

Prediabetes-Associated Changes in Skeletal Muscle Function and Their Possible Links with Diabetes: A Literature Review

Abstract
1. Introduction
2. Role of Satellite Cells in Skeletal Muscle
3. Prediabetes
3.1. Effects of PreDM on Skeletal Muscle Glucose Homeostasis
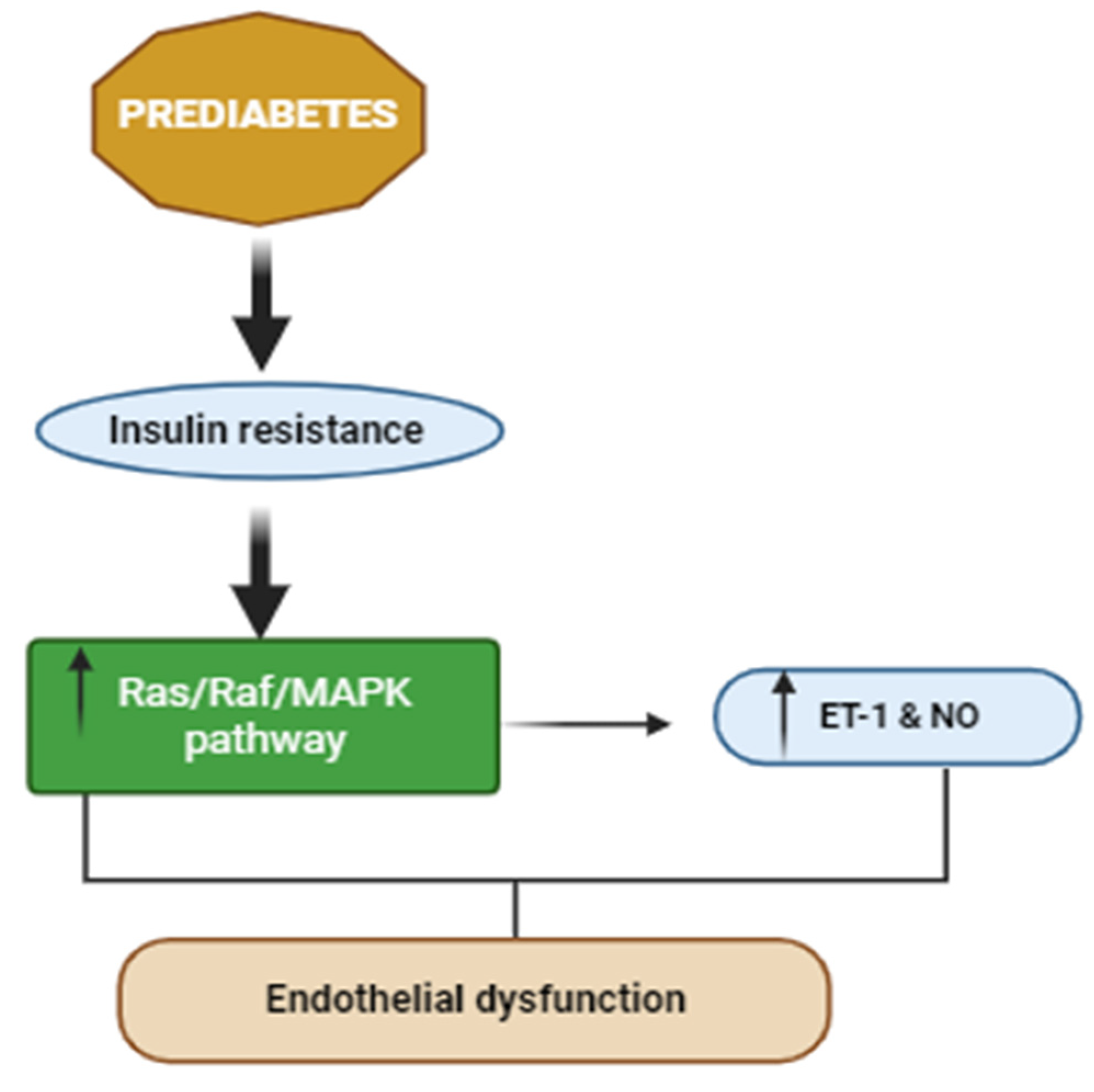
3.2. Effects of PreDM on Skeletal Muscle Vasculature and Extracellular Matrix
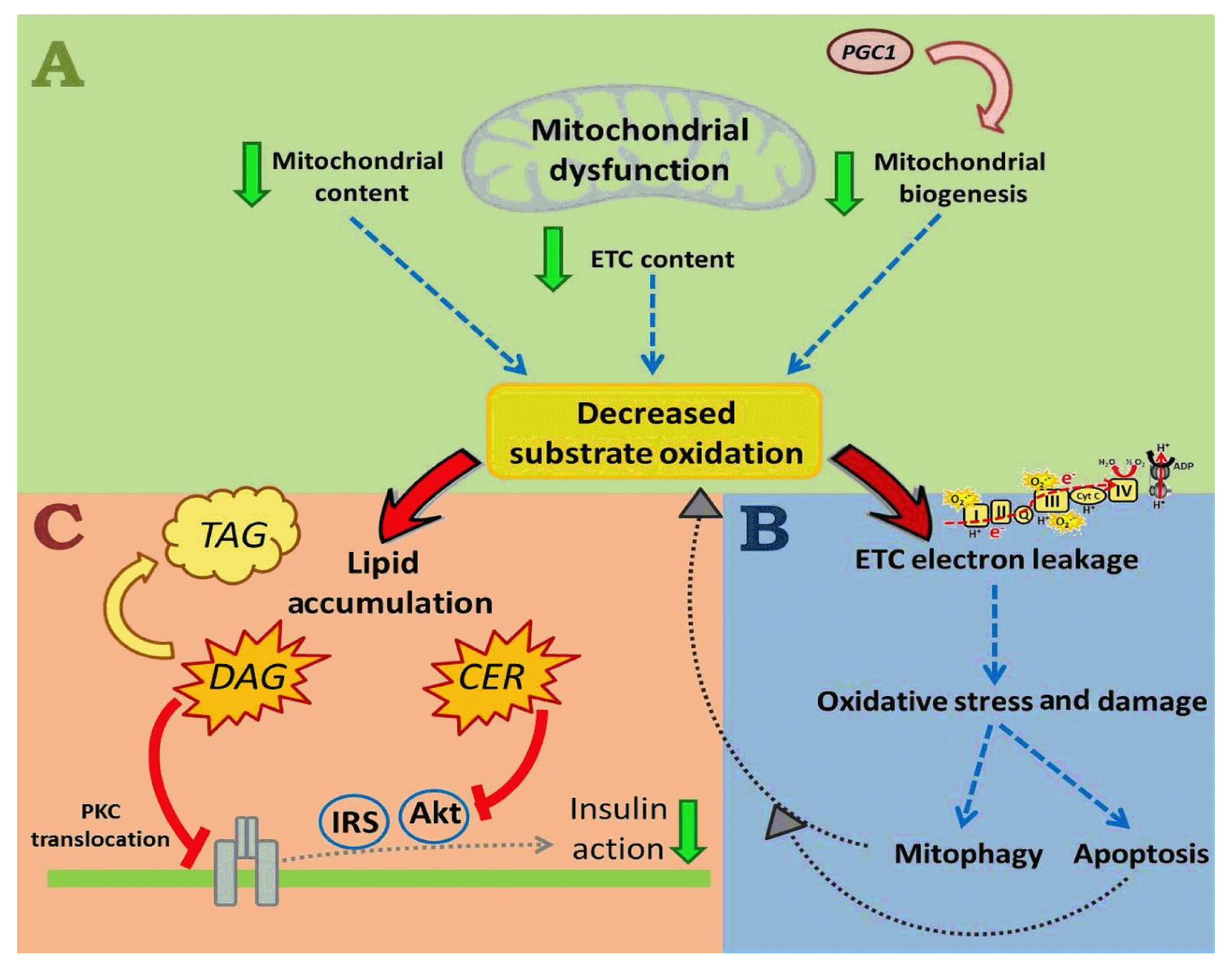
3.3. Effects of PreDM on Skeletal Muscle Satellite Cells
3.4. Effects of PreDM on Skeletal Muscle Strength
4. Type 2 Diabetes Mellitus
4.1. T2DM-Associated Complication: Diabetic Myopathy
4.2. Effects of T2DM on Skeletal Muscle Strength
4.3. Effect of T2DM on Skeletal Muscle Satellite Cells
4.4. Effects of T2DM on Skeletal Muscle Extracellular Matrix
5. PreDM-Associated Changes in Skeletal Muscle and Their Mechanistic Links to T2DM
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pfl?gers Arch. Eur. J. Physiol. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.C.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diabetes. 2022. Available online: https://www.who.int/health-topics/diabetes#tab=tab_1 (accessed on 25 February 2022).
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine 2014, 42, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Sicree, R.; Zimmet, P. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pr. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2006, 29 (Suppl. 1), S43–S48.
- Bergman, M. Pathophysiology of prediabetes and treatment implications for the prevention of type 2 diabetes mellitus. Endocrine 2012, 43, 504–513. [Google Scholar] [CrossRef]
- Andersen, H.; Gadeberg, P.C.; Brock, B.; Jakobsen, J. Muscular atrophy in diabetic neuropathy: A stereological magnetic resonance imaging study. Diabetologia 1997, 40, 1062–1069. [Google Scholar] [CrossRef]
- Andersen, H.; Gjerstad, M.D.; Jakobsen, J. Atrophy of foot muscles: A measure of diabetic neuropathy. Diabetes Care 2004, 27, 2382–2385. [Google Scholar] [CrossRef]
- Andersen, H.; Schmitz, O.; Nielsen, S. Decreased isometric muscle strength after acute hyperglycaemia in Type 1 diabetic patients. Diabet. Med. 2005, 22, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Schmalbruch, H. The morphology of regeneration of skeletal muscles in the rat. Tissue Cell 1976, 8, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Yang, Z.; Tapscott, S.J.; Olwin, B.B. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J. Cell Biol. 2007, 177, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; Jaenisch, R. The MyoD family of transcription factors and skeletal myogenesis. BioEssays 1995, 17, 203–209. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Schnegelsberg, P.N.; Stead, R.H.; Braun, T.; Arnold, H.-H.; Jaenisch, R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esuml, E.; Li, S.; Nonaka, I.; Nabeshima, Y.-I. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Maltin, C.; Delday, M.; Sinclair, K.; Steven, J.; Sneddon, A. Impact of manipulations of myogenesis in utero on the performance of adult skeletal muscle. Reproduction 2001, 122, 359–374. [Google Scholar] [CrossRef]
- Torres-Machorro, A.L. Homodimeric and Heterodimeric Interactions among Vertebrate Basic Helix–Loop–Helix Transcription Factors. Int. J. Mol. Sci. 2021, 22, 12855. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Muscle stem cells at a glance. J. Cell Sci. 2014, 127, 4543–4548. [Google Scholar] [CrossRef] [PubMed]
- Stepanek, L.; Horakova, D.; Nakladalova, M.; Cibickova, L.; Karasek, D.; Zadrazil, J. Significance of prediabetes as a nosological entity. Biomed. Pap. 2018, 162, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2014, 38 (Suppl. 1), S8–S16. [Google Scholar] [CrossRef] [PubMed]
- Hostalek, U. Global epidemiology of prediabetes - present and future perspectives. Clin. Diabetes Endocrinol. 2019, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003, 26 (Suppl. 1), S5–S20. [CrossRef]
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar]
- The Emerging Risk Factors Collaboration. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841.
- Goyal, S.N.; Sharma, A.K.; Haiderali, S.; Navya, R.M..; Arya, D.S.; Patil, C.R. Correction: Prediabetes: Grounds of pitfall signalling alteration for cardiovascular disease. RSC Adv. 2014, 5, 1619. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Matteo Ciccone, M. Endothelial Function in Pre-diabetes, Diabetes and Diabetic Cardiomyopathy: A Review. J. Diabetes Metab. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Liu, Z. Vascular function, insulin action, and exercise: An intricate interplay. Trends Endocrinol. Metab. 2015, 26, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Breslin, J.W.; Perrin, R.; Gaudreault, N.; Guo, M.; Kargozaran, H.; Wu, M.H. Microvascular Permeability in Diabetes and Insulin Resistance. Microcirculation 2007, 14, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Clark, M.G. Role of blood flow in the regulation of muscle glucose uptake. Annu. Rev. Nutr. 1997, 17, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Koh, K.K.; Quon, M.J. The union of vascular and metabolic actions of insulin in sickness and in health. Am. Heart Assoc. 2005, 25, 889–891. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Liu, Z. Muscle Insulin Resistance and the Inflamed Microvasculature: Fire from Within. Int. J. Mol. Sci. 2019, 20, 562. [Google Scholar] [CrossRef]
- Wasserman, D.H.; Wang, T.J.; Brown, N.J. The Vasculature in Prediabetes. Circ. Res. 2018, 122, 1135–1150. [Google Scholar] [CrossRef]
- Keogh, J.B.; Grieger, J.A.; Noakes, M.; Clifton, P.M. Flow-Mediated Dilatation Is Impaired by a High–Saturated Fat Diet but Not by a High-Carbohydrate Diet. Arter. Thromb. Vasc. Biol. 2005, 25, 1274–1279. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a Single High-Fat Meal on Endothelial Function in Healthy Subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.-X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free Fatty Acids Inhibit Insulin Signaling–Stimulated Endothelial Nitric Oxide Synthase Activation Through Upregulating PTEN or Inhibiting Akt Kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Lynch, J.J.; Ferro, T.J.; Blumenstock, F.A.; Brockenauer, A.M.; Malik, A.B. Increased endothelial albumin permeability mediated by protein kinase C activation. J. Clin. Investig. 1990, 85, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Koya, D.; Jirousek, M.R.; Lin, Y.W.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J. Clin. Investig. 1997, 100, 115–126. [Google Scholar] [CrossRef]
- Kim, J.-A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I.; et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef]
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine Phosphorylation of Insulin Receptor Substrate 1 by Inhibitor κB Kinase Complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-Induced Muscle Insulin Resistance Is Associated With Extracellular Matrix Remodeling and Interaction With Integrin α2β1 in Mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; De Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol. Metab. 2006, 290, E560–E565. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan Accumulates With High-Fat Feeding and Contributes to Insulin Resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef]
- Chajara, A.; Raoudi, M.; Delpech, B.; Leroy, M.; Basuyau, J.P.; Levesque, H. Increased Hyaluronan and Hyaluronidase Production and Hyaluronan Degradation in Injured Aorta of Insulin-Resistant Rats. Arter. Thromb. Vasc. Biol. 2000, 20, 1480–1487. [Google Scholar] [CrossRef]
- Kang, L.; Mayes, W.H.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice. Diabetologia 2013, 57, 603–613. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E. Effect of High-Fat Diets on Oxidative Stress, Cellular Inflammatory Response and Cognitive Function. Nutrients 2019, 11, 2579. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef]
- Draicchio, F.; Behrends, V.; Tillin, N.A.; Hurren, N.M.; Sylow, L.; Mackenzie, R. Involvement of the extracellular matrix and integrin signalling proteins in skeletal muscle glucose uptake. J. Physiol. 2022, 600, 4393–4408. [Google Scholar] [CrossRef]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P. Endothelial dysfunction in insulin resistance and type 2 diabetes. J. Intern. Med. 2007, 262, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.S.; Lantier, L.; Hasenour, C.M.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Muscle-Specific Vascular Endothelial Growth Factor Deletion Induces Muscle Capillary Rarefaction Creating Muscle Insulin Resistance. Diabetes 2013, 62, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular Inflammation, Insulin Resistance, and Reduced Nitric Oxide Production Precede the Onset of Peripheral Insulin Resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired Insulin Signaling in Endothelial Cells Reduces Insulin-Induced Glucose Uptake by Skeletal Muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef]
- Chung, A.W.; Hsiang, Y.N.; Matzke, L.A.; McManus, B.M.; van Breemen, C.; Okon, E.B. Reduced Expression of Vascular Endothelial Growth Factor Paralleled With the Increased Angiostatin Expression Resulting From the Upregulated Activities of Matrix Metalloproteinase-2 and -9 in Human Type 2 Diabetic Arterial Vasculature. Circ. Res. 2006, 99, 140–148. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.-S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.-Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Z.; Wang, X.; Yang, L.; Han, S.; Cao, K.; Xu, J.; Zhao, L.; Zhang, Y.; Liu, J.; et al. O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia 2016, 59, 1287–1296. [Google Scholar] [CrossRef]
- Xu, D.; Jiang, Z.; Sun, Z.; Wang, L.; Zhao, G.; Hassan, H.M.; Fan, S.; Zhou, W.; Han, S.; Zhang, L.; et al. Mitochondrial dysfunction and inhibition of myoblast differentiation in mice with high-fat-diet-induced pre-diabetes. J. Cell Physiol. 2019, 234, 7510–7523. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of Sphingosine Kinase 1 Prevents Ceramide Accumulation and Ameliorates Muscle Insulin Resistance in High-Fat Diet–Fed Mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Galadari, A.; Thayyullathil, F. Role of ceramide in diabetes mellitus: Evidence and mechanisms. Lipids Heal. Dis. 2013, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Grzelkowska-Kowalczyk, K.; Wieteska-Skrzeczyńska, W.; Grabiec, K.; Tokarska, J. High glucose-mediated alterations of mechanisms important in myogenesis of mouse C2C12 myoblasts. Cell Biol. Int. 2012, 37, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.L.; Castro, A.V.; Iyer, M.; Paszkiewicz, R.L.; Bediako, I.A.; Szczepaniak, L.S.; Szczepaniak, E.W.; Bergman, R.N.; Kolka, C.M. Insulin access to skeletal muscle is impaired during the early stages of diet-induced obesity. Obesity 2016, 24, 1922–1928. [Google Scholar] [CrossRef]
- Nagata, Y.; Partridge, T.A.; Matsuda, R.; Zammit, P.S. Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. J. Cell Biol. 2006, 174, 245–253. [Google Scholar] [CrossRef]
- Mebarek, S.; Komati, H.; Naro, F.; Zeiller, C.; Alvisi, M.; Lagarde, M.; Prigent, A.-F.; Némoz, G. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J. Cell Sci. 2007, 120, 407–416. [Google Scholar] [CrossRef]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: Structure, function, regulation, and role in disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef]
- Tamilarasan, K.P.; Temmel, H.; Das, S.K.; Al Zoughbi, W.; Schauer, S.; Vesely, P.W.; Hoefler, G. Skeletal muscle damage and impaired regeneration due to LPL-mediated lipotoxicity. Cell Death Dis. 2012, 3, e354. [Google Scholar] [CrossRef]
- Ryall, J.G.; Dell'Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Sartorelli, V.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell. Biochem. 2010, 349, 139–147. [Google Scholar] [CrossRef]
- Mathieu-Costello, O.A.; Tsuchiya, N.; Filburn, C.; Hansford, R.G.; Kim, B.; Kim, J.-S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.-Y.; et al. Mitochondrial biogenesis during cellular differentiation. Am. J. Physiol. Physiol. 1997, 272, C1345–C1351. [Google Scholar] [CrossRef]
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389. [Google Scholar] [CrossRef]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef]
- Joseph, J.; Doles, J.D. Disease-associated metabolic alterations that impact satellite cells and muscle regeneration: Perspectives and therapeutic outlook. Nutr. Metab. 2021, 18, 1–8. [Google Scholar] [CrossRef]
- Xu, D.; Wang, L.; Jiang, Z.; Zhao, G.; Hassan, H.M.; Sun, L.; Fan, S.; Zhou, Z.; Zhang, L.; Wang, T. A new hypoglycemic mechanism of catalpol revealed by enhancing MyoD/MyoG-mediated myogenesis. Life Sci. 2018, 209, 313–323. [Google Scholar] [CrossRef]
- Li, S.; Mao, J.; Zhou, W. Prediabetes is associated with loss of appendicular skeletal muscle mass and sarcopenia. Front. Nutr. 2023, 10, 1109824. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, S.; Wang, W.; Zhou, C.; Xu, L.; Qiu, J.; Wang, J.; Meng, X.; Liang, Y.; Niu, K.; et al. Poor Lower Extremity Function Was Associated with Pre-Diabetes and Diabetes in Older Chinese People. PLoS ONE 2014, 9, e115883. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, X.-H.; Feng, W.-M.; Ye, G.-F.; Qiu, W.; Li, B. Analysis of Inflammatory Mediators in Prediabetes and Newly Diagnosed Type 2 Diabetes Patients. J. Diabetes Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Alrabiah, M.; Al-Aali, K.A.; Al-Sowygh, Z.H.; Binmahfooz, A.M.; Mokeem, S.A.; Abduljabbar, T. Association of advanced glycation end products with peri-implant inflammation in prediabetes and type 2 diabetes mellitus patients. Clin. Implant. Dent. Relat. Res. 2018, 20, 535–540. [Google Scholar] [CrossRef]
- Pereira, S.; Marliss, E.B.; Morais, J.A.; Chevalier, S.; Gougeon, R. Insulin Resistance of Protein Metabolism in Type 2 Diabetes. Diabetes 2008, 57, 56–63. [Google Scholar] [CrossRef]
- Tabara, Y.; Ikezoe, T.; Yamanaka, M.; Setoh, K.; Segawa, H.; Kawaguchi, T.; Kosugi, S.; Nakayama, T.; Ichihashi, N.; Tsuboyama, T.; et al. Advanced Glycation End Product Accumulation Is Associated With Low Skeletal Muscle Mass, Weak Muscle Strength, and Reduced Bone Density: The Nagahama Study. Journals Gerontol. Ser. A 2018, 74, 1446–1453. [Google Scholar] [CrossRef]
- Mesinovic, J.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sarcopenia and type 2 diabetes mellitus: A bidirectional relationship. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1057–1072. [Google Scholar] [CrossRef]
- Sayer, A.A.; Dennison, E.M.; Syddall, H.E.; Gilbody, H.J.; Phillips, D.I.; Cooper, C. Type 2 diabetes, muscle strength, and impaired physical function: The tip of the iceberg? Diabetes Care 2005, 28, 2541–2543. [Google Scholar] [CrossRef]
- Perreault, L.; Bergman, B.C.; Hunerdosse, D.M.; Eckel, R.H. Altered Intramuscular Lipid Metabolism Relates to Diminished Insulin Action in Men, but Not Women, in Progression to Diabetes. Obesity 2010, 18, 2093–2100. [Google Scholar] [CrossRef]
- Mahler, R.J.; Adler, M.L. Type 2 Diabetes Mellitus: Update on Diagnosis, Pathophysiology, and Treatment. J. Clin. Endocrinol. Metab. 1999, 84, 1165–1171. [Google Scholar] [CrossRef]
- Authors/Task Force Members; Ryden, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.P.; et al. ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The task force on diabetes, pre-diabetes, and cardiovascular diseases of the european society of cardiology (ESC) and developed in collaboration with the european association for the study of diabetes (EASD). Eur. Heart, J. 2013, 34, 3035–3087. [Google Scholar]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Huang, B.K.; Monu, J.U.V.; Doumanian, J. Diabetic Myopathy: MRI Patterns and Current Trends. Am. J. Roentgenol. 2010, 195, 198–204. [Google Scholar] [CrossRef]
- Kapur, S.; Brunet, J.A.; McKendry, R.J. Diabetic muscle infarction: Case report and review. J. Rheumatol. 2004, 31, 190–194. [Google Scholar]
- Horton, W.B.; Taylor, J.S.; Ragland, T.J.; Subauste, A.R. Diabetic muscle infarction: A systematic review. BMJ Open Diabetes Res. Care 2015, 3, e000082. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia–reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Sran, S.; Sran, M.; Ferguson, N.; Anand, P. Diabetic Myonecrosis: Uncommon Complications in Common Diseases. Case Rep. Endocrinol. 2014, 2014, 1–3. [Google Scholar] [CrossRef]
- Chester, C.S.; Banker, B.Q. Focal Infarction of Muscle in Diabetics. Diabetes Care 1986, 9, 623–630. [Google Scholar] [CrossRef]
- Trujillo-Santos, A.J. Diabetic Muscle Infarction: An underdiagnosed complication of long-standing diabetes. Diabetes Care 2003, 26, 211–215. [Google Scholar] [CrossRef]
- Saikia, U.; Choudhury, B.; Sarma, D.; Saikia, M.; Choudhury, S.; Bhuyan, D. Diabetic myonecrosis: An underreported complication of diabetes mellitus. Indian J. Endocrinol. Metab. 2011, 15, S58–S61. [Google Scholar] [CrossRef]
- Jelinek, J.S.; Murphey, M.D.; Aboulafia, A.J.; Dussault, R.G.; Kaplan, P.A.; Snearly, W.N. Muscle Infarction in Patients with Diabetes Mellitus: MR Imaging Findings. Radiology 1999, 211, 241–247. [Google Scholar] [CrossRef]
- De Vlieger, G.; Bammens, B.; Claus, F.; Vos, R.; Claes, K. Diabetic Muscle Infarction: A Rare Cause of Acute Limb Pain in Dialysis Patients. Case Rep. Nephrol. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Goswami, P.; Baruah, M.P. The Role of MRI in Diagnosis of Diabetic Muscle Infarction: An Underdiagnosed Entity. Int. J. Endocrinol. Metab. 2012, 9, 353–355. [Google Scholar] [CrossRef]
- Morcuende, J.A.; Dobbs, M.B.; Crawford, H.; Buckwalter, J.A. Diabetic muscle infarction. Iowa Orthop. J. 2000, 20, 65–74. [Google Scholar] [PubMed]
- Ijzerman, T.H.; Schaper, N.C.; Melai, T.; Meijer, K.; Willems, P.J.; Savelberg, H.H. Lower extremity muscle strength is reduced in people with type 2 diabetes, with and without polyneuropathy, and is associated with impaired mobility and reduced quality of life. Diabetes Res. Clin. Pr. 2011, 95, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yan, J.; Zhu, H.; Zhang, Z.; Jiang, Y.; Zhang, X.; Wu, Y.; Lu, J.; Cun, H.; He, B. Low thigh muscle strength in relation to myosteatosis in patients with type 2 diabetes mellitus. Sci. Rep. 2023, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Nielsen, S.; Mogensen, C.E.; Jakobsen, J. Muscle Strength in Type 2 Diabetes. Diabetes 2004, 53, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Meex, R.; Ouwens, D.M.; Sparks, L.; Hoeks, J.; Schaart, G.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. High Oxidative Capacity Due to Chronic Exercise Training Attenuates Lipid-Induced Insulin Resistance. Diabetes 2012, 61, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.; Yule, V.; Syddall, H.; Dennison, E.; Cooper, C.; Sayer, A.A. Is Hand-Held Dynamometry Useful for the Measurement of Quadriceps Strength in Older People? A Comparison with the Gold Standard Biodex Dynamometry. Gerontology 2006, 52, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Jung, S.M.; Bang, H.; Kim, H.S.; Kim, Y.B. Association between muscle strength and type 2 diabetes mellitus in adults in Korea: Data from the Korea national health and nutrition examination survey (KNHANES) VI. Medicine 2018, 97, e10984. [Google Scholar] [CrossRef]
- Li, J.J.; Wittert, G.A.; Vincent, A.; Atlantis, E.; Shi, Z.; Appleton, S.L.; Hill, C.L.; Jenkins, A.J.; Januszewski, A.S.; Adams, R.J. Muscle grip strength predicts incident type 2 diabetes: Population-based cohort study. Metabolism 2016, 65, 883–892. [Google Scholar] [CrossRef]
- McGrath, R.; Vincent, B.M.; Al Snih, S.; Markides, K.S.; Peterson, M.D. The Association Between Muscle Weakness and Incident Diabetes in Older Mexican Americans. J. Am. Med Dir. Assoc. 2017, 18, 452.e7–452.e12. [Google Scholar] [CrossRef]
- Peterson, M.D.; Zhang, P.; Choksi, P.; Markides, K.S.; Al Snih, S. Muscle Weakness Thresholds for Prediction of Diabetes in Adults. Sports Med. 2016, 46, 619–628. [Google Scholar] [CrossRef] [PubMed]
- van der Kooi, A.-L.L.F.; Snijder, M.B.; Peters, R.J.G.; van Valkengoed, I.G.M. The Association of Handgrip Strength and Type 2 Diabetes Mellitus in Six Ethnic Groups: An Analysis of the HELIUS Study. PLoS ONE 2015, 10, e0137739. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.P.; Teo, K.K.; Rangarajan, S.; Lopez-Jaramillo, P.; Avezum, A., Jr.; Orlandini, A.; Seron, P.; Ahmed, S.H.; Rosergen, A.; Kelishad, R.; et al. Prognostic value of grip strength: Findings from the Prospective Urban Rural Epidemiology (PURE) study. Lancet 2015, 386, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hao, Y.T. Effect of handgrip on coronary artery disease and myocardial infarction: A Mendelian randomization study. Sci. Rep. 2017, 7, 954. [Google Scholar] [CrossRef] [PubMed]
- Schaap, L.A.; Pluijm, S.M.F.; Deeg, D.J.H.; Harris, T.B.; Kritchevsky, S.B.; Newman, A.B.; Colbert, L.H.; Pahor, M.; Rubin, S.M.; Tylavsky, F.A.; et al. Higher Inflammatory Marker Levels in Older Persons: Associations With 5-Year Change in Muscle Mass and Muscle Strength. Journals Gerontol. Ser. A 2009, 64A, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef]
- Hickey, M.S.; Carey, J.O.; Azevedo, J.L.; Houmard, J.A.; Pories, W.J.; Israel, R.G.; Dohm, G.L. Skeletal muscle fiber composition is related to adiposity and in vitro glucose transport rate in humans. Am. J. Physiol. Metab. 1995, 268, E453–E457. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.J.; Barakat, H.A.; Dohm, G.L.; Pories, W.J.; MacDonald, K.G.; Cunningham, P.R.G.; Swanson, M.S.; Houmard, J.A. Muscle fiber type is associated with obesity and weight loss. Am. J. Physiol. Metab. 2002, 282, E1191–E1196. [Google Scholar] [CrossRef]
- Stuart, C.A.; McCurry, M.P.; Marino, A.; South, M.A.; Howell, M.E.A.; Layne, A.S.; Ramsey, M.W.; Stone, M.H. Slow-Twitch Fiber Proportion in Skeletal Muscle Correlates With Insulin Responsiveness. J. Clin. Endocrinol. Metab. 2013, 98, 2027–2036. [Google Scholar] [CrossRef]
- Albers, P.H.; Pedersen, A.J.; Birk, J.B.; Kristensen, D.E.; Vind, B.F.; Baba, O.; Nøhr, J.; Højlund, K.; Wojtaszewski, J.F. Human Muscle Fiber Type–Specific Insulin Signaling: Impact of Obesity and Type 2 Diabetes. Diabetes 2014, 64, 485–497. [Google Scholar] [CrossRef]
- Kelley, D.E.; Robinson, M.M.; Sather, B.K.; Burney, E.R.; Ehrlicher, S.E.; Stierwalt, H.D.; Franco, M.C.; Newsom, S.A.; Dasari, S.; Miller, B.F.; et al. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J. Appl. Physiol. 1997, 83, 166–171. [Google Scholar] [CrossRef]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schön, M.R.; Blüher, M.; Punkt, K. Altered Fiber Distribution and Fiber-Specific Glycolytic and Oxidative Enzyme Activity in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef]
- Giebelstein, J.; Poschmann, G.; Højlund, K.; Schechinger, W.; Dietrich, J.; Levin, K.; Beck-Nielson, H.; Podwojski, K.; Stuhler, K.; Meyer, H.E.; et al. The proteomic signature of insulin-resistant human skeletal muscle reveals increased glycolytic and decreased mitochondrial enzymes. Diabetologia 2012, 55, 1114–1127. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R.; Catalano, M.G.; Brignardello, E.; Danni, O.; Boccuzzi, G. Oxidative Stress Impairs Skeletal Muscle Repair in Diabetic Rats. Diabetes 2004, 53, 1082–1088. [Google Scholar] [CrossRef]
- Woo, M.; Isganaitis, E.; Cerletti, M.; Fitzpatrick, C.; Wagers, A.J.; Jimenez-Chillaron, J.; Patti, M.E.; Domingues-Faria, C.; Vasson, M.-P.; Goncalves-Mendes, N.; et al. Early Life Nutrition Modulates Muscle Stem Cell Number: Implications for Muscle Mass and Repair. Stem Cells Dev. 2011, 20, 1763–1769. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, H.; Lee, I.H.; Modi, S.; Wang, X.; Du, J.; Mitch, W.E. PTEN Inhibition Improves Muscle Regeneration in Mice Fed a High-Fat Diet. Diabetes 2010, 59, 1312–1320. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bryner, R.W.; Alway, S.E.; Acevedo, L.M.; Raya, A.I.; Ríos, R.; Aguilera-Tejero, E.; Rivero, J.-L.L.; Bennett, B.T.; Wilson, J.C.; et al. Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. Am. J. Physiol. Physiol. 2008, 295, C521–C528. [Google Scholar] [CrossRef]
- Nguyen, M.-H.; Cheng, M.; Koh, T.J. Impaired Muscle Regeneration in Ob/ob and Db/db Mice. Sci. World, J. 2011, 11, 1525–1535. [Google Scholar] [CrossRef]
- Amacher, S.L.; Buskin, J.N.; Hauschka, S.D. Multiple Regulatory Elements Contribute Differentially to Muscle Creatine Kinase Enhancer Activity in Skeletal and Cardiac Muscle. Mol. Cell. Biol. 1993, 13. [Google Scholar] [CrossRef]
- Wheeler, M.T.; Snyder, E.C.; Patterson, M.N.; Swoap, S.J.; Miura, S.; Kai, Y.; Tadaishi, M.; Tokutake, Y.; Sakamoto, K.; Bruce, C.R.; et al. An E-box within the MHC IIB gene is bound by MyoD and is required for gene expression in fast muscle. Am. J. Physiol. Physiol. 1999, 276, C1069–C1078. [Google Scholar] [CrossRef]
- Wentworth, B.M.; Donoghue, M.; Engert, J.C.; Berglund, E.B.; Rosenthal, N. Paired MyoD-binding sites regulate myosin light chain gene expression. Proc. Natl. Acad. Sci. USA 1991, 88, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Vettor, R.; Fabris, R.; Serra, R.; Lombardi, A.; Tonello, C.; Granzotto, M.; Marzolo, M.; Carruba, M.; Ricquier, D.; Federspil, G.; et al. Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int. J. Obes. 2002, 26, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Cheng, P.F.; Weintraub, H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc. Natl. Acad. Sci. USA. [CrossRef] [PubMed]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Knoblauch, A.; Will, C.; Goncharenko, G.; Ludwig, S.; Wixler, V. The binding of Mss4 to α-integrin subunits regulates matrix metalloproteinase activation and fibronectin remodeling. FASEB J. 2006, 21, 497–510. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Zong, H.; Bastie, C.C.; Xu, J.; Fassler, R.; Campbell, K.P.; Kurland, I.J.; Pessin, J.E. Insulin resistance in striated muscle-specific integrin receptor beta1-deficient mice. J. Biol. Chem. 2009, 284, 4679–4688. [Google Scholar] [CrossRef]
- Tam, C.S.; Chaudhuri, R.; Hutchison, A.T.; Samocha-Bonet, D.; Heilbronn, L.K. Skeletal muscle extracellular matrix remodeling after short-term overfeeding in healthy humans. Metabolism 2017, 67, 26–30. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y. Role of matrix metalloproteinases in skeletal muscle: Migration, differentiation, regeneration and fibrosis. Cell Adhes. Migr. 2009, 3, 337–341. [Google Scholar] [CrossRef]
- Leeman, M.F.; Curran, S.; Murray, G.I. The Structure, Regulation, and Function of Human Matrix Metalloproteinase-13. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Choi, I.; Lee, Y.-H. Implications of Skeletal Muscle Extracellular Matrix Remodeling in Metabolic Disorders: Diabetes Perspective. Int. J. Mol. Sci. 2020, 21, 3845. [Google Scholar] [CrossRef] [PubMed]
- Eringa, E.C.; Serne, E.H.; Meijer, R.I.; Schalkwijk, C.G.; Houben, A.J.H.M.; Stehouwer, C.D.A.; Smulders, Y.M.; van Hinsbergh, V.W.M. Endothelial dysfunction in (pre)diabetes: Characteristics, causative mechanisms and pathogenic role in type 2 diabetes. Rev. Endocr. Metab. Disord. 2013, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Wong, B.W.; De Bock, K. Treating Diabetes by Blocking a Vascular Growth Factor. Cell Metab. 2012, 16, 553–555. [Google Scholar] [CrossRef][Green Version]
- Hagberg, C.E.; Mehlem, A.; Falkevall, A.; Muhl, L.; Fam, B.C.; Ortsäter, H.; Scotney, P.; Nyqvist, D.; Samén, E.; Lu, L.; et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 2012, 490, 426–430. [Google Scholar] [CrossRef]
- Musale, V.; Wasserman, D.H.; Kang, L. Extracellular matrix remodelling in obesity and metabolic disorders. Life Metab. 2023, 2. [Google Scholar] [CrossRef]
- Aleksandrowicz, R.; Strączkowski, M. Link between insulin resistance and skeletal muscle extracellular matrix remodeling. Endocr. Connect. 2023, 12. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PreDM | T2DM |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dlamini, M.; Khathi, A. Prediabetes-Associated Changes in Skeletal Muscle Function and Their Possible Links with Diabetes: A Literature Review. Int. J. Mol. Sci. 2024, 25, 469. https://doi.org/10.3390/ijms25010469
Dlamini M, Khathi A. Prediabetes-Associated Changes in Skeletal Muscle Function and Their Possible Links with Diabetes: A Literature Review. International Journal of Molecular Sciences. 2024; 25(1):469. https://doi.org/10.3390/ijms25010469
Chicago/Turabian StyleDlamini, Mandlakazi, and Andile Khathi. 2024. "Prediabetes-Associated Changes in Skeletal Muscle Function and Their Possible Links with Diabetes: A Literature Review" International Journal of Molecular Sciences 25, no. 1: 469. https://doi.org/10.3390/ijms25010469
APA StyleDlamini, M., & Khathi, A. (2024). Prediabetes-Associated Changes in Skeletal Muscle Function and Their Possible Links with Diabetes: A Literature Review. International Journal of Molecular Sciences, 25(1), 469. https://doi.org/10.3390/ijms25010469