
To Gain Insights into the Pathophysiological Mechanisms of the Thrombo-Inflammatory Process in the Atherosclerotic Plaque

Abstract
:1. Introduction
Search Strategy
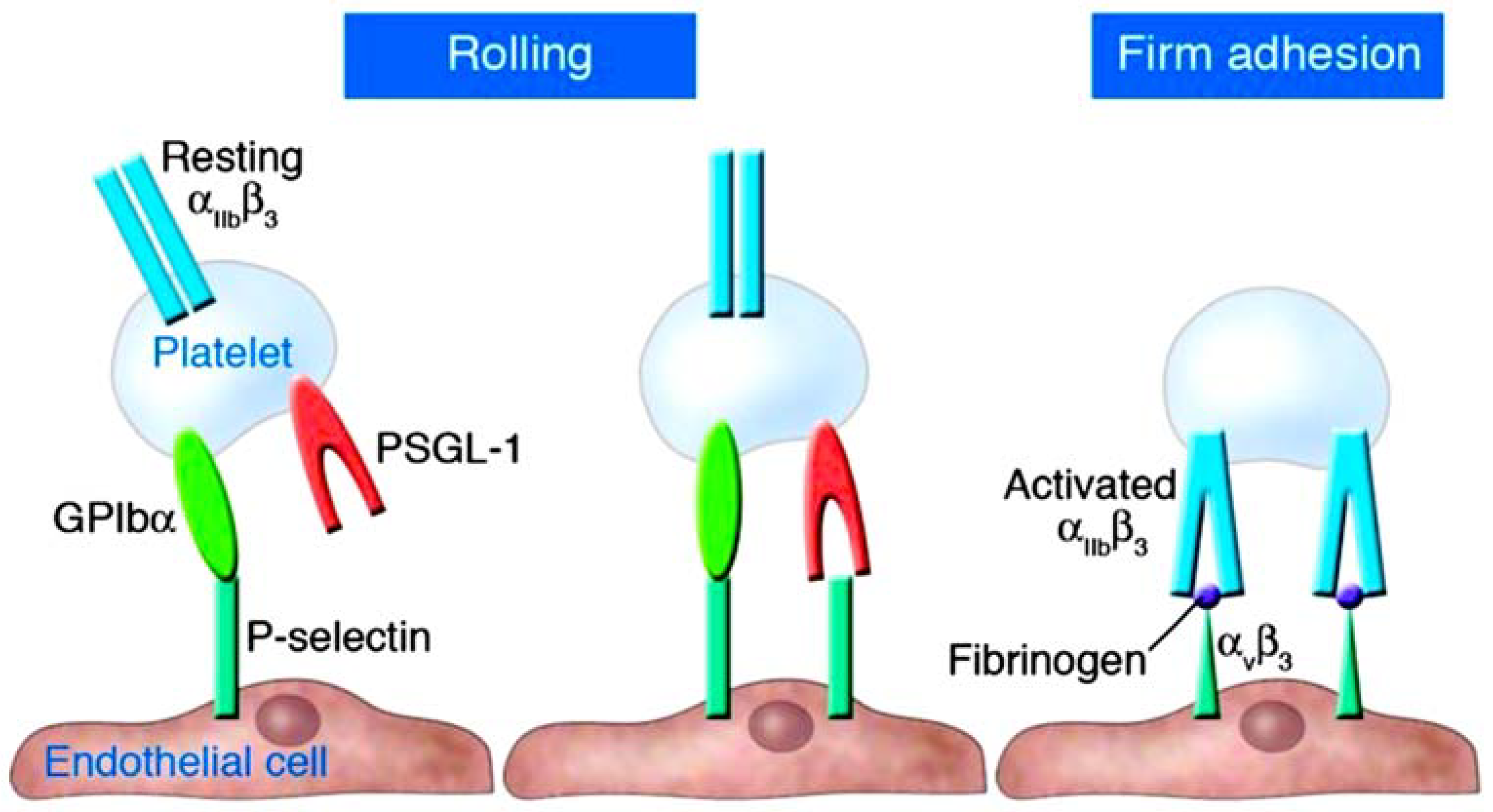
2. Understanding the Role of Platelets and P-Selectin as Key Actors in Thromboinflammation
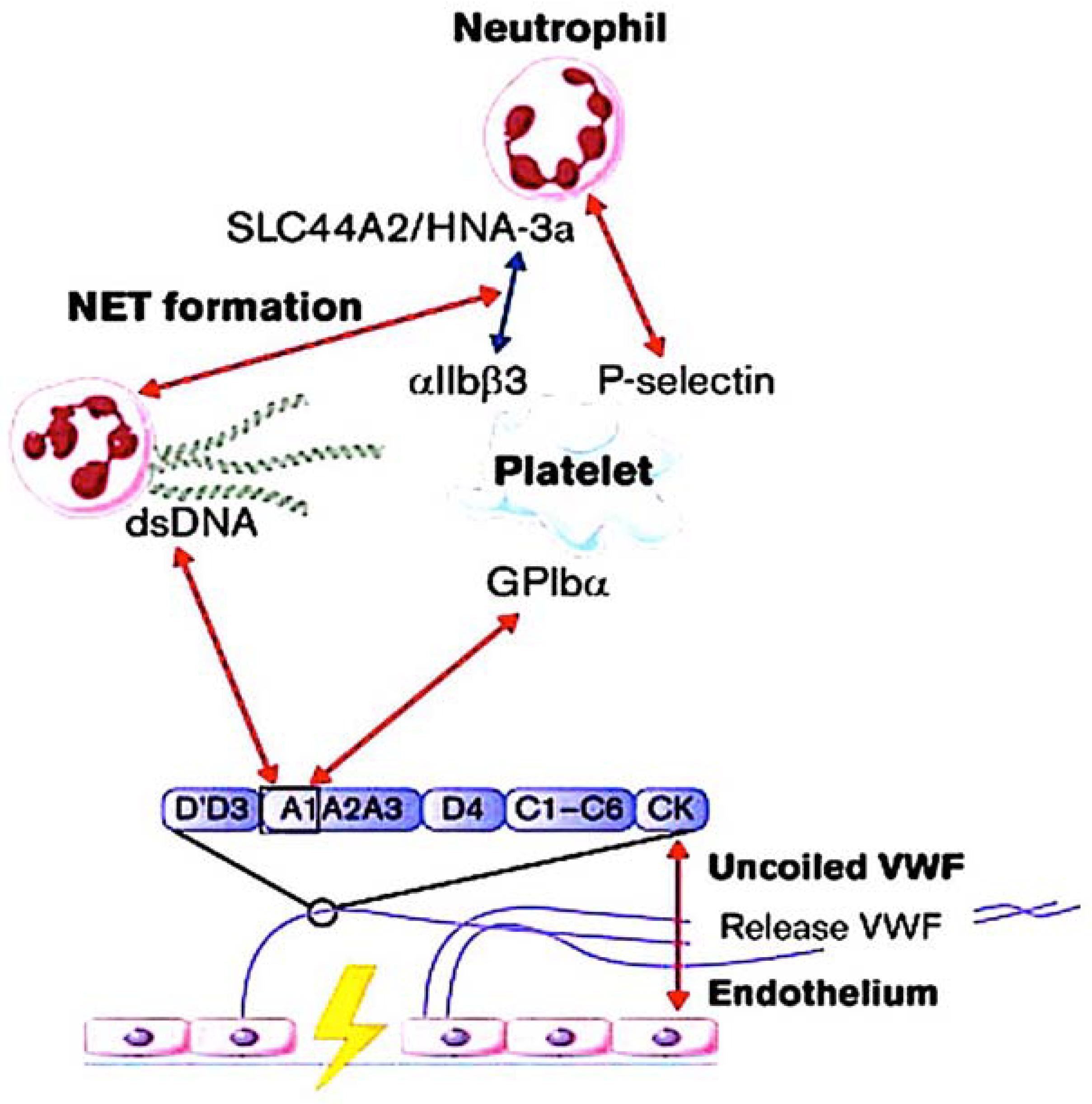
3. Interaction of NETs with Ultra-Large VWF in Thromboinflammatory Vasculopathy
3.1. Identifying the Role Played by NETs
3.2. The Role of VWF and ADAMTS13
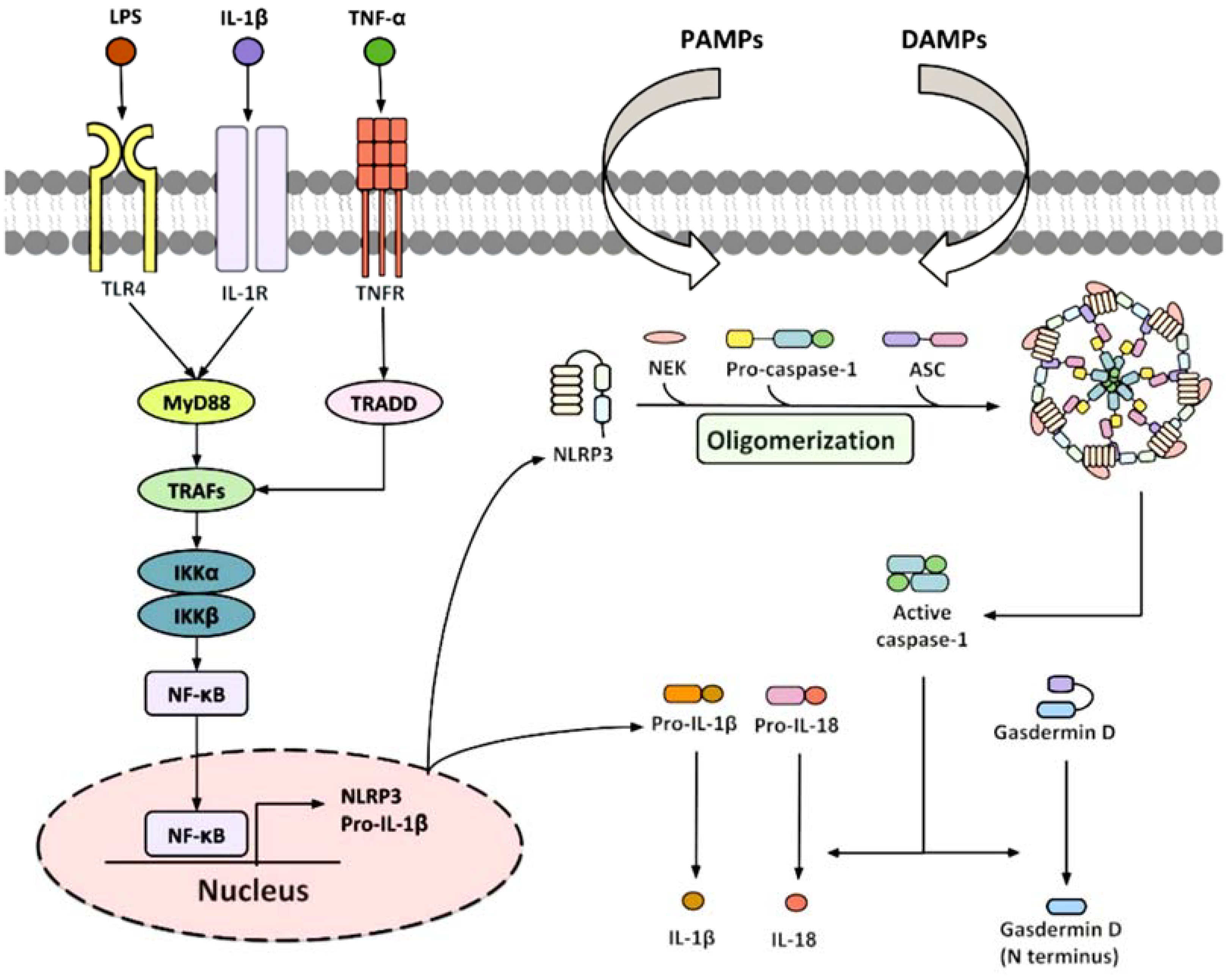
3.3. Inflammasone to the Direction of All Actors
4. Focusing on the Thromboinflammation Process in Atherosclerosis and COVID-19
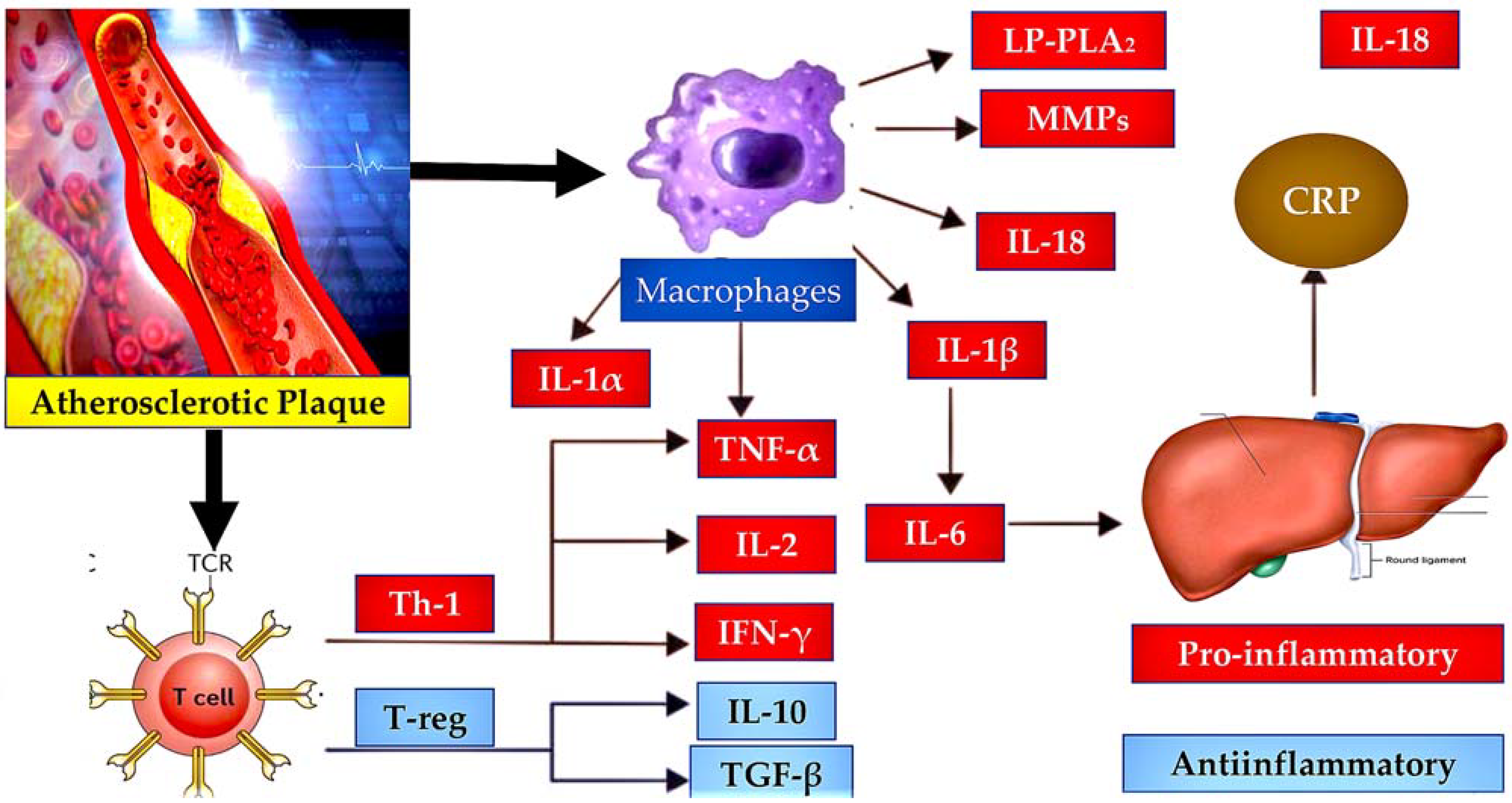
4.1. Implication of Atherosclerosis
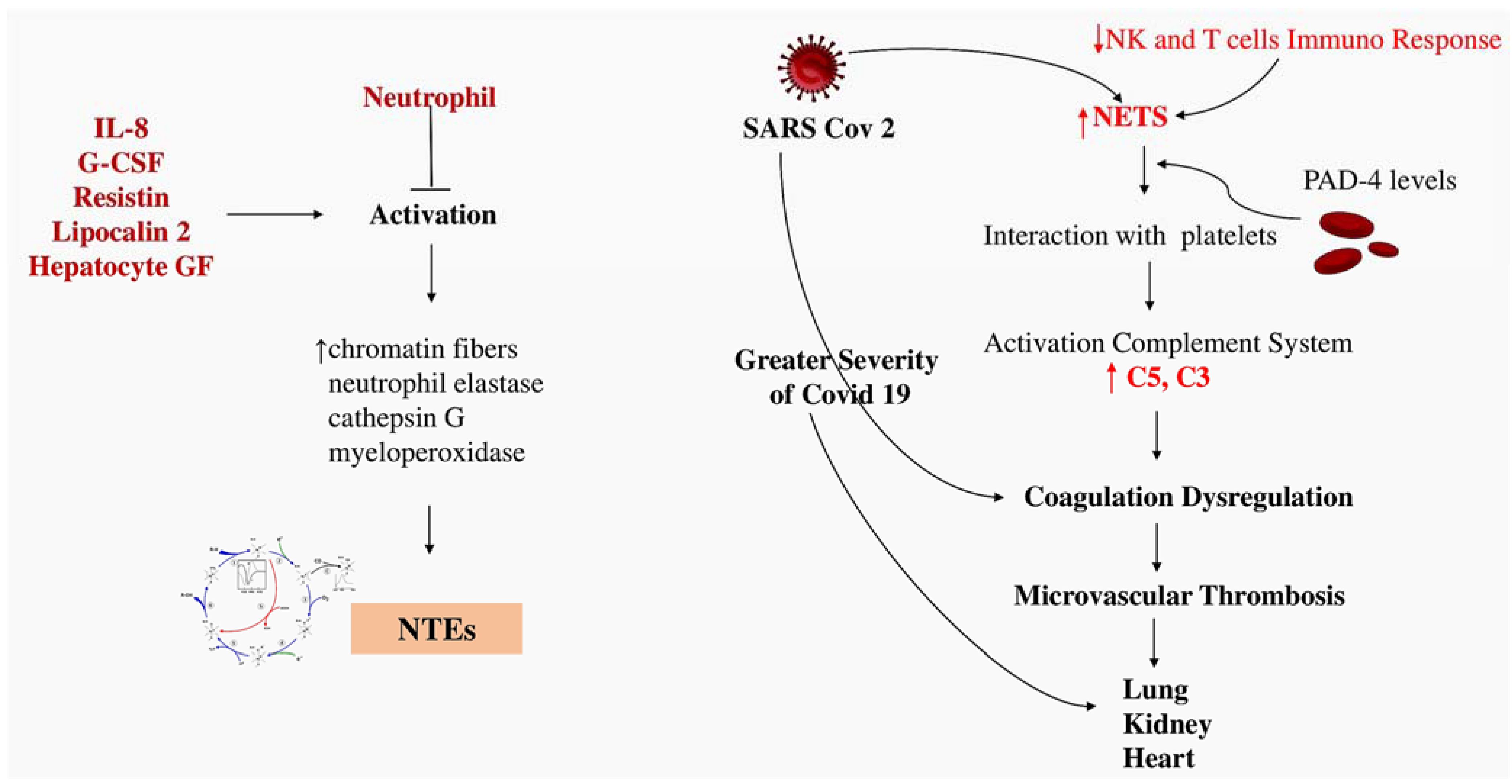
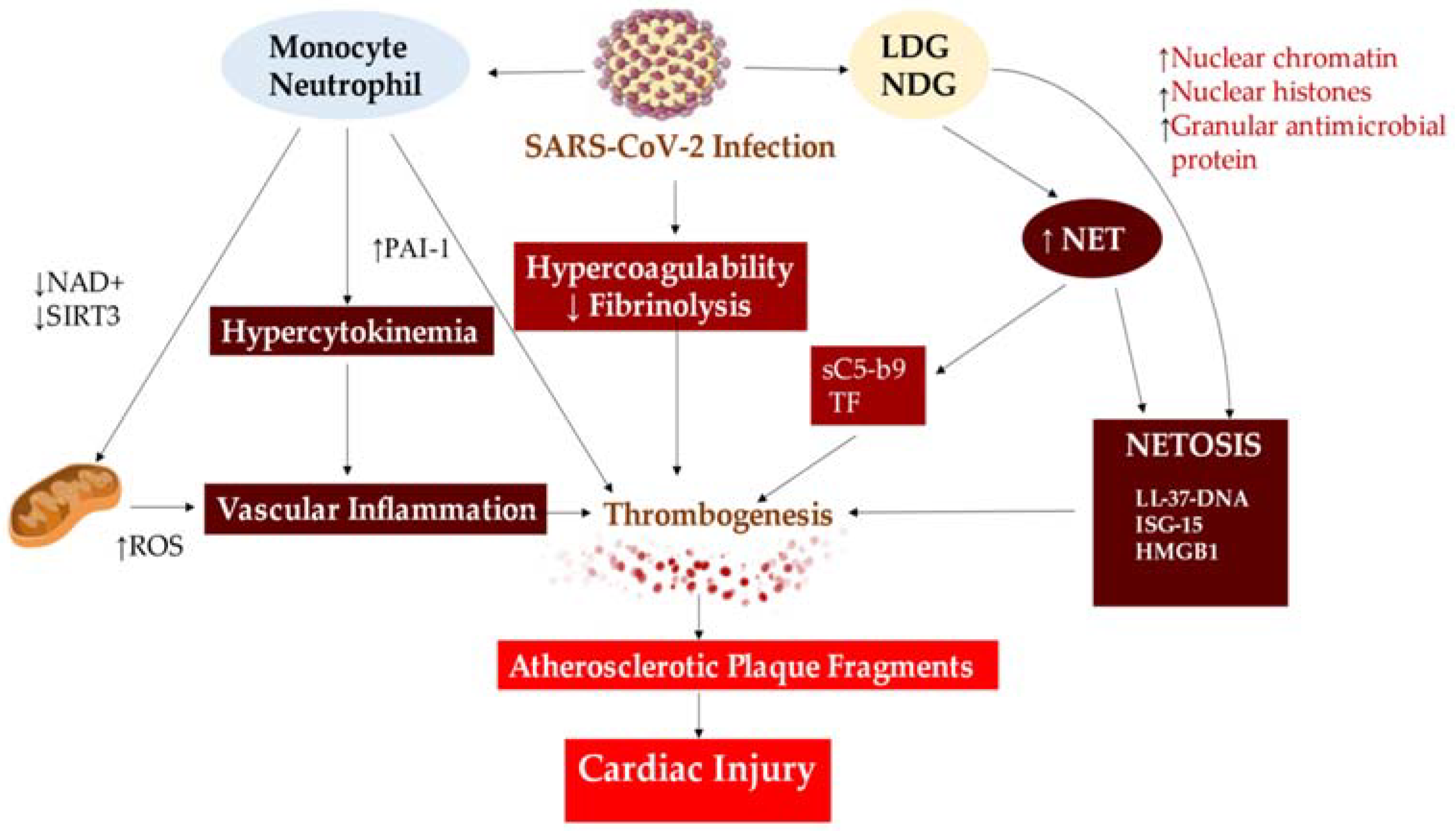
4.2. Implication of Thromboinflammation in COVID-19
5. Nex Steps
6. Limitation
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Bellomo, F.; Avtaar Singh, S.S. Worsening Thrombotic Complication of Atherosclerotic Plaques Due to Neutrophils Extracellular Traps: A Systematic Review. Biomedicines 2023, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Trivigno, S.M.G.; Guidetti, G.F.; Barbieri, S.S.; Zarà, M. Blood Platelets in Infection: The Multiple Roles of the Platelet Signalling Machinery. Int. J. Mol. Sci. 2023, 24, 7462. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Ma, J.; Wu, D.; Fang, C.; Wang, Z.; Guo, T.; Mo, J. Neutrophil extracellular traps mediate deep vein thrombosis: From mechanism to therapy. Front. Immunol. 2023, 14, 1198952. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Gollomp, K.; Kim, M.; Johnston, I.; Hayes, V.; Welsh, J.; Arepally, G.M.; Kahn, M.; Lambert, M.P.; Cuker, A.; Cines, D.B.; et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight 2018, 3, e99445. [Google Scholar] [CrossRef] [PubMed]
- Krott, K.J.; Feige, T.; Elvers, M. Flow Chamber Analyses in Cardiovascular Research: Impact of Platelets and the Intercellular Crosstalk with Endothelial Cells, Leukocytes, and Red Blood Cells. Hamostaseologie 2023, 43, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Wagner, D.D.; Burger, P.C. Platelets in inflammation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef]
- Solomon, S.D.; Lowenstein, C.J.; Bhatt, A.S.; Peikert, A.; Vardeny, O.; Kosiborod, M.N.; Berger, J.S.; Reynolds, H.R.; Mavromichalis, S.; Barytol, A.; et al. Effect of the P-Selectin Inhibitor Crizanlizumab on Survival Free of Organ Support in Patients Hospitalized for COVID-19: A Randomized Controlled Trial. Circulation 2023, 148, 381–390. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Johnson, R.C.; Rayburn, H.; Hynes, R.O.; Wagner, D.D. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell 1993, 74, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Hrachovinová, I.; Cambien, B.; Hafezi-Moghadam, A.; Kappelmayer, J.; Camphausen, R.T.; Widom, A.; Xia, L.; Kazazian, H.H., Jr.; Schaub, R.G.; McEver, R.P.; et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat. Med. 2003, 9, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Polgar, J.; Matuskova, J.; Wagner, D.D. The P-selectin, tissue factor, coagulation triad. J. Thromb. Haemost. 2005, 3, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Braun, O.O.; Slotta, J.E.; Menger, M.D.; Erlinge, D.; Thorlacius, H. Primary and secondary capture of platelets onto inflamed femoral artery endothelium is dependent on P-selectin and PSGL-1. Eur. J. Pharmacol. 2008, 592, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Dole, V.S.; Bergmeier, W.; Patten, I.S.; Hirahashi, J.; Mayadas, T.N.; Wagner, D.D. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb. Haemost. 2007, 98, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Dole, V.S.; Bergmeier, W.; Mitchell, H.A.; Eichenberger, S.C.; Wagner, D.D. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: Role of P-selectin. Blood 2005, 106, 2334–2339. [Google Scholar] [CrossRef]
- Blann, A.D.; Nadar, S.K.; Lip, G.Y. The adhesion molecule P-selectin and cardiovascular disease. Eur. Heart J. 2003, 24, 2166–2179. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N. Soluble P-selectin and the risk of future cardiovascular events. Circulation 2001, 103, 491–495. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Kisucka, J.; Chauhan, A.K.; Zhao, B.Q.; Patten, I.S.; Yesilaltay, A.; Krieger, M.; Wagner, D.D. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood 2009, 113, 6015–6022. [Google Scholar] [CrossRef]
- NIHR Global Health Unit on Global Surgery. Elective surgery system strengthening development, measurement, and validation of the surgical preparedness index across 1632 hospitals in 119 countries. COVIDSurg Collaborative. Lancet 2022, 400, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- COVIDSurg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 infection and venous thromboembolism after surgery: An international prospective cohort study. Anaesthesia 2022, 77, 28–39. [Google Scholar] [CrossRef] [PubMed]
- COVIDSurg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 vaccination modelling for safe surgery to save lives: Data from an international prospective cohort study. Br. J. Surg. 2021, 108, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Timing of surgery following SARS-CoV-2 infection: An international prospective cohort study. Anaesthesia 2021, 76, 748–758. [Google Scholar] [CrossRef] [PubMed]
- COVIDSurg Collaborative; GlobalSurg Collaborative. Effects of pre-operative isolation on postoperative pulmonary complications after elective surgery: An international prospective cohort study. Anaesthesia 2021, 76, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Giacinto, O.; Ellouze, O.; Nenna, A.; Avtaar Singh, S.S.; Chello, M.; Bouzguenda, A.; Copie, X. Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines 2022, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Rosati, C.M.; El-Dalati, S. Acute Type A Aortic Dissection During the COVID-19 Outbreak. Ann. Thorac. Surg. 2020, 110, e405–e407. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F. Incertitude Pathophysiology and Management During the First Phase of the COVID-19 Pandemic. Ann. Thorac. Surg. 2022, 113, 693. [Google Scholar] [CrossRef]
- Troisi, R.; Balasco, N.; Autiero, I.; Sica, F.; Vitagliano, L. New insight into the traditional model of the coagulation cascade and its regulation: Illustrated review of a three-dimensional view. Res Pract Thromb. Haemost. 2023, 7, 102160. [Google Scholar] [CrossRef]
- Rolling, C.C.; Barrett, T.J.; Berger, J.S. Platelet-monocyte aggregates: Molecular mediators of thromboinflammation. Front. Cardiovasc. Med. 2023, 10, 960398. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Heestermans, M.; Poenou, G.; Duchez, A.C.; Hamzeh-Cognasse, H.; Bertoletti, L.; Cognasse, F. Immunothrombosis and the Role of Platelets in Venous Thromboembolic Diseases. Int. J. Mol. Sci. 2022, 23, 13176. [Google Scholar] [CrossRef] [PubMed]
- Scholz, T.; Temmler, U.; Krause, S.; Heptinstall, S.; Lösche, W. Transfer of tissue factor from platelets to monocytes: Role of platelet-derived microvesicles and CD62P. Thromb. Haemost. 2002, 88, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Hartwell, D.; Hrachovinová, I.; Saffaripour, S.; Wagner, D.D. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc. Natl. Acad. Sci. USA 2000, 97, 13835–13840. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.C.; Wagner, D.D. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003, 101, 2661–2666. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef]
- Libby, P.; Everett, B.M. Novel antiatherosclerotic therapies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 538–545. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, J.; Deng, L.; Ishimwe, N.; Pauli, J.; Wu, W.; Shan, S.; Kempf, W.; Ballantyne, M.D.; Kim, D.; et al. INKILN is a Novel Long Noncoding RNA Promoting Vascular Smooth Muscle Inflammation via Scaffolding MKL1 and USP10. Circulation 2023, 148, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Wang, X.H.; Zhao, Y. Platelet factor 4 (PF4) induces cluster of differentiation 40 (CD40) expression in human aortic endothelial cells (HAECs) through the SIRT1/NF-κB/p65 signaling pathway. Vitr. Cell. Dev. Biol. Anim. 2023, 59, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Rux, A.H.; Ma, P.; Bdeir, K.; Sachais, B.S. Endothelial expression of E-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood 2005, 105, 3545–3551. [Google Scholar] [CrossRef] [PubMed]
- Stadtmann, A.; Brinkhaus, L.; Mueller, H.; Rossaint, J.; Bolomini-Vittori, M.; Bergmeier, W.; Van Aken, H.; Wagner, D.D.; Laudanna, C.; Ley, K.; et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 2011, 41, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Aisan, A.; Maheshati, T.; Tian, R.; Li, Y.; Chen, Y. Predictive value of combining leucocyte and platelet counts for mortality in ST-segment elevation myocardial infarction patients after percutaneous coronary intervention treatment in Chinese population: A retrospective cohort study. BMJ Open 2023, 13, e060756. [Google Scholar] [CrossRef] [PubMed]
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet activation: The mechanisms and potential biomarkers. Biomed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef]
- Goerge, T.; Ho-Tin-Noe, B.; Carbo, C.; Benarafa, C.; Remold-O’Donnell, E.; Zhao, B.Q.; Cifuni, S.M.; Wagner, D.D. Inflammation induces hemorrhage in thrombocytopenia. Blood 2008, 111, 4958–4964. [Google Scholar] [CrossRef]
- Ho-Tin-Noé, B.; Goerge, T.; Cifuni, S.M.; Duerschmied, D.; Wagner, D.D. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008, 68, 6851–6858. [Google Scholar] [CrossRef]
- Graca, F.A.; Stephan, A.; Minden-Birkenmaier, B.A.; Shirinifard, A.; Wang, Y.D.; Demontis, F.; Labelle, M. Platelet-derived chemokines promote skeletal muscle regeneration by guiding neutrophil recruitment to injured muscles. Nat. Commun. 2023, 14, 2900. [Google Scholar] [CrossRef] [PubMed]
- Hook, J.S.; Cao, M.; Potera, R.M.; Alsmadi, N.Z.; Schmidtke, D.W.; Moreland, J.G. Nox2 Regulates Platelet Activation and NET Formation in the Lung. Front. Immunol. 2019, 10, 1472. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Nappi, P.; Gambardella, I.; Avtaar Singh, S.S. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites 2022, 12, 889. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef]
- Nappi, F.; Bellomo, F.; Avtaar Singh, S.S. Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. J. Clin. Med. 2022, 11, 2460. [Google Scholar] [CrossRef]
- Nappi, F.; Iervolino, A.; Avtaar Singh, S.S. Thromboembolic Complications of SARS-CoV-2 and Metabolic Derangements: Suggestions from Clinical Practice Evidence to Causative Agents. Metabolites 2021, 11, 341. [Google Scholar] [CrossRef]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, fj201800691R. [Google Scholar] [CrossRef]
- Münzer, P.; Negro, R.; Fukui, S.; di Meglio, L.; Aymonnier, K.; Chu, L.; Cherpokova, D.; Gutch, S.; Sorvillo, N.; Shi, L.; et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under Sterile Conditions. Front. Immunol. 2021, 12, 683803. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. Reply to Liu: The disassembly of the actin cytoskeleton is an early event during NETosis. Proc. Natl. Acad. Sci. USA 2020, 117, 22655–22656. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, N.; Cherpokova, D.; Martinod, K.; Wagner, D.D. Extracellular DNA NET-works with dire consequences for health. Circ. Res. 2019, 125, 470–488. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Brüggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. National Heart, Lung, and Blood Institute Severe Asthma Research Program-3 Investigators. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3, eaao4747. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Gutch, S.; Fukui, S.; Cherpokova, D.; Aymonnier, K.; Sheehy, C.E.; Chu, L.; Wagner, D.D. The prominent role of hematopoietic peptidyl arginine deiminase 4 in arthritis: Collagen- and granulocyte colony-stimulating factor-induced arthritis model in C57BL/6 MICE. Arthritis Rheumatol. 2022, 74, 1139–1146. [Google Scholar] [CrossRef]
- Morrell, C.N.; Hilt, Z.T.; Pariser, D.N.; Maurya, P. PAD4 and von Willebrand Factor Link Inflammation and Thrombosis. Circ. Res. 2019, 125, 520–522. [Google Scholar] [CrossRef]
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ. Res. 2019, 125, 507–519. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef]
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. J. Clin. Med. 2019, 8, E2072. [Google Scholar] [CrossRef] [PubMed]
- Liman, T.G.; Bachelier-Walenta, K.; Neeb, L.; Rosinski, J.; Reuter, U.; Böhm, M.; Endres, M. Circulating endothelial microparticles in female migraineurs with aura. Cephalalgia 2015, 35, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-dependent coagulation activation in sepsis. Front. Immunol. 2021, 12, 641750. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil extracellular traps participate in cardiovascular diseases: Recent experimental and clinical insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Novotny, J.; Oberdieck, P.; Titova, A.; Pelisek, J.; Chandraratne, S.; Nicol, P.; Hapfelmeier, A.; Joner, M.; Maegdefessel, L.; Poppert, H.; et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology 2020, 94, e2346–e2360. [Google Scholar] [CrossRef]
- Ducroux, C.; DiMeglio, L.; Loyau, S.; Delbosc, S.; Boisseau, W.; Deschildre, C.; BenMaacha, M.; Blanc, R.; Redjem, H.; Ciccio, G.; et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke 2018, 49, 754–757. [Google Scholar] [CrossRef]
- Blasco, A.; Coronado, M.-J.; Hernández-Terciado, F.; Martín, P.; Royuela, A.; Ramil, E.; García, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol. 2021, 6, 469. [Google Scholar] [CrossRef]
- Blasco, A.; Bellas, C.; Goicolea, L.; Muñiz, A.; Abraira, V.; Royuela, A.; Mingo, S.; Oteo, J.F.; García-Touchard, A.; Goicolea, F.J. Immunohistological analysis of intracoronary thrombus aspirate in STEMI patients: Clinical implications of pathological findings. Rev. Esp. Cardiol. (Engl. Ed.) 2017, 70, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Langseth, M.S.; Helseth, R.; Ritschel, V.; Hansen, C.H.; Andersen, G.; Eritsland, J.; Halvorsen, S.; Fagerland, M.W.; Solheim, S.; Arnesen, H.; et al. Double-Stranded DNA and NETs Components in Relation to Clinical Outcome After ST-Elevation Myocardial Infarction. Sci. Rep. 2020, 10, 5007. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights into immunothrombosis: The interplay among neutrophil extracellular trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 610696. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Walsh, M.T.; Zhu, G.; Ginsburg, D.; Wagner, D.D.; Motto, D.G. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 2008, 111, 3452–3457. [Google Scholar] [CrossRef]
- Sadler, J.E. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017, 130, 1181–1188. [Google Scholar] [CrossRef]
- Frenette, P.S.; Atweh, G.F. Sickle cell disease: Old discoveries, new concepts, and future promise. J. Clin. Investig. 2007, 117, 850–858. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Springer, T.A. von Willebrand factor, Jedi knight of the bloodstream. Blood 2014, 124, 1412–1425. [Google Scholar] [CrossRef]
- Arya, M.; Anvari, B.; Romo, G.M.; Cruz, M.A.; Dong, J.F.; McIntire, L.V.; Moake, J.L.; López, J.A. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: Studies using optical tweezers. Blood 2002, 99, 3971–3977. [Google Scholar] [CrossRef] [PubMed]
- Peetermans, M.; Meyers, S.; Liesenborghs, L.; Vanhoorelbeke, K.; De Meyer, S.F.; Vandenbriele, C.; Lox, M.; Hoylaerts, M.F.; Martinod, K.; Jacquemin, M.; et al. Von Willebrand factor and ADAMTS13 impact on the outcome of Staphylococcus aureus sepsis. J. Thromb. Haemost. 2020, 18, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Vischer, U.M. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J. Thromb. Haemost. 2006, 4, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Jood, K.; Karlsson, S.; Nilsson, S.; Blomstrand, C.; Jern, C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J. Thromb. Haemost. 2011, 9, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.K.; Kisucka, J.; Brill, A.; Walsh, M.T.; Scheiflinger, F.; Wagner, D.D. ADAMTS13: A new link between thrombosis and inflammation. J. Exp. Med. 2008, 205, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.; Motto, D.G.; Jensen, M.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5224–5230. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, S.F.; Savchenko, A.S.; Haas, M.S.; Schatzberg, D.; Carroll, M.C.; Schiviz, A.; Dietrich, B.; Rottensteiner, H.; Scheiflinger, F.; Wagner, D.D. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5217–5223. [Google Scholar] [CrossRef]
- Zhao, B.Q.; Chauhan, A.K.; Canault, M.; Patten, I.S.; Yang, J.J.; Dockal, M.; Scheiflinger, F.; Wagner, D.D. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood 2009, 114, 3329–3334. [Google Scholar] [CrossRef]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef]
- Matsui, T.; Hamako, J. Structure and function of snake venom toxins interacting with human von Willebrand factor. Toxicon 2005, 45, 1075–1087. [Google Scholar] [CrossRef]
- Colicchia, M.; Perrella, G.; Gant, P.; Rayes, J. Novel mechanisms of thrombo-inflammation during infection: Spotlight on neutrophil extracellular trap-mediated platelet activation. Res. Pract. Thromb. Haemost. 2023, 7, 100116. [Google Scholar] [CrossRef] [PubMed]
- Courson, J.A.; Lam, F.W.; Langlois, K.W.; Rumbaut, R.E. Histone-stimulated platelet adhesion to mouse cremaster venules in vivo is dependent on von Willebrand factor. Microcirculation 2022, 29, e12782. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Tetaz, T.J.; Andrews, R.K.; Berndt, M.C. Binding of the von Willebrand factor A1 domain to histone. Thromb. Res. 1997, 86, 469–477. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, V.; Singh, K.; Kretz, C.A. Mechanisms of ADAMTS13 regulation. J. Thromb. Haemost. 2022, 20, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Goverman, J.; Staudinger, C.; Wagner, D.D. Recombinant human ADAMTS13 treatment and anti-NET strategies enhance skin allograft survival in mice. Am. J. Transplant. 2020, 20, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Gajendran, C.; Fukui, S.; Sadhu, N.M.; Zainuddin, M.; Rajagopal, S.; Gosu, R.; Gutch, S.; Fukui, S.; Sheehy, C.E.; Chu, L.; et al. Alleviation of arthritis through prevention of neutrophil extracellular traps by an orally available inhibitor of protein arginine deiminase 4. Sci. Rep. 2023, 13, 3189. [Google Scholar] [CrossRef]
- Arisz, R.A.; de Vries, J.J.; Schols, S.E.M.; Eikenboom, J.C.J.; de Maat, M.P.M. Interaction of von Willebrand factor with blood cells in flow models: A systematic review. Blood Adv. 2022, 6, 3979–3990. [Google Scholar] [CrossRef]
- Black, R.A.; Kronheim, S.R. Sleath Activation of interleukin-1 beta by a co-induced protease. PR. FEBS Lett. 1989, 247, 386–390. [Google Scholar] [CrossRef]
- Black, R.A.; Kronheim, S.R.; Merriam, J.E.; March, C.J.; Hopp, T.P. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 1989, 264, 5323–5326. [Google Scholar] [CrossRef]
- Krieger, T.J.; Hook, V.Y. Purification and characterization of a novel thiol protease involved in processing the enkephalin precursor. J. Biol. Chem. 1991, 266, 8376–8383. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Targeting inflammatory pathways in cardiovascular disease: The inflammasome, Interleukin-1, Interleukin-6 and BEYOND. Cells 2021, 10, 951. [Google Scholar] [CrossRef] [PubMed]
- Kocatürk, B.; Lee, Y.; Nosaka, N.; Abe, M.; Martinon, D.; Lane, M.E.; Moreira, D.; Chen, S.; Fishbein, M.C.; Porritt, R.A.; et al. Platelets exacerbate cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 2023, 8, e169855. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, V.; Ribeiro, L.S.; Hawwari, I.; Böttcher, L.; Rosero, N.; Maasewerd, S.; Santos, M.L.S.; Pröchnicki, T.; Silva, C.M.S.; Wanderley, C.W.S.; et al. Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. 2020, 31, 107615. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Wang, X.; Feuerstein, G.Z.; Clark, R.K.; Yue, T.L. Enhanced leucocyte adhesion to interleukin-1 beta stimulated vascular smooth muscle cells is mainly through intercellular adhesion molecule-1. Cardiovasc. Res. 1994, 28, 1808–1814. [Google Scholar] [CrossRef]
- Wang, X.; Feuerstein, G.Z.; Gu, J.L.; Lysko, P.G.; Yue, T.L. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis 1995, 115, 89–98. [Google Scholar] [CrossRef]
- González-Carnicero, Z.; Hernanz, R.; Martínez-Casales, M.; Barrús, M.T.; Martín, Á.; Alonso, M.J. Regulation by Nrf2 of IL-1β-induced inflammatory and oxidative response in VSMC and its relationship with TLR4. Front. Pharmacol. 2023, 14, 1058488. [Google Scholar] [CrossRef] [PubMed]
- Stackowicz, J.; Gaudenzio, N.; Serhan, N.; Conde, E.; Godon, O.; Marichal, T.; Starkl, P.; Balbino, B.; Roers, A.; Bruhns, P.; et al. Neutrophil-specific gain- of-function mutations in Nlrp3 promote development of cryopyrin- associated periodic syndrome. J. Exp. Med. 2021, 218, e20201466. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Cheung, S.W.; Cheng, K.K. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 4184. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Schwerdtner, L.; Sams, K.; Mondal, S.; Ahmad, F.; Schmidt, R.E.; Coonrod, S.A.; Thompson, P.R.; Lerch, M.M.; Bossaller, L. Cutting edge: Protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome-dependent IL-1β maturation and ASC speck formation in macrophages. J. Immunol. 2019, 203, 795–800. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.L.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021, 19, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar] [CrossRef]
- Koh, T.W.; DeSouza, A.C.; Pepper, J.R. The adhesion molecule P-selectin and cardiovascular disease--cardiac surgical implications. Eur. Heart J. 2004, 25, 993. [Google Scholar] [CrossRef]
- Wagner, D.D.; Heger, L.A. Thromboinflammation: From Atherosclerosis to COVID-19. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Woollard, K.J.; Chin-Dusting, J. Therapeutic targeting of p-selectin in atherosclerosis. Inflamm. Allergy Drug Targets 2007, 6, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Shim, C.Y.; Liu, Y.N.; Atkinson, T.; Xie, A.; Foster, T.; Davidson, B.P.; Treible, M.; Qi, Y.; López, J.A.; Munday, A.; et al. Molecular Imaging of Platelet-Endothelial Interactions and Endothelial von Willebrand Factor in Early and Mid-Stage Atherosclerosis. Circ. Cardiovasc. Imaging 2015, 8, e002765. [Google Scholar] [CrossRef] [PubMed]
- Methia, N.; André, P.; Denis, C.V.; Economopoulos, M.; Wagner, D.D. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood 2001, 98, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Doddapattar, P.; Dev, R.; Jain, M.; Dhanesha, N.; Chauhan, A.K. Differential Roles of Endothelial Cell-Derived and Smooth Muscle Cell-Derived Fibronectin Containing Extra Domain A in Early and Late Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.; Khan, M.M.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood 2012, 119, 2385–2391. [Google Scholar] [CrossRef]
- Jin, S.Y.; Tohyama, J.; Bauer, R.C.; Cao, N.N.; Rader, D.J.; Zheng, X.L. Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1817–1823. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: Implications for superficial erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Violi, F.; Cangemi, R.; Calvieri, C. Hospitalization for pneumonia and risk of cardiovascular disease. JAMA 2015, 313, 1753. [Google Scholar] [CrossRef]
- Dalager-Pedersen, M.; Søgaard, M.; Schønheyder, H.C.; Nielsen, H.; Thomsen, R.W. Risk for myocardial infarction and stroke after community-acquired bacteremia: A 20-year population-based cohort study. Circulation 2014, 129, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.F.; Salmon, M.; Su, G.; Lu, G.; Stone, M.L.; Zhao, Y.; Owens, G.K.; Upchurch, G.R., Jr.; Ailawadi, G. Genetic and pharmacologic disruption of interleukin-1β signaling inhibits experimental aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Popa-Fotea, N.M.; Ferdoschi, C.E.; Micheu, M.M. Molecular and cellular mechanisms of inflammation in atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1200341. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Avtaar Singh, S.S. Endothelial Dysfunction in SARS-CoV-2 Infection. Biomedicines 2022, 10, 654. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Avtaar Singh, S.S. Distinctive Signs of Disease as Deterrents for the Endothelial Function: A Systematic Review. Metabolites 2023, 13, 430. [Google Scholar] [CrossRef] [PubMed]
- Msemburi, W.; Karlinsky, A.; Knutson, V.; Aleshin-Guendel, S.; Chatterji, S.; Wakefield, J. The WHO estimates of excess mortality associated with the COVID-19 pandemic. Nature 2023, 613, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef]
- Levy, J.H.; Iba, T.; Olson, L.B.; Corey, K.M.; Ghadimi, K.; Connors, J.M. COVID-19: Thrombosis, thromboinflammation, and anticoagulation considerations. Int. J. Lab. Hematol. 2021, 43 (Suppl. 1), 29–35. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Group, R.C. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2022, 399, 143–151. [Google Scholar]
- Investigators R-CWCftR-C; Bradbury, C.A.; Lawler, P.R.; Stanworth, S.J.; McVerry, B.J.; McQuilten, Z.; Higgins, A.M.; Mouncey, S.J.; Al-Beidh, F.; Rowan, K.M.; et al. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: A randomized clinical trial. JAMA 2022, 327, 1247–1259. [Google Scholar]
- Kander, T. Coagulation disorder in COVID-19. Lancet Haematol. 2020, 7, e630–e632. [Google Scholar] [CrossRef] [PubMed]
- Sinkovits, G.; Réti, M.; Müller, V.; Iványi, Z.; Gál, J.; Gopcsa, L.; Reményi, P.; Szathmáry, B.; Lakatos, B.; Szlávik, J.; et al. Associations between the von Willebrand Factor-ADAMTS13 Axis, Complement Activation, and COVID-19 Severity and Mortality. Thromb. Haemost. 2022, 122, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Lonati, P.; Grossi, C.; Borghi, M.O.; Novembrino, C.; et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 2021, 116, 102560. [Google Scholar] [CrossRef] [PubMed]
- Pascreau, T.; Zia-Chahabi, S.; Zuber, B.; Tcherakian, C.; Farfour, E.; Vasse, M. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb. Res. 2021, 204, 138–140. [Google Scholar] [CrossRef]
- Afzali, B.; Noris, M.; Lambrecht, B.N.; Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 2022, 22, 77–84. [Google Scholar] [CrossRef]
- The Lancet Haematology. COVID-19 coagulopathy: An evolving story. Lancet Haematol. 2020, 7, e425. [Google Scholar] [CrossRef]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Yatim, N.; Boussier, J.; Chocron, R.; Hadjadj, J.; Philippe, A.; Gendron, N.; Barnabei, L.; Charbit, B.; Szwebel, T.A.; Carlier, N.; et al. Platelet activation in critically ill COVID-19 patients. Ann. Intensive Care. 2021, 11, 113. [Google Scholar] [CrossRef]
- Gue, Y.X.; Pula, G.; Lip, G.Y.H. Crizanlizumab: A CRITICAL Drug During a CRITICAL Time? JACC Basic Transl. Sci. 2021, 6, 946–947. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef]
- Aymonnier, K.; Ng, J.; Fredenburgh, L.E.; Zambrano-Vera, K.; Münzer, P.; Gutch, S.; Fukui, S.; Desjardins, M.; Subramaniam, M.; Baron, R.M.; et al. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. 2022, 6, 2001–2013. [Google Scholar] [CrossRef]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Köllnberger, M.; Wakefield, T.W.; Lämmle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hägele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 2012, 23, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Bozza, P.T. Platelet-leukocyte interactions in COVID-19: Contributions to hypercoagulability, inflammation, and disease severity. Res. Pract. Thromb. Haemost. 2022, 6, e12709. [Google Scholar] [CrossRef] [PubMed]
- Damiana, T.; Damgaard, D.; Sidelmann, J.J.; Nielsen, C.H.; de Maat, M.P.M.; Münster, A.B.; Palarasah, Y. Citrullination of fibrinogen by peptidylarginine deiminase 2 impairs fibrin clot structure. Clinica Chimica Acta 2020, 501, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Osca-Verdegal, R.; Beltrán-García, J.; Paes, A.B.; Nacher-Sendra, E.; Novella, S.; Hermenegildo, C.; Carbonell, N.; García-Giménez, J.L.; Pallardó, F.V. Histone Citrullination Mediates a Protective Role in Endothelium and Modulates Inflammation. Cells 2022, 11, 4070. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Leopizzi, M.; Dominici, M.; d’Amati, G.; Bartimoccia, S.; Nocella, C.; Cammisotto, V.; D’Amico, A.; Castellani, V.; Baratta, F.; et al. PAD4-Induced NETosis Via Cathepsin G-Mediated Platelet-Neutrophil Interaction in ChAdOx1 Vaccine-Induced Thrombosis-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e396–e403. [Google Scholar] [CrossRef]
- Taifour, T.; Attalla, S.S.; Zuo, D.; Gu, Y.; Sanguin-Gendreau, V.; Proud, H.; Solymoss, E.; Bui, T.; Kuasne, H.; Papavasiliou, V.; et al. The tumor-derived cytokine Chi3l1 induces neutrophil extracellular traps that promote T cell exclusion in triple-negative breast cancer. Immunity 2023, 56, 2755–2772. [Google Scholar] [CrossRef]
- Keir, H.R.; Richardson, H.; Fillmore, C.; Shoemark, A.; Lazaar, A.L.; Miller, B.E.; Tal-Singer, R.; Chalmers, J.D.; Mohan, D. CXCL-8-dependent and -independent neutrophil activation in COPD: Experiences from a pilot study of the CXCR2 antagonist danirixin. ERJ Open Res. 2020, 6, 00583–2020. [Google Scholar] [CrossRef]
- Dundee, United Kingdom, DD1 9SY GSK Investigational Site Randomized Study Evaluating the Effect of Danirixin on Neutrophil Extracellular Traps (NETs) in Chronic Obstructive Pulmonary Disease (COPD). Available online: https://www.clinicaltrials.gov/study/NCT03250689 (accessed on 15 December 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent/Drug Name | Indication | Study No./Status | Preclinical/Clinical Study Outcome | Refs. |
|---|---|---|---|---|
| Crizanlizumab | P-selectin inhibitor | NCT03814746 | No improvement in clinical outcomes | [161] |
| Pulmozyme | Reduces NETosis in neutrophils. | NCT04402944 | Phase 2 | [166] |
| Targeting Chi3l1 | Advanced cancer. Enhance anti-tumor immunity | “ ” “ ” | Reduced levels of helper and cytotoxic T cells in TNBC tumors led to decreased tumor infiltration. | [177] |
| Danirixin hydrobromide | Reduces NETs formation | NCT02453022 | Phase 1 | [178,179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F. To Gain Insights into the Pathophysiological Mechanisms of the Thrombo-Inflammatory Process in the Atherosclerotic Plaque. Int. J. Mol. Sci. 2024, 25, 47. https://doi.org/10.3390/ijms25010047
Nappi F. To Gain Insights into the Pathophysiological Mechanisms of the Thrombo-Inflammatory Process in the Atherosclerotic Plaque. International Journal of Molecular Sciences. 2024; 25(1):47. https://doi.org/10.3390/ijms25010047
Chicago/Turabian StyleNappi, Francesco. 2024. "To Gain Insights into the Pathophysiological Mechanisms of the Thrombo-Inflammatory Process in the Atherosclerotic Plaque" International Journal of Molecular Sciences 25, no. 1: 47. https://doi.org/10.3390/ijms25010047
APA StyleNappi, F. (2024). To Gain Insights into the Pathophysiological Mechanisms of the Thrombo-Inflammatory Process in the Atherosclerotic Plaque. International Journal of Molecular Sciences, 25(1), 47. https://doi.org/10.3390/ijms25010047