, F.; Rudnicki, M.A. GLI3 regulates muscle stem cell entry into G(Alert) and self-renewal. Nat. Commun. 2022, 13, 3961. [Google Scholar] [CrossRef]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 2013, 280, 3991–4003. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Rudnicki, M.A. Satellite cells: The architects of skeletal muscle. Curr. Top. Dev. Biol. 2014, 107, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Yablonka-Reuveni, Z.; Rudnicki, M.A.; Rivera, A.J.; Primig, M.; Anderson, J.E.; Natanson, P. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev. Biol. 1999, 210, 440–455. [Google Scholar] [CrossRef] [PubMed]
- White, J.D.; Scaffidi, A.; Davies, M.; McGeachie, J.; Rudnicki, M.A.; Grounds, M.D. Myotube formation is delayed but not prevented in MyoD-deficient skeletal muscle: Studies in regenerating whole muscle grafts of adult mice. J. Histochem. Cytochem. 2000, 48, 1531–1544. [Google Scholar] [CrossRef]
- Sabourin, L.A.; Girgis-Gabardo, A.; Seale, P.; Asakura, A.; Rudnicki, M.A. Reduced differentiation potential of primary MyoD-/- myogenic cells derived from adult skeletal muscle. J. Cell Biol. 1999, 144, 631–643. [Google Scholar] [CrossRef]
- Fujita, R.; Mizuno, S.; Sadahiro, T.; Hayashi, T.; Sugasawa, T.; Sugiyama, F.; Ono, Y.; Takahashi, S.; Ieda, M. Generation of a MyoD knock-in reporter mouse line to study muscle stem cell dynamics and heterogeneity. iScience 2023, 26, 106592. [Google Scholar] [CrossRef]
- Weintraub, H.; Dwarki, V.J.; Verma, I.; Davis, R.; Hollenberg, S.; Snider, L.; Lassar, A.; Tapscott, S.J. Muscle-specific transcriptional activation by MyoD. Genes Dev. 1991, 5, 1377–1386. [Google Scholar] [CrossRef]
- Weintraub, H.; Davis, R.; Tapscott, S.; Thayer, M.; Krause, M.; Benezra, R.; Blackwell, T.K.; Turner, D.; Rupp, R.; Hollenberg, S.; et al. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science 1991, 251, 761–766. [Google Scholar] [CrossRef]
- Tapscott, S.J.; Weintraub, H. MyoD and the regulation of myogenesis by helix-loop-helix proteins. J. Clin. Investig. 1991, 87, 1133–1138. [Google Scholar] [CrossRef]
- Zhang, K.; Sha, J.; Harter, M.L. Activation of Cdc6 by MyoD is associated with the expansion of quiescent myogenic satellite cells. J. Cell Biol. 2010, 188, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Choi, S.; Liu, X.; Zhang, M.; Schageman, J.J.; Lee, S.Y.; Hart, R.; Lin, L.; Thurmond, F.A.; Williams, R.S. Highly coordinated gene regulation in mouse skeletal muscle regeneration. J. Biol. Chem. 2003, 278, 8826–8836. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. The Basis of Muscle Regeneration. Adv. Biol. 2014, 2014, 16. [Google Scholar] [CrossRef]
- Hernandez-Hernandez, J.M.; Garcia-Gonzalez, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. 1994, 77, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Edstrom, L.; Lindegren, B.; Gorza, L.; Schiaffino, S. MHC composition and enzyme-histochemical and physiological properties of a novel fast-twitch motor unit type. Am. J. Physiol. 1991, 261, C93–C101. [Google Scholar] [CrossRef]
- Jakubiec-Puka, A.; Kordowska, J.; Catani, C.; Carraro, U. Myosin heavy chain isoform composition in striated muscle after denervation and self-reinnervation. Eur. J. Biochem. 1990, 193, 623–628. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Konishi, C.T.; Perez Carbajal, E.E.; Wang, T.T.; Walsh, R.A.; Gan, Q.; Wagner, M.W.; Rando, T.A. Mesenchymal Stromal Cells Are Required for Regeneration and Homeostatic Maintenance of Skeletal Muscle. Cell Rep. 2019, 27, 2029–2035.e2025. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Migliavacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433–446.e437. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Li, Y.L. Inflammation balance in skeletal muscle damage and repair. Front. Immunol. 2023, 14, 1133355. [Google Scholar] [CrossRef] [PubMed]
- Tarban, N.; Papp, A.B.; Deak, D.; Szentesi, P.; Halasz, H.; Patsalos, A.; Csernoch, L.; Sarang, Z.; Szondy, Z. Loss of adenosine A3 receptors accelerates skeletal muscle regeneration in mice following cardiotoxin-induced injury. Cell Death Dis. 2023, 14, 706. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, A.; Kawashima, M.; Nagata, I.; Miyoshi, M.; Miyakawa, M.; Sugiyama, M.; Sakuraya, T.; Sonomura, T.; Arakawa, T. Icing after skeletal muscle injury decreases M1 macrophage accumulation and TNF-alpha expression during the early phase of muscle regeneration in rats. Histochem. Cell Biol. 2023, 159, 77–89. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Wu, Y.; Wang, L.; Wang, X.; Du, J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem. 2013, 288, 1489–1499. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-kappaB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 2011, 96, 85–119. [Google Scholar] [CrossRef]
- Chen, S.E.; Jin, B.; Li, Y.P. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol.-Cell Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef]
- Zhan, M.; Jin, B.; Chen, S.E.; Reecy, J.M.; Li, Y.P. TACE release of TNF-alpha mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J. Cell Sci. 2007, 120, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Xiao, J.; Wei, Y.; Li, S.; Liu, Y.; Yin, J.; Sun, K.; Sun, H.; Wang, H.; Zhang, Z.; et al. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015, 25, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Suttles, J. Functional plasticity of macrophages: Reversible adaptation to changing microenvironments. J. Leukoc. Biol. 2004, 76, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 2011, 20, 790–805. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 2007, 578, 327–336. [Google Scholar] [CrossRef]
- Fried, L.P.; Tangen, C.M.; Walston, J.; Newman, A.B.; Hirsch, C.; Gottdiener, J.; Seeman, T.; Tracy, R.; Kop, W.J.; Burke, G.; et al. Frailty in older adults: Evidence for a phenotype. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, M146–M156. [Google Scholar] [CrossRef] [PubMed]
- Clegg, A.; Young, J.; Iliffe, S.; Rikkert, M.O.; Rockwood, K. Frailty in elderly people. Lancet 2013, 381, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Sukkriang, N.; Punsawad, C. Comparison of geriatric assessment tools for frailty among community elderly. Heliyon 2020, 6, e04797. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Correa-de-Araujo, R.; Addison, O.; Miljkovic, I.; Goodpaster, B.H.; Bergman, B.C.; Clark, R.V.; Elena, J.W.; Esser, K.A.; Ferrucci, L.; Harris-Love, M.O.; et al. Myosteatosis in the Context of Skeletal Muscle Function Deficit: An Interdisciplinary Workshop at the National Institute on Aging. Front. Physiol. 2020, 11, 963. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Fleckenstein, J.; Schleip, R.; Hoppe, K.; Wearing, S.; Klingler, W. Structural and Functional Changes in the Coupling of Fascial Tissue, Skeletal Muscle, and Nerves During Aging. Front. Physiol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Ouchi, N.; Izumiya, Y.; Bernardo, B.L.; Lebrasseur, N.K.; Walsh, K. Glycolytic fast-twitch muscle fiber restoration counters adverse age-related changes in body composition and metabolism. Aging Cell 2014, 13, 80–91. [Google Scholar] [CrossRef]
- Roth, S.M.; Martel, G.F.; Ivey, F.M.; Lemmer, J.T.; Metter, E.J.; Hurley, B.F.; Rogers, M.A. Skeletal muscle satellite cell populations in healthy young and older men and women. Anat. Rec. 2000, 260, 351–358. [Google Scholar] [CrossRef]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 2010, 340, 330–343. [Google Scholar] [CrossRef]
- Renault, V.; Thornell, L.E.; Eriksson, P.O.; Butler-Browne, G.; Mouly, V. Regenerative potential of human skeletal muscle during aging. Aging Cell 2002, 1, 132–139. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Smythe, G.M.; Rando, T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science 2003, 302, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Van de Mark, D.P.; Richardson, J.B.; Yablonka-Reuveni, Z. Satellite-cell pool size does matter: Defining the myogenic potency of aging skeletal muscle. Dev. Biol. 2006, 294, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, J.V.; Jones, K.M.; Basson, M.A.; Brack, A.S. The aged niche disrupts muscle stem cell quiescence. Nature 2012, 490, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; Gutarra, S.; Garcia-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardi, M.; Ballestar, E.; Gonzalez, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Pont, A.R.; Sadri, N.; Hsiao, S.J.; Smith, S.; Schneider, R.J. mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol. Cell 2012, 47, 5–15. [Google Scholar] [CrossRef]
- Lavasani, M.; Robinson, A.R.; Lu, A.; Song, M.; Feduska, J.M.; Ahani, B.; Tilstra, J.S.; Feldman, C.H.; Robbins, P.D.; Niedernhofer, L.J.; et al. Muscle-derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat. Commun. 2012, 3, 608. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef]
- Beggs, M.L.; Nagarajan, R.; Taylor-Jones, J.M.; Nolen, G.; Macnicol, M.; Peterson, C.A. Alterations in the TGFbeta signaling pathway in myogenic progenitors with age. Aging Cell 2004, 3, 353–361. [Google Scholar] [CrossRef]
- Yarasheski, K.E.; Bhasin, S.; Sinha-Hikim, I.; Pak-Loduca, J.; Gonzalez-Cadavid, N.F. Serum myostatin-immunoreactive protein is increased in 60-92 year old women and men with muscle wasting. J. Nutr. Health Aging 2002, 6, 343–348. [Google Scholar] [PubMed]
- Scicchitano, B.M.; Rizzuto, E.; Musaro, A. Counteracting muscle wasting in aging and neuromuscular diseases: The critical role of IGF-1. Aging 2009, 1, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Shoturma, D.I.; Musaro, A.; Rosenthal, N.; Sweeney, H.L. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc. Natl. Acad. Sci. USA 1998, 95, 15603–15607. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H.; Skinhoj, P.; Pedersen, A.N.; Schroll, M.; Pedersen, B.K. Ageing, tumour necrosis factor-alpha (TNF-alpha) and atherosclerosis. Clin. Exp. Immunol. 2000, 121, 255–260. [Google Scholar] [CrossRef]
- Harris, T.B.; Ferrucci, L.; Tracy, R.P.; Corti, M.C.; Wacholder, S.; Ettinger, W.H., Jr.; Heimovitz, H.; Cohen, H.J.; Wallace, R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 1999, 106, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.R.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef]
- Lukjanenko, L.; Jung, M.J.; Hegde, N.; Perruisseau-Carrier, C.; Migliavacca, E.; Rozo, M.; Karaz, S.; Jacot, G.; Schmidt, M.; Li, L.; et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 2016, 22, 897–905. [Google Scholar] [CrossRef]
- Rozo, M.; Li, L.; Fan, C.M. Targeting beta1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat. Med. 2016, 22, 889–896. [Google Scholar] [CrossRef]
- Schuler, S.C.; Kirkpatrick, J.M.; Schmidt, M.; Santinha, D.; Koch, P.; Di Sanzo, S.; Cirri, E.; Hemberg, M.; Ori, A.; von Maltzahn, J. Extensive remodeling of the extracellular matrix during aging contributes to age-dependent impairments of muscle stem cell functionality. Cell Rep. 2021, 35, 109223. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fujita, H.; Seino, Y.; Hattori, S.; Hidaka, S.; Miyakawa, T.; Suzuki, A.; Waki, H.; Yabe, D.; Seino, Y.; et al. Gastric inhibitory polypeptide receptor antagonism suppresses intramuscular adipose tissue accumulation and ameliorates sarcopenia. J. Cachexia Sarcopenia Muscle 2023, 14, 2703–2718. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.J.; Vukovic, J.; Dunlop, S.; Grounds, M.D.; Shavlakadze, T. Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS ONE 2011, 6, e28090. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Ade, C.; Schmalbruch, H. Satellite cells and myonuclei in long-term denervated rat muscles. Anat. Rec. 1995, 243, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Jejurikar, S.S.; Marcelo, C.L.; Kuzon, W.M., Jr. Skeletal muscle denervation increases satellite cell susceptibility to apoptosis. Plast. Reconstr. Surg. 2002, 110, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Sandri, M.; Tessari, P. Anabolic Resistance in the Pathogenesis of Sarcopenia in the Elderly: Role of Nutrition and Exercise in Young and Old People. Nutrients 2023, 15, 4073. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.; Morley, J.E.; Cruz-Jentoft, A.J.; Arai, H.; Kritchevsky, S.B.; Guralnik, J.; Bauer, J.M.; Pahor, M.; Clark, B.C.; Cesari, M.; et al. International Clinical Practice Guidelines for Sarcopenia (ICFSR): Screening, Diagnosis and Management. J. Nutr. Health Aging 2018, 22, 1148–1161. [Google Scholar] [CrossRef]
- Borde, R.; Hortobagyi, T.; Granacher, U. Dose-Response Relationships of Resistance Training in Healthy Old Adults: A Systematic Review and Meta-Analysis. Sports Med. 2015, 45, 1693–1720. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Wakabayashi, H.; Yamada, M.; Kim, H.; Harada, A.; Arai, H. Interventions for Treating Sarcopenia: A Systematic Review and Meta-Analysis of Randomized Controlled Studies. J. Am. Med. Dir. Assoc. 2017, 18, 553.e1–553.e16. [Google Scholar] [CrossRef]
- Darr, K.C.; Schultz, E. Exercise-induced satellite cell activation in growing and mature skeletal muscle. J. Appl. Physiol. 1987, 63, 1816–1821. [Google Scholar] [CrossRef]
- Walker, D.K.; Fry, C.S.; Drummond, M.J.; Dickinson, J.M.; Timmerman, K.L.; Gundermann, D.M.; Jennings, K.; Volpi, E.; Rasmussen, B.B. PAX7+ satellite cells in young and older adults following resistance exercise. Muscle Nerve 2012, 46, 51–59. [Google Scholar] [CrossRef]
- Cisterna, B.; Giagnacovo, M.; Costanzo, M.; Fattoretti, P.; Zancanaro, C.; Pellicciari, C.; Malatesta, M. Adapted physical exercise enhances activation and differentiation potential of satellite cells in the skeletal muscle of old mice. J. Anat. 2016, 228, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Rauner, G.; Yablonka-Reuveni, Z.; Benayahu, D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS ONE 2010, 5, e13307. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Barton, E.R.; Sweeney, H.L.; Farrar, R.P. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J. Appl. Physiol. 2004, 96, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardi, M.; Munoz-Canoves, P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Guidelines on Physical Activity and Sedentary Behaviour; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Carter, J.C.; Sheehan, D.W.; Prochoroff, A.; Birnkrant, D.J. Muscular Dystrophies. Clin. Chest Med. 2018, 39, 377–389. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Monaco, A.P.; Feener, C.C.; Kunkel, L.M. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 1987, 238, 347–350. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy-Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Villa, C.; Tripodi, L.; Legato, M.; Torrente, Y. Role of Immunoglobulins in Muscular Dystrophies and Inflammatory Myopathies. Front. Immunol. 2021, 12, 666879. [Google Scholar] [CrossRef] [PubMed]
- Renjini, R.; Gayathri, N.; Nalini, A.; Srinivas Bharath, M.M. Oxidative damage in muscular dystrophy correlates with the severity of the pathology: Role of glutathione metabolism. Neurochem. Res. 2012, 37, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Careccia, G.; Saclier, M.; Tirone, M.; Ruggieri, E.; Principi, E.; Raffaghello, L.; Torchio, S.; Recchia, D.; Canepari, M.; Gorzanelli, A.; et al. Rebalancing expression of HMGB1 redox isoforms to counteract muscular dystrophy. Sci. Transl. Med. 2021, 13, eaay8416. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Forcina, L.; Nicoletti, C.; Scicchitano, B.M.; Musaro, A. Increased Circulating Levels of Interleukin-6 Induce Perturbation in Redox-Regulated Signaling Cascades in Muscle of Dystrophic Mice. Oxidative Med. Cell. Longev. 2017, 2017, 1987218. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.; Blau, H.M. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: Implications for cell and gene therapy. Somat. Cell Mol. Genet. 1990, 16, 557–565. [Google Scholar] [CrossRef]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef]
- Kottlors, M.; Kirschner, J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef]
- Bankole, L.C.; Feasson, L.; Ponsot, E.; Kadi, F. Fibre type-specific satellite cell content in two models of muscle disease. Histopathology 2013, 63, 826–832. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, M.R.F.; Mournetas, V.; Borczyk, M.; Verma, S.; Occhipinti, A.; Rog, J.; Bozycki, L.; Korostynski, M.; Robson, S.C.; Angione, C.; et al. Loss of full-length dystrophin expression results in major cell-autonomous abnormalities in proliferating myoblasts. Elife 2022, 11, e75521. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, F.; Santoso, J.W.; Rabichow, L.; Jin, R.; Little, I.; Nelson, S.F.; McCain, M.L.; Miceli, M.C. Modeling Patient-Specific Muscular Dystrophy Phenotypes and Therapeutic Responses in Reprogrammed Myotubes Engineered on Micromolded Gelatin Hydrogels. Front. Cell Dev. Biol. 2022, 10, 830415. [Google Scholar] [CrossRef] [PubMed]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Zimmerman, J.F.; Rao, J.; Sieiro, D.; McNamara, H.M.; Cherrier, T.; Rodriguez-delaRosa, A.; Hick-Colin, A.; Bousson, F.; Fugier-Schmucker, C.; et al. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell-derived skeletal muscle in vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2022960118. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef]
- Reimann, M.; Lee, S.; Loddenkemper, C.; Dorr, J.R.; Tabor, V.; Aichele, P.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell 2010, 17, 262–272. [Google Scholar] [CrossRef]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-beta1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214. [Google Scholar] [CrossRef]
- Allen, R.E.; Boxhorn, L.K. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J. Cell. Physiol. 1987, 133, 567–572. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, H.; Teramoto, N.; Nakamura, K.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Ogasawara, M.; Nishino, I.; Matsuwaki, T.; Nishihara, M.; et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 16385. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, V.; Cisneros, A.; Sica, V.; Deryagin, O.; Lai, Y.; Jung, S.; Andres, E.; An, J.; Segales, J.; Ortet, L.; et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 2023, 613, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Kefi, K.; Rivera, L.; Bergiers, O.; Cardone, N.; Coulpier, F.; Gioftsidi, S.; Drayton-Libotte, B.; Hou, C.; Authier, F.J.; et al. Thyroid-stimulating hormone receptor signaling restores skeletal muscle stem cell regeneration in rats with muscular dystrophy. Sci. Transl. Med. 2023, 15, eadd5275. [Google Scholar] [CrossRef] [PubMed]
- Biressi, S.; Miyabara, E.H.; Gopinath, S.D.; Carlig, P.M.; Rando, T.A. A Wnt-TGFbeta2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 2014, 6, 267ra176. [Google Scholar] [CrossRef] [PubMed]
- Pessina, P.; Kharraz, Y.; Jardi, M.; Fukada, S.; Serrano, A.L.; Perdiguero, E.; Munoz-Canoves, P. Fibrogenic Cell Plasticity Blunts Tissue Regeneration and Aggravates Muscular Dystrophy. Stem Cell Rep. 2015, 4, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Saleh, K.K.; Xi, H.; Switzler, C.; Skuratovsky, E.; Romero, M.A.; Chien, P.; Gibbs, D.; Gane, L.; Hicks, M.R.; Spencer, M.J.; et al. Single cell sequencing maps skeletal muscle cellular diversity as disease severity increases in dystrophic mouse models. iScience 2022, 25, 105415. [Google Scholar] [CrossRef]
- Hicks, M.R.; Saleh, K.K.; Clock, B.; Gibbs, D.E.; Yang, M.; Younesi, S.; Gane, L.; Gutierrez-Garcia, V.; Xi, H.; Pyle, A.D. Regenerating human skeletal muscle forms an emerging niche in vivo to support PAX7 cells. Nat. Cell Biol. 2023, 25, 1758–1773. [Google Scholar] [CrossRef]
- Wang, Y.X.; Feige, P.; Brun, C.E.; Hekmatnejad, B.; Dumont, N.A.; Renaud, J.M.; Faulkes, S.; Guindon, D.E.; Rudnicki, M.A. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell 2019, 24, 419–432.e416. [Google Scholar] [CrossRef]
- Ieko, T.; Fujiki, J.; Hasegawa, Y.; Iwasaki, T.; Iwano, H.; Maeda, N. Mechanism of skeletal muscle atrophy by muscle fiber types in male rats under long-term fasting stress. Steroids 2023, 200, 109328. [Google Scholar] [CrossRef]
- Priez, A.; Duchene, J.; Goubel, F. Duchenne muscular dystrophy quantification: A multivariate analysis of surface EMG. Med. Biol. Eng. Comput. 1992, 30, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Theroux, M.C.; Olivant, A.; Akins, R.E. C Histomorphology of neuromuscular junction in Duchenne muscular dystrophy. Pediatr. Anesth. 2008, 18, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.B.; Chamberlain, J.S.; Froehner, S.C. Truncated dystrophins can influence neuromuscular synapse structure. Mol. Cell. Neurosci. 2009, 40, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Pisani, C.; Strimpakos, G.; Gabanella, F.; Di Certo, M.G.; Onori, A.; Severini, C.; Luvisetto, S.; Farioli-Vecchioli, S.; Carrozzo, I.; Esposito, A.; et al. Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.J.P.; Shah, S.B.; Ward, C.W.; Inacio, M.P.; Stains, J.P.; Lovering, R.M. Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Physiol. 2013, 591, 559–570. [Google Scholar] [CrossRef]
- Pratt, S.J.P.; Shah, S.B.; Ward, C.W.; Kerr, J.P.; Stains, J.P.; Lovering, R.M. Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell. Mol. Life Sci. 2015, 72, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Kumar, A. TRAF6 regulates satellite stem cell self-renewal and function during regenerative myogenesis. J. Clin. Investig. 2016, 126, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Reano, S.; Angelino, E.; Ferrara, M.; Malacarne, V.; Sustova, H.; Sabry, O.; Agosti, E.; Clerici, S.; Ruozi, G.; Zentilin, L.; et al. Unacylated Ghrelin Enhances Satellite Cell Function and Relieves the Dystrophic Phenotype in Duchenne Muscular Dystrophy mdx Model. Stem Cells 2017, 35, 1733–1746. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef]
- Boyer, J.G.; Huo, J.; Han, S.; Havens, J.R.; Prasad, V.; Lin, B.L.; Kass, D.A.; Song, T.; Sadayappan, S.; Khairallah, R.J.; et al. Depletion of skeletal muscle satellite cells attenuates pathology in muscular dystrophy. Nat. Commun. 2022, 13, 2940. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Bonfanti, C.; Antonini, S.; Bastoni, M.; Monteverde, S.; Innocenzi, A.; Saclier, M.; Taglietti, V.; Messina, G. Silencing Nfix rescues muscular dystrophy by delaying muscle regeneration. Nat. Commun. 2017, 8, 1055. [Google Scholar] [CrossRef]
- Chen, G.; Wei, T.; Yang, H.; Li, G.; Li, H. CRISPR-Based Therapeutic Gene Editing for Duchenne Muscular Dystrophy: Advances, Challenges and Perspectives. Cells 2022, 11, 2964. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.B.; Ettyreddy, A.R.; Vankara, A.; Bohning, J.D.; Devlin, G.; Hauschka, S.D.; Asokan, A.; Gersbach, C.A. In Vivo Gene Editing of Muscle Stem Cells with Adeno-Associated Viral Vectors in a Mouse Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 19, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.E.; Shi, R.; Hakim, C.H.; Wasala, N.B.; Yue, Y.; Pan, X.; Zhang, T.; Robinson, C.A.; Duan, S.X.; Yao, G.; et al. AAV9 Edits Muscle Stem Cells in Normal and Dystrophic Adult Mice. Mol. Ther. 2019, 27, 1568–1585. [Google Scholar] [CrossRef] [PubMed]
- Domenig, S.A.; Bundschuh, N.; Lenardic, A.; Ghosh, A.; Kim, I.; Qabrati, X.; D’Hulst, G.; Bar-Nur, O. CRISPR/Cas9 editing of directly reprogrammed myogenic progenitors restores dystrophin expression in a mouse model of muscular dystrophy. Stem Cell Rep. 2022, 17, 321–336. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Keithley, J.K.; Swanson, B. HIV-associated wasting. J. Assoc. Nurses AIDS Care 2013, 24, S103–S111. [Google Scholar] [CrossRef]
- Cheung, W.W.; Paik, K.H.; Mak, R.H. Inflammation and cachexia in chronic kidney disease. Pediatr. Nephrol. 2010, 25, 711–724. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Cancer-Associated Cachexia: A Systemic Consequence of Cancer Progression. Annu. Rev. Cancer Biol. 2020, 4, 391–411. [Google Scholar] [CrossRef]
- Ferrara, M.; Samaden, M.; Ruggieri, E.; Venereau, E. Cancer cachexia as a multiorgan failure: Reconstruction of the crime scene. Front. Cell Dev. Biol. 2022, 10, 960341. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Lowry, S.F.; Cerami, A. Cachectin: A hormone that triggers acute shock and chronic cachexia. J. Infect. Dis. 1988, 157, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Narsale, A.A.; Carson, J.A. Role of interleukin-6 in cachexia: Therapeutic implications. Curr. Opin. Support. Palliat. Care 2014, 8, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zheng, H.; Zhou, Y.; Tang, X.; Yu, B.; Li, J. Association of IL-1beta gene polymorphism with cachexia from locally advanced gastric cancer. BMC Cancer 2007, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Llovera, M.; Carbo, N.; Garcia-Martinez, C.; Lopez-Sorianoq, F.J.; Argiles, J.M. Interleukin-1 receptor antagonist (IL-1ra) is unable to reverse cachexia in rats bearing an ascites hepatoma (Yoshida AH-130). Cancer Lett. 1995, 95, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Serrano, A.L.; Calabria, E.; Pallafacchina, G.; Lomo, T.; Schiaffino, S. Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat. Cell Biol. 2000, 2, 142–147. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef]
- Sartori, R.; Hagg, A.; Zampieri, S.; Armani, A.; Winbanks, C.E.; Viana, L.R.; Haidar, M.; Watt, K.I.; Qian, H.; Pezzini, C.; et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci. Transl. Med. 2021, 13, eaay9592. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Metzger, G.A.; He, W.A.; Guttridge, D.C. Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J. Cachexia Sarcopenia Muscle 2014, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Borisov, A.B.; Dedkov, E.I.; Carlson, B.M. Differentiation of activated satellite cells in denervated muscle following single fusions in situ and in cell culture. Histochem. Cell Biol. 2005, 124, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Donohue, M.; Buck, M. Decreased Jun-D and myogenin expression in muscle wasting of human cachexia. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E392–E401. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.M.; Nosacka, R.L.; Delitto, D.; Gerber, M.H.; Cameron, M.E.; Trevino, J.G.; Judge, A.R. Skeletal Muscle Fibrosis in Pancreatic Cancer Patients with Respect to Survival. JNCI Cancer Spectr. 2018, 2, pky043. [Google Scholar] [CrossRef] [PubMed]
- Banduseela, V.; Ochala, J.; Lamberg, K.; Kalimo, H.; Larsson, L. Muscle paralysis and myosin loss in a patient with cancer cachexia. Acta Myol. 2007, 26, 136–144. [Google Scholar]
- Mu, X.; Agarwal, R.; March, D.; Rothenberg, A.; Voigt, C.; Tebbets, J.; Huard, J.; Weiss, K. Notch Signaling Mediates Skeletal Muscle Atrophy in Cancer Cachexia Caused by Osteosarcoma. Sarcoma 2016, 2016, 3758162. [Google Scholar] [CrossRef]
- Nosacka, R.L.; Delitto, A.E.; Delitto, D.; Patel, R.; Judge, S.M.; Trevino, J.G.; Judge, A.R. Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J. Cachexia Sarcopenia Muscle 2020, 11, 820–837. [Google Scholar] [CrossRef]
- Berardi, E.; Aulino, P.; Murfuni, I.; Toschi, A.; Padula, F.; Scicchitano, B.M.; Coletti, D.; Adamo, S. Skeletal muscle is enriched in hematopoietic stem cells and not inflammatory cells in cachectic mice. Neurol. Res. 2008, 30, 160–169. [Google Scholar] [CrossRef]
- Inaba, S.; Hinohara, A.; Tachibana, M.; Tsujikawa, K.; Fukada, S.I. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS ONE 2018, 13, e0205467. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, D.; Duelen, R.; Penna, F.; Neumann, D.; Costelli, P.; Sampaolesi, M. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 2020, 11, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Neuparth, M.J.; Ascensao, A.; Magalhaes, J.; Vitorino, R.; Duarte, J.A.; Amado, F. Skeletal muscle atrophy increases cell proliferation in mice gastrocnemius during the first week of hindlimb suspension. Eur. J. Appl. Physiol. 2006, 97, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Olwin, B.B. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: A potential mechanism for self-renewal. Dev. Biol. 2004, 275, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Pisconti, A. Marking the tempo for myogenesis: Pax7 and the regulation of muscle stem cell fate decisions. J. Cell. Mol. Med. 2012, 16, 1013–1025. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Kuzon, W.M., Jr. Satellite cell depletion in degenerative skeletal muscle. Apoptosis 2003, 8, 573–578. [Google Scholar] [CrossRef]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Ballaro, R.; Penna, F.; Pin, F.; Gomez-Cabrera, M.C.; Vina, J.; Costelli, P. Moderate Exercise Improves Experimental Cancer Cachexia by Modulating the Redox Homeostasis. Cancers 2019, 11, 285. [Google Scholar] [CrossRef] [PubMed]
- Tsitkanou, S.; Murach, K.A.; Washington, T.A.; Greene, N.P. Exercise Counteracts the Deleterious Effects of Cancer Cachexia. Cancers 2022, 14, 2512. [Google Scholar] [CrossRef] [PubMed]
- Montero-Bullon, J.F.; Melo, T.; Ferreira, R.; Padrao, A.I.; Oliveira, P.A.; Domingues, M.R.M.; Domingues, P. Exercise training counteracts urothelial carcinoma-induced alterations in skeletal muscle mitochondria phospholipidome in an animal model. Sci. Rep. 2019, 9, 13423. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Sanchis-Gomar, F.; Martinez-Bello, V.; Gomez-Cabrera, M.C. Exercise acts as a drug; the pharmacological benefits of exercise. Br. J. Pharmacol. 2012, 167, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Davis, J.M.; Barrilleaux, T.L.; McClellan, J.L.; Steiner, J.L.; Carmichael, M.D.; Pena, M.M.; Hebert, J.R.; Green, J.E. Benefits of exercise training on breast cancer progression and inflammation in C3(1)SV40Tag mice. Cytokine 2011, 55, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Mader, T.; Chaillou, T.; Alves, E.S.; Jude, B.; Cheng, A.J.; Kenne, E.; Mijwel, S.; Kurzejamska, E.; Vincent, C.T.; Rundqvist, H.; et al. Exercise reduces intramuscular stress and counteracts muscle weakness in mice with breast cancer. J. Cachexia Sarcopenia Muscle 2022, 13, 1151–1163. [Google Scholar] [CrossRef]
- Solsona, R.; Pavlin, L.; Bernardi, H.; Sanchez, A.M. Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training. Int. J. Mol. Sci. 2021, 22, 2741. [Google Scholar] [CrossRef]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef]
- Hoffmann, C.; Weigert, C. Skeletal Muscle as an Endocrine Organ: The Role of Myokines in Exercise Adaptations. Cold Spring Harb. Perspect. Med. 2017, 7, a029793. [Google Scholar] [CrossRef]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- Ljubicic, V.; Miura, P.; Burt, M.; Boudreault, L.; Khogali, S.; Lunde, J.A.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 2011, 20, 3478–3493. [Google Scholar] [CrossRef] [PubMed]
- Kotelnikova, E.; Shkrob, M.A.; Pyatnitskiy, M.A.; Ferlini, A.; Daraselia, N. Novel approach to meta-analysis of microarray datasets reveals muscle remodeling-related drug targets and biomarkers in Duchenne muscular dystrophy. PLoS Comput. Biol. 2012, 8, e1002365. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Careccia, G.; Mangiavini, L.; Cirillo, F. Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2024, 25, 512. https://doi.org/10.3390/ijms25010512
Careccia G, Mangiavini L, Cirillo F. Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions. International Journal of Molecular Sciences. 2024; 25(1):512. https://doi.org/10.3390/ijms25010512
Chicago/Turabian StyleCareccia, Giorgia, Laura Mangiavini, and Federica Cirillo. 2024. "Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions" International Journal of Molecular Sciences 25, no. 1: 512. https://doi.org/10.3390/ijms25010512
APA StyleCareccia, G., Mangiavini, L., & Cirillo, F. (2024). Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions. International Journal of Molecular Sciences, 25(1), 512. https://doi.org/10.3390/ijms25010512