
3-O-Methyltolcapone and Its Lipophilic Analogues Are Potent Inhibitors of Transthyretin Amyloidogenesis with High Permeability and Low Toxicity

 , , , ,
, , , ,  , , ,
, , ,  ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Design of 3-OMT Lipophilic Analogues
2.2. In Silico Docking of 3-OMT Lipophilic Analogues
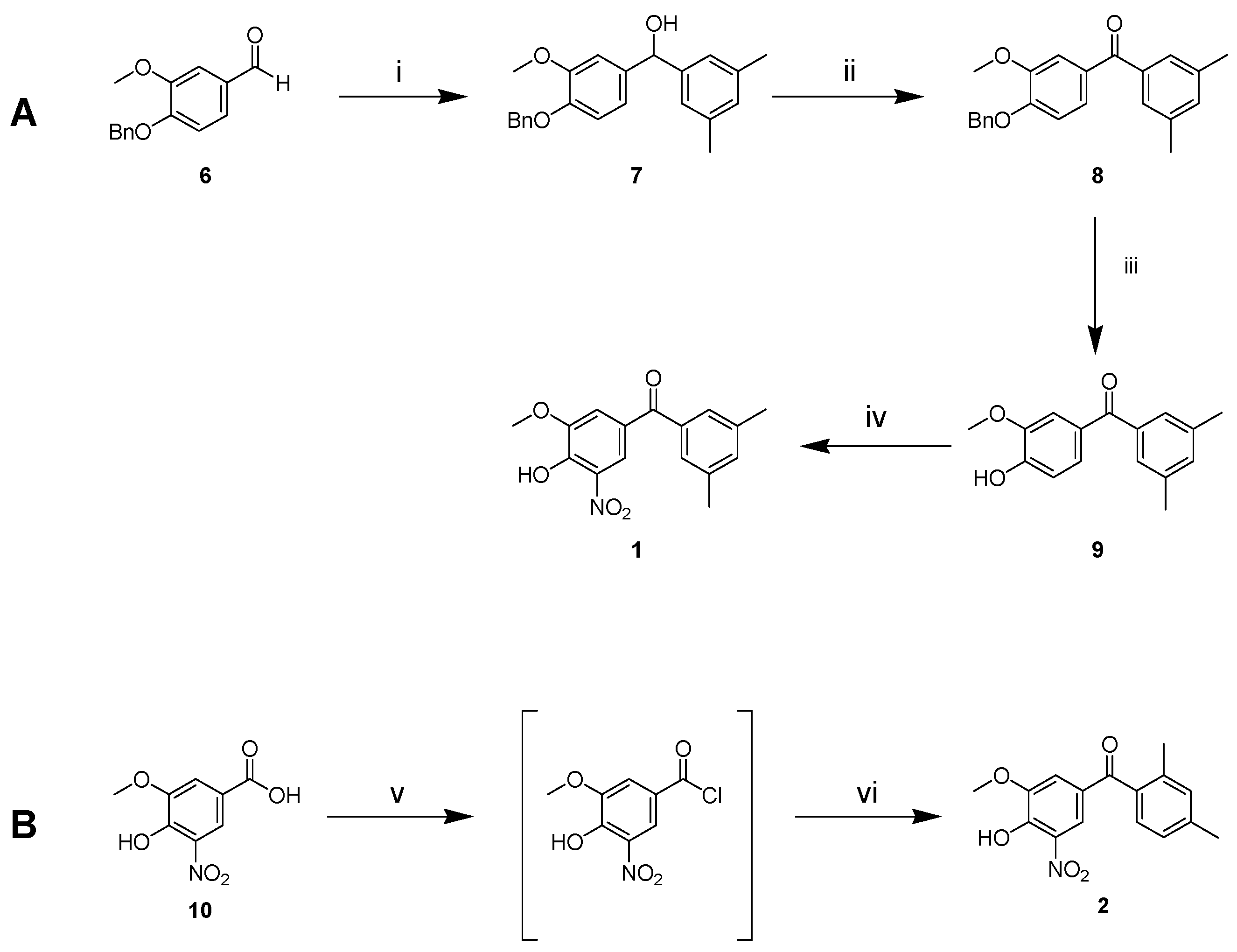
2.3. Synthesis of 1 and 2
2.4. Chromatographic Hydrophobicity Index (CHI)
2.5. 3-OMT and Its Lipophilic Analogues Have Reduced In Vitro Neuronal- and Hepato-Toxicity Compared to Tolcapone
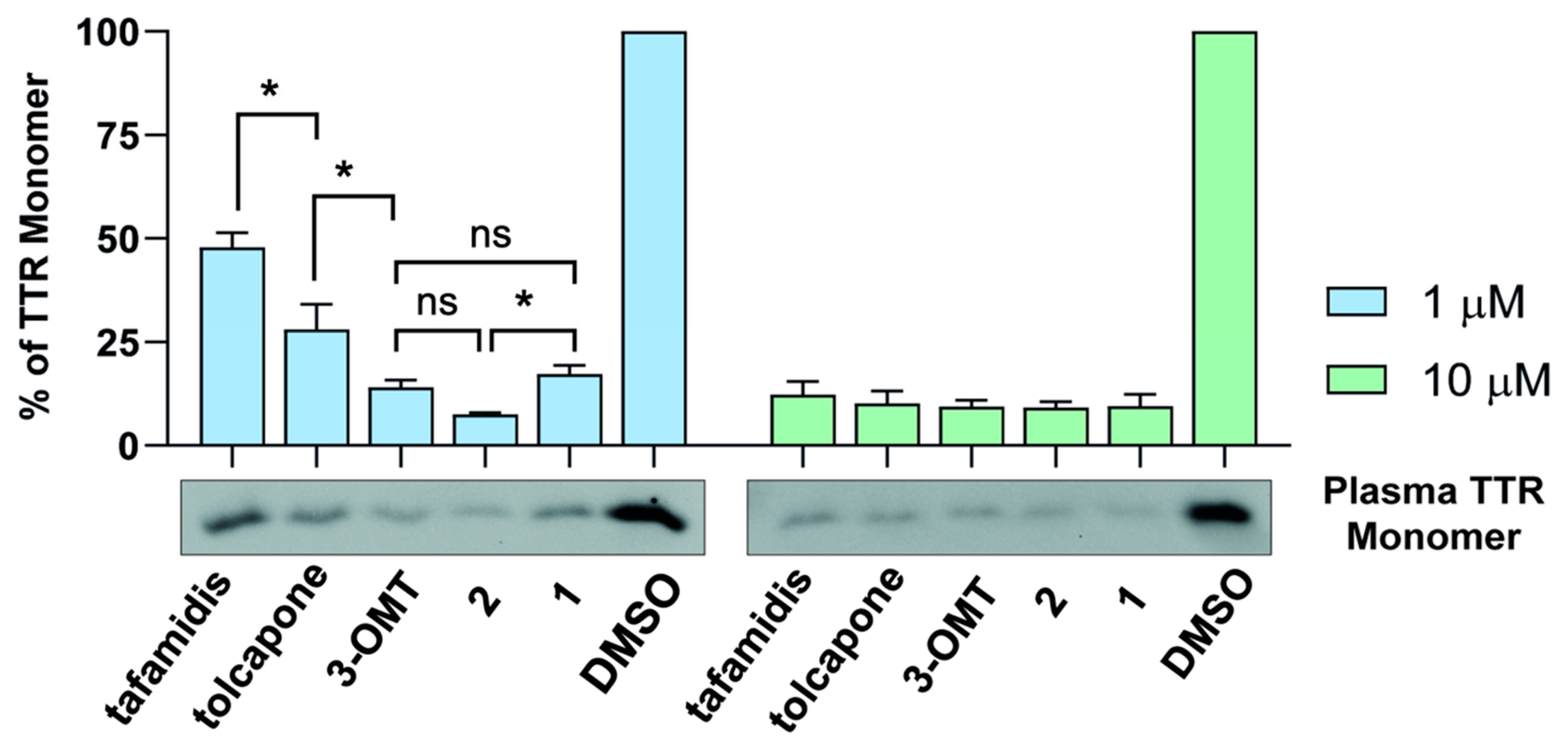
2.6. 3-OMT and Its Lipophilic Analogues Stabilise TTR in Human Plasma
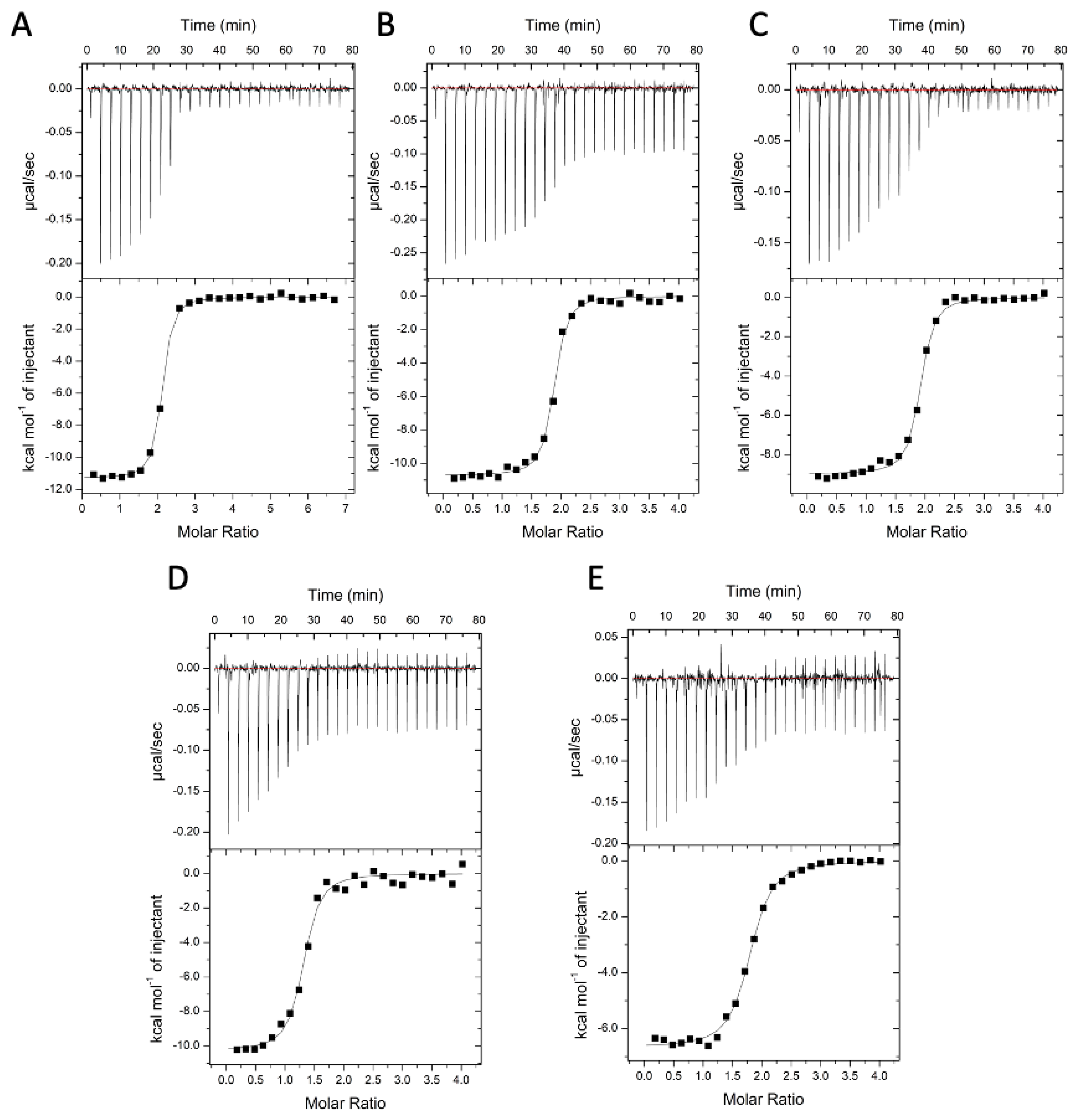
2.7. Binding Affinities and Thermodynamics of Interactions
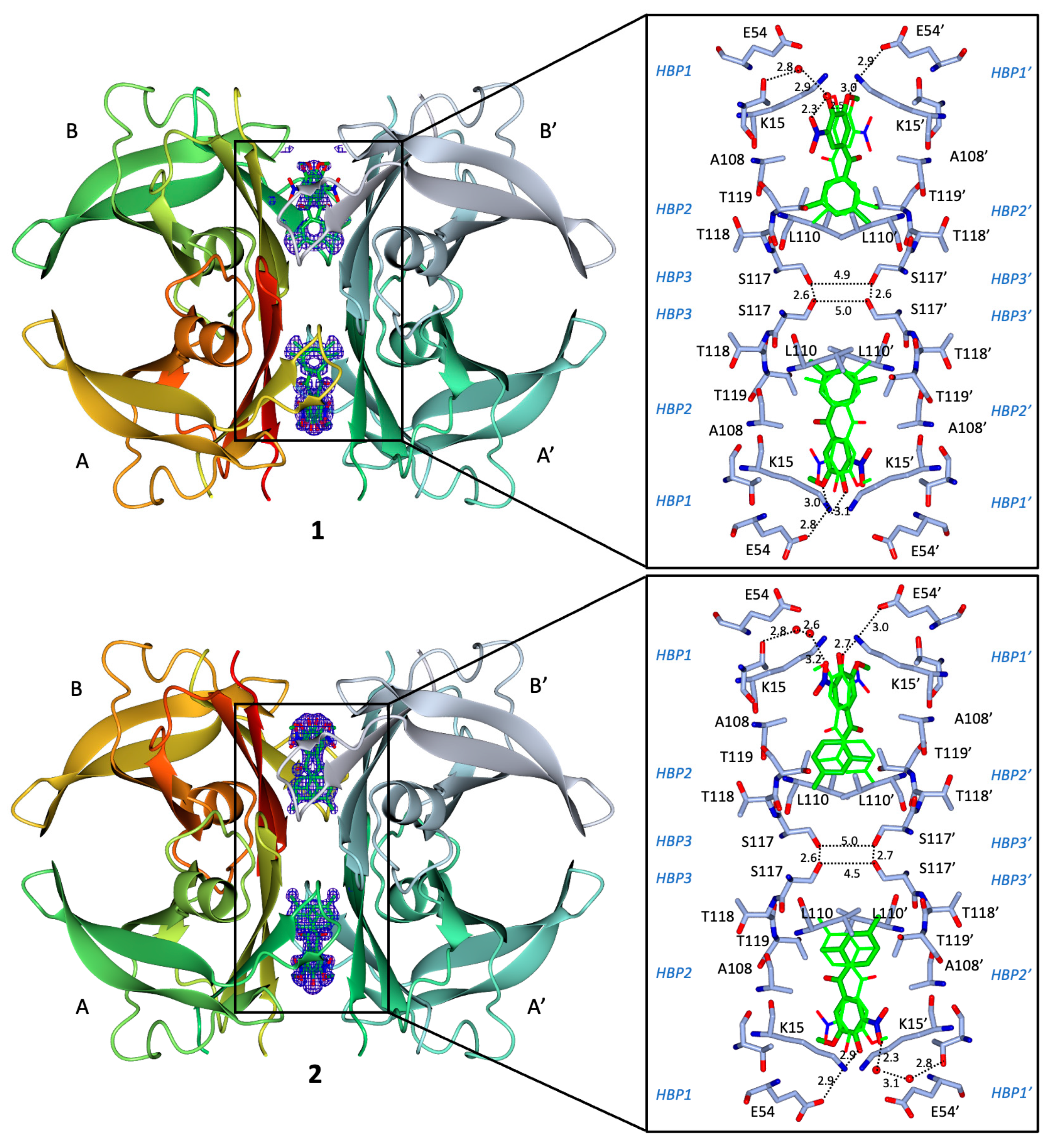
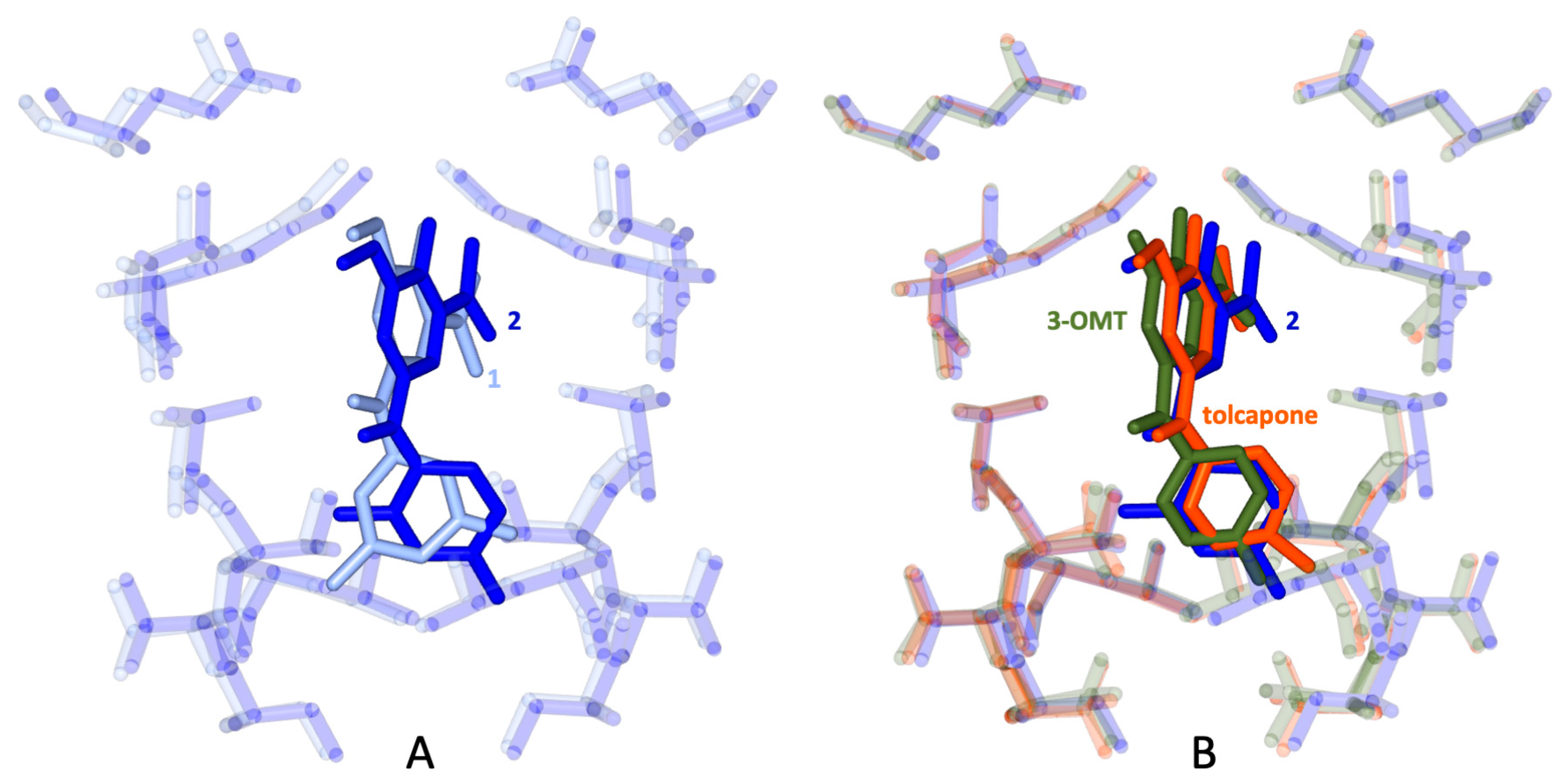
2.8. Binding Interactions between 1 and WT Human TTR
2.9. Binding Interactions between 2 and WT Human TTR
2.10. In Vitro Intestinal and Blood-Brain Barrier Permeability, and Solubility
2.11. Overall Performance of 3-OMT and Its Lipophilic Analogues in Stabilising Human Transthyretin
3. Conclusions
4. Experimental Section
4.1. In Silico Structural and Docking Studies
4.2. Chemistry: General
4.3. (4-(Benzyloxy)-3-methoxyphenyl)-1-(3,5-dimethylphenyl)methanol (7)
4.4. (4-(Benzyloxy)-3-methoxyphenyl)-1-(3,5-dimethylphenyl)methanone (8)
4.5. (4-Hydroxy-3-methoxyphenyl)-1-(3,5-dimethylphenyl)methanone (9)
4.6. (4-Hydroxy-3-methoxy-5-nitrophenyl)-1-(3,5-dimethylphenyl)methanone (1)
4.7. (4-Hydroxy-3-methoxy-5-nitrophenyl)-1-(2,4-dimethylphenyl)methanone (2)
4.8. Key Compounds Purity
4.9. Chromatographic Hydrophobicity Index (CHI)
4.10. Protein Expression and Purification
4.11. Solubility (Kinetic) and Blood Brain Barrier Permeability (PAMPA-BBB)
4.12. Caco-2 Intestinal Permeability
4.13. Cytotoxicity in Neuronal-Derived SH-SY5Y and Hepatic-Derived Hepg2 Human Cell Lines: (ATP Viability Assay)
4.14. Isothermal Titration Calorimetry (ITC)
4.15. Stability Studies of TTR in Serum by Immunoblotting
4.16. Crystallisation
4.17. Data Collection and Crystal Structure Determination
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zanotti, G.; Berni, R. Plasma Retinol-Binding Protein: Structure and Interactions with Retinol, Retinoids, and Transthyretin. Vitam. Horm. 2004, 69, 271–295. [Google Scholar] [PubMed]
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of Human Transthyretin Complexed with Thyroxine at 2.0 Å Resolution and 3′,5′-Dinitro-N-Acetyl-L-Thyronine at 2.2 Å Resolution. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Trovato, A.; Seno, F.; Folli, C.; Alfieri, B.; Zanotti, G.; Berni, R. Amyloidogenic Potential of Transthyretin Variants. J. Biol. Chem. 2009, 284, 25832–25841. [Google Scholar] [CrossRef] [PubMed]
- Galant, N.J.; Westermark, P.; Higaki, J.N.; Chakrabartty, A. Transthyretin Amyloidosis: An under-Recognized Neuropathy and Cardiomyopathy. Clin. Sci. 2017, 131, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the Treatment of Hereditary Transthyretin Amyloidosis: A Review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [PubMed]
- Connelly, S.; Choi, S.; Johnson, S.M.; Kelly, J.W.; Wilson, I.A. Structure-Based Design of Kinetic Stabilizers That Ameliorate the Transthyretin Amyloidoses. Curr. Opin. Struct. Biol. 2010, 20, 54–62. [Google Scholar] [CrossRef]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM Structure of a Transthyretin-Derived Amyloid Fibril from a Patient with Hereditary ATTR Amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef]
- Haupt, M.; Blakeley, M.P.; Fisher, S.J.; Mason, S.A.; Cooper, J.B.; Mitchell, E.P.; Forsyth, V.T. Binding Site Asymmetry in Human Transthyretin: Insights from a Joint Neutron and X-ray Crystallographic Analysis Using Perdeuterated Protein. IUCrJ 2014, 1, 429–438. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; de Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A Molecular Mechanism for Transthyretin Amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi Therapeutic for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- Buxbaum, J.N. Oligonucleotide Drugs for Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Bulawa, C.E.; Connelly, S.; De Vit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [PubMed]
- Penchala, S.C.; Connelly, S.; Wang, Y.; Park, M.S.; Zhao, L.; Baranczak, A.; Rappley, I.; Vogel, H.; Liedtke, M.; Witteles, R.M.; et al. AG10 Inhibits Amyloidogenesis and Cellular Toxicity of the Familial Amyloid Cardiomyopathy-Associated V122I Transthyretin. Proc. Natl. Acad. Sci. USA 2013, 110, 9992–9997. [Google Scholar] [CrossRef] [PubMed]
- Verona, G.; Mangione, P.P.; Raimondi, S.; Giorgetti, S.; Faravelli, G.; Porcari, R.; Corazza, A.; Gillmore, J.D.; Hawkins, P.N.; Pepys, M.B.; et al. Inhibition of the Mechano-Enzymatic Amyloidogenesis of Transthyretin: Role of Ligand Affinity, Binding Cooperativity and Occupancy of the Inner Channel. Sci. Rep. 2017, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Rosário Almeida, M.; et al. Repositioning Tolcapone as a Potent Inhibitor of Transthyretin Amyloidogenesis and Associated Cellular Toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef]
- Florio, P.; Folli, C.; Cianci, M.; del Rio, D.; Zanotti, G.; Berni, R. Transthyretin Binding Heterogeneity and Antiamyloidogenic Activity of Natural Polyphenols and Their Metabolites. J. Biol. Chem. 2015, 290, 29769–29780. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Nakagawa, Y.; Inui, K.; Katayama, W.; Sawai, Y.; Shimane, A.; Kitakami, R.; Okada, T.; Nabeshima, Y.; Yokoyama, T.; et al. Chlorinated Naringenin Analogues as Potential Inhibitors of Transthyretin Amyloidogenesis. J. Med. Chem. 2022, 65, 16218–16233. [Google Scholar] [CrossRef]
- Pinheiro, F.; Pallarès, I.; Peccati, F.; Sánchez-Morales, A.; Varejão, N.; Bezerra, F.; Ortega-Alarcon, D.; Gonzalez, D.; Osorio, M.; Navarro, S.; et al. Development of a Highly Potent Transthyretin Amyloidogenesis Inhibitor: Design, Synthesis, and Evaluation. J. Med. Chem. 2022, 65, 14673–14691. [Google Scholar] [CrossRef]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Natural Compounds as Inhibitors of Transthyretin Amyloidosis and Neuroprotective Agents: Analysis of Structural Data for Future Drug Design. J. Enzyme Inhib. Med. Chem. 2020, 35, 1145–1162. [Google Scholar] [CrossRef]
- Waddington Cruz, M.; Amass, L.; Keohane, D.; Schwartz, J.; Li, H.; Gundapaneni, B. Early Intervention with Tafamidis Provides Long-Term (5.5-Year) Delay of Neurologic Progression in Transthyretin Hereditary Amyloid Polyneuropathy. Amyloid 2016, 23, 178–183. [Google Scholar] [CrossRef]
- Keohane, D.; Schwartz, J.; Gundapaneni, B.; Stewart, M.; Amass, L. Tafamidis Delays Disease Progression in Patients with Early Stage Transthyretin Familial Amyloid Polyneuropathy: Additional Supportive Analyses from the Pivotal Trial. Amyloid 2017, 24, 30–36. [Google Scholar] [CrossRef]
- Sultan, M.B.; Gundapaneni, B.; Schumacher, J.; Schwartz, J.H. Treatment with Tafamidis Slows Disease Progression in Early-Stage Transthyretin Cardiomyopathy. Clin. Med. Insights Cardiol. 2017, 11, 117954681773032. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N.; Deeks, E.D. Tafamidis: A Review in Transthyretin Amyloidosis with Polyneuropathy. Drugs 2019, 79, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS—The Transthyretin Amyloidosis Outcomes Survey: Initial Report on Clinical Manifestations in Patients with Hereditary and Wild-Type Transthyretin Amyloidosis. Curr. Med. Res. Opin. 2013, 29, 63–76. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef]
- Hammarström, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-Dependent Denaturation Energetics: A Major Determinant in Amyloid Disease Diversity. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16427–16432. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Dendle, M.T.; Wiseman, R.L.; White, J.T.; D’haeze, W.; Kelly, J.W. R104H May Suppress Transthyretin Amyloidogenesis by Thermodynamic Stabilization, but Not by the Kinetic Mechanism Characterizing T119 Interallelic Trans-Suppression. Amyloid 2006, 13, 57–66. [Google Scholar] [CrossRef]
- Russ, H.; Müller, T.; Woitalla, D.; Rahbar, A.; Hahn, J.; Kuhn, W. Detection of Tolcapone in the Cerebrospinal Fluid of Parkinsonian Subjects. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 360, 719–720. [Google Scholar] [CrossRef]
- Pinheiro, F.; Varejão, N.; Esperante, S.; Santos, J.; Velázquez-Campoy, A.; Reverter, D.; Pallarès, I.; Ventura, S. Tolcapone, a Potent Aggregation Inhibitor for the Treatment of Familial Leptomeningeal Amyloidosis. FEBS J. 2021, 288, 310–324. [Google Scholar] [CrossRef]
- Maia, L.F.; Magalhães, R.; Freitas, J.; Taipa, R.; Pires, M.M.; Osório, H.; Dias, D.; Pessegueiro, H.; Correia, M.; Coelho, T. CNS Involvement in V30M Transthyretin Amyloidosis: Clinical, Neuropathological and Biochemical Findings. J. Neurol. Neurosurg. Psychiatry 2015, 86, 159–167. [Google Scholar] [CrossRef]
- Oshima, T.; Kawahara, S.; Ueda, M.; Kawakami, Y.; Tanaka, R.; Okazaki, T.; Misumi, Y.; Obayashi, K.; Yamashita, T.; Ohya, Y.; et al. Changes in Pathological and Biochemical Findings of Systemic Tissue Sites in Familial Amyloid Polyneuropathy More than 10 Years after Liver Transplantation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ando, Y.; Ueda, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Hayashi, M.; Yamamoto, M.; Mukai, E.; Nakamura, T.; et al. Distinct Characteristics of Amyloid Deposits in Early- and Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy. J. Neurol. Sci. 2009, 287, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary Transthyretin Amyloidosis: A Model of Medical Progress for a Fatal Disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Assal, F.; Spahr, L.; Hadengue, A.; Rubbia-Brandt, L.; Burkhard, P.R. Tolcapone and Fulminant Hepatitis. Lancet 1998, 352, 958. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W. Tolcapone and Hepatotoxic Effects. Arch. Neurol. 2000, 57, 263. [Google Scholar] [CrossRef] [PubMed]
- Spahr, L.; Rubbia-Brandt, L.; Burkhard, P.R.; Assal, F.; Hadengue, A. Tolcapone-Related Fulminant Hepatitis: Electron Microscopy Shows Mitochondrial Alterations. Dig. Dis. Sci. 2000, 45, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Jorga, K.; Fotteler, B.; Heizmann, P.; Gasser, R. Metabolism and Excretion of Tolcapone, a Novel Inhibitor of Catechol-O-Methyltransferase. Br. J. Clin. Pharmacol. 1999, 48, 513–520. [Google Scholar] [CrossRef]
- Bethesda, M.D. National Institute of Diabetes and Digestive and Kidney Diseases: Tolcapone. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548573/ (accessed on 25 October 2021).
- Loconte, V.; Cianci, M.; Menozzi, I.; Sbravati, D.; Sansone, F.; Casnati, A.; Berni, R. Interactions of Tolcapone Analogues as Stabilizers of the Amyloidogenic Protein Transthyretin. Bioorg Chem. 2020, 103, 104144. [Google Scholar] [CrossRef]
- Hörnberg, A.; Eneqvist, T.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, A.E. A Comparative Analysis of 23 Structures of the Amyloidogenic Protein Transthyretin. J. Mol. Biol. 2000, 302, 649–669. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Chapter 10—Blood-Brain Barrier. In Drug-Like Properties, 2nd ed.; Di, L., Kerns, E.H., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 141–159. ISBN 978-0-12-801076-1. [Google Scholar]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, Virtual High Throughput Screening and in Silico Fragment-Based Drug Design. J. Cell Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef]
- Kim, B.; Park, H.; Lee, S.K.; Park, S.J.; Koo, T.-S.; Kang, N.S.; Hong, K.B.; Choi, S. Systemic Optimization and Structural Evaluation of Quinoline Derivatives as Transthyretin Amyloidogenesis Inhibitors. Eur. J. Med. Chem. 2016, 123, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Simões, C.J.V.; Almeida, Z.L.; Costa, D.; Jesus, C.S.H.; Cardoso, A.L.; Almeida, M.R.; Saraiva, M.J.; Pinho e Melo, T.M.V.D.; Brito, R.M.M. A Novel Bis-Furan Scaffold for Transthyretin Stabilization and Amyloid Inhibition. Eur. J. Med. Chem. 2016, 121, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, E.Y.; Santos, L.M.; Rivas, J.; Blasi, D.; Leite, J.P.; Liz, M.A.; Busquets, M.A.; Planas, A.; Prohens, R.; Gimeno, A.; et al. Targeting Transthyretin in Alzheimer’s Disease: Drug Discovery of Small-Molecule Chaperones as Disease-Modifying Drug Candidates for Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 226, 113847. [Google Scholar] [CrossRef] [PubMed]
- Loconte, V.; Menozzi, I.; Ferrari, A.; Folli, C.; Imbimbo, B.P.; Zanotti, G.; Berni, R. Structure-Activity Relationships of Flurbiprofen Analogues as Stabilizers of the Amyloidogenic Protein Transthyretin. J. Struct. Biol. 2019, 208, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Learmonth, D.A.; Vieira-Coelho, M.A.; Benes, J.; Alves, P.C.; Borges, N.; Freitas, A.P.; Soares-da-Silva, P. Synthesis of 1-(3,4-Dihydroxy-5-Nitrophenyl)-2-Phenyl-Ethanone and Derivatives as Potent and Long-Acting Peripheral Inhibitors of Catechol-O-Methyltransferase. J. Med. Chem. 2002, 45, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Ferreira, H.S.; Torrão, L.; Bonifácio, M.J.; Palma, P.N.; Soares-Da-Silva, P.; Learmonth, D.A. Discovery of a Long-Acting, Peripherally Selective Inhibitor of Catechol-O-Methyltransferase. J. Med. Chem. 2010, 53, 3396–3411. [Google Scholar] [CrossRef] [PubMed]
- Valko, K. Physicochemical and Biomimetic Properties in Drug Discovery: Chromatographic Techniques for Lead Optimization; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Korlipara, L.P.; Cooper, J.M.; Schapira, A.H. Differences in Toxicity of the Catechol-O-Methyl Transferase Inhibitors, Tolcapone and Entacapone to Cultured Human Neuroblastoma Cells. Neuropharmacology 2004, 46, 562–569. [Google Scholar] [CrossRef]
- Maser, T.; Rich, M.; Hayes, D.; Zhao, P.; Nagulapally, A.B.; Bond, J.; Saulnier Sholler, G. Tolcapone Induces Oxidative Stress Leading to Apoptosis and Inhibition of Tumor Growth in Neuroblastoma. Cancer Med. 2017, 6, 1341–1352. [Google Scholar] [CrossRef]
- Miller, M.; Pal, A.; Albusairi, W.; Joo, H.; Pappas, B.; Haque Tuhin, M.T.; Liang, D.; Jampala, R.; Liu, F.; Khan, J.; et al. Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis. J. Med. Chem. 2018, 61, 7862–7876. [Google Scholar] [CrossRef]
- Iakovleva, I.; Brännström, K.; Nilsson, L.; Gharibyan, A.L.; Begum, A.; Anan, I.; Walfridsson, M.; Sauer-Eriksson, A.E.; Olofsson, A. Enthalpic Forces Correlate with the Selectivity of Transthyretin-Stabilizing Ligands in Human Plasma. J. Med. Chem. 2015, 58, 6507–6515. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High Throughput Artificial Membrane Permeability Assay for Blood–Brain Barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.N.; Martínez-González, L.; Pérez, D.I.; Martínez, A.; Valente, M.J.; Garrido, J.; Uriarte, E.; et al. Development of Blood–Brain Barrier Permeable Nitrocatechol-Based Catechol O-Methyltransferase Inhibitors with Reduced Potential for Hepatotoxicity. J. Med. Chem. 2016, 59, 7584–7597. [Google Scholar] [CrossRef] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Soares-da-Silva, P.; Falcão, A. A New PAMPA Model Using an In-House Brain Lipid Extract for Screening the Blood–Brain Barrier Permeability of Drug Candidates. Int. J. Pharm. 2016, 501, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Freire, E. Finding a Better Path to Drug Selectivity. Drug Discov. Today 2011, 16, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Sebastião, M.P.; Lamzin, V.; Saraiva, M.J.; Damas, A.M. Transthyretin Stability as a Key Factor in Amyloidogenesis: X-ray Analysis at Atomic Resolution. J. Mol. Biol. 2001, 306, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wei, C.; Piao, Y.; Lian, F.; Wu, H.; Zhou, A.; Wang, F.; Zuo, X.; Han, Y.; Lyu, J.; et al. Current Review of Leptomeningeal Amyloidosis Associated with Transthyretin Mutations. Neurologist 2021, 26, 189–195. [Google Scholar] [CrossRef]
- McColgan, P.; Viegas, S.; Gandhi, S.; Bull, K.; Tudor, R.; Sheikh, F.; Pinney, J.; Fontana, M.; Rowczenio, D.; Gillmore, J.D.; et al. Oculoleptomeningeal Amyloidosis Associated with Transthyretin Leu12Pro in an African Patient. J. Neurol. 2015, 262, 228–234. [Google Scholar] [CrossRef]
- Sinha, A.; Chang, J.C.; Xu, P.; Gindinova, K.; Cho, Y.; Sun, W.; Wu, X.; Li, Y.M.; Greengard, P.; Kelly, J.W.; et al. Brain Permeable Tafamidis Amide Analogs for Stabilizing TTR and Reducing APP Cleavage. ACS Med. Chem. Lett. 2020, 11, 1973–1979. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast Docking Using the CHARMM Force Field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Haberthür, U.; Caflisch, A. FACTS: Fast Analytical Continuum Treatment of Solvation. J. Comput. Chem. 2008, 29, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mu, Y.; Dong, H.; Yan, H.; Hao, C.; Wang, X.; Zhang, L. Chemical Constituents of the Ethyl Acetate Extract from Diaphragma Juglandis Fructus and Their Inhibitory Activity on Nitric Oxide Production In Vitro. Molecules 2018, 23, 72. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Larsson, A.; Begum, A.; Iakovleva, I.; Carlsson, M.; Brännström, K.; Sauer-Eriksson, A.E.; Olofsson, A. Modifications of the 7-Hydroxyl Group of the Transthyretin Ligand Luteolin Provide Mechanistic Insights into Its Binding Properties and High Plasma Specificity. PLoS ONE 2016, 11, e0153112. [Google Scholar] [CrossRef] [PubMed]
- Cianci, M.; Bourenkov, G.; Pompidor, G.; Karpics, I.; Kallio, J.; Bento, I.; Roessle, M.; Cipriani, F.; Fiedler, S.; Schneider, T.R. P13, the EMBL Macromolecular Crystallography Beamline at the Low-Emittance PETRA III Ring for High- and Low-Energy Phasing with Variable Beam Focusing. J. Synchrotron Radiat. 2017, 24, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and Assessment of Data Quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Keegan, R.M.; Winn, M.D. MrBUMP: An Automated Pipeline for Molecular Replacement. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 119–124. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHI (pH 2.0) | CHI (pH 7.4) | CHI (pH 10.5) | |
|---|---|---|---|
| tolcapone | 78.2 | n.d. | 49.3 |
| 3-OMT | 89.2 | 50.2 | 46.0 |
| 1 | 97.8 | 55.5 | 51.7 |
| 2 | 96.1 | 53.8 | 50.2 |
| Compound | SH-SY5Y EC50 (µM) | HepG2 EC50 (µM) |
|---|---|---|
| tolcapone | 29.8 ± 1.1 | 17.5 ± 2.4 |
| 3-OMT | 226.3 ± 1.3 | 262.3 ± 20.7 |
| 1 | 172.1 ± 5 | 410 ± 33 |
| 2 | 174.4 ± 3.7 | 215.6 ± 2.2 |
| Ligand | n | Kd (nM) | ∆H (kcal mol−1) | ∆S (kcal mol−1 T−1) |
|---|---|---|---|---|
| tafamidis | 2.4 ± 0.1 | 128 ± 63 | −3.2 ± 0.3 | 20.9 ± 2.1 |
| tolcapone | 2.0 ± 0.1 | 26 ± 4 | −11.8 ± 0.5 | −4.8 ± 1.9 |
| 3-OMT | 1.9 ± 0.2 | 33 ± 9 | −8.9 ± 0.3 | 4.6 ± 0.5 |
| 1 | 1.8 ± 0.1 | 71 ± 26 | −11.5 ± 1.3 | −2.9 ± 0.9 |
| 2 | 2.1 ± 0.3 | 25 ± 5 | −10.5 ± 0.2 | −0.4 ± 0.1 |
| Compound | Direction/±Inhibitor | Papp (nm/s) [Expected Values] | Recovery % | Efflux Ratio (Reduction %) |
|---|---|---|---|---|
| Atenolol † | A→B | <10 [<10] | 86 ± 1 | |
| Metoprolol † | A→B | >400 [>400] | 105 ± 5 | |
| Digoxin † (P-gp substrate) | A→B | 2 ± 1 [2] | 67 ± 5 | |
| B→A | 141 ± 26 [139] | 71 ± 6 | >10 | |
| A→B + EL * 2 µM | 35 ± 5 [26] | 72 ± 1 | ||
| B→A + EL * 2 µM | 83 ± 31 [63] | 75 ± 3 | 2.3 (96%) | |
| tolcapone | A→B | 101 ± 12 | 53 ± 1 | 2 |
| B→A | 206 ± 58 | 70 ± 4 | ||
| 3-OMT | A→B | 160 ± 29 | 66 ± 4 | 1.5 |
| B→A | 243 ± 30 | 83 ± 8 | ||
| 1 | A→B | 240 ± 21 | 64 ± 4 | 1 |
| B→A | 231 ± 23 | 77 ± 2 | ||
| 2 | A→B | 250 ± 3 | 69 ± 8 | 1 |
| B→A | 268 ± 25 | 80 ± 9 |
| Compound | BBB-Pe (10−6 cm/s) [Expected Values] | 1-Rm | CNS Class [Expected Values] | Solubility (µM) |
|---|---|---|---|---|
| Verapamil † | 12.0 (±2.4) [>10] | 0.8 | CNS+ [CNS+] | >500 |
| Caffeine † | 1.9 (±0.1) [1.3] | 0.9 | CNS− [CNS−] | >500 |
| Theophylline † | 0.2 (±0.1) [0.12] | 1.0 | CNS− [CNS−] | >500 |
| tolcapone | 5.6 (±1.2) | 0.9 | CNS+ | >500 |
| 3-OMT | 14.4 (±0.2) | 0.9 | CNS+ | >500 |
| 1 | 15.3 (±2.0) | 0.9 | CNS+ | >500 |
| 2 | 13.5 (±3.9) | 1.0 | CNS+ | >500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poonsiri, T.; Dell’Accantera, D.; Loconte, V.; Casnati, A.; Cervoni, L.; Arcovito, A.; Benini, S.; Ferrari, A.; Cipolloni, M.; Cacioni, E.; et al. 3-O-Methyltolcapone and Its Lipophilic Analogues Are Potent Inhibitors of Transthyretin Amyloidogenesis with High Permeability and Low Toxicity. Int. J. Mol. Sci. 2024, 25, 479. https://doi.org/10.3390/ijms25010479
Poonsiri T, Dell’Accantera D, Loconte V, Casnati A, Cervoni L, Arcovito A, Benini S, Ferrari A, Cipolloni M, Cacioni E, et al. 3-O-Methyltolcapone and Its Lipophilic Analogues Are Potent Inhibitors of Transthyretin Amyloidogenesis with High Permeability and Low Toxicity. International Journal of Molecular Sciences. 2024; 25(1):479. https://doi.org/10.3390/ijms25010479
Chicago/Turabian StylePoonsiri, Thanalai, Davide Dell’Accantera, Valentina Loconte, Alessandro Casnati, Laura Cervoni, Alessandro Arcovito, Stefano Benini, Alberto Ferrari, Marco Cipolloni, Elisa Cacioni, and et al. 2024. "3-O-Methyltolcapone and Its Lipophilic Analogues Are Potent Inhibitors of Transthyretin Amyloidogenesis with High Permeability and Low Toxicity" International Journal of Molecular Sciences 25, no. 1: 479. https://doi.org/10.3390/ijms25010479
APA StylePoonsiri, T., Dell’Accantera, D., Loconte, V., Casnati, A., Cervoni, L., Arcovito, A., Benini, S., Ferrari, A., Cipolloni, M., Cacioni, E., De Franco, F., Giacchè, N., Rinaldo, S., Folli, C., Sansone, F., Berni, R., & Cianci, M. (2024). 3-O-Methyltolcapone and Its Lipophilic Analogues Are Potent Inhibitors of Transthyretin Amyloidogenesis with High Permeability and Low Toxicity. International Journal of Molecular Sciences, 25(1), 479. https://doi.org/10.3390/ijms25010479