
BAY-3827 and SBI-0206965: Potent AMPK Inhibitors That Paradoxically Increase Thr172 Phosphorylation


Abstract
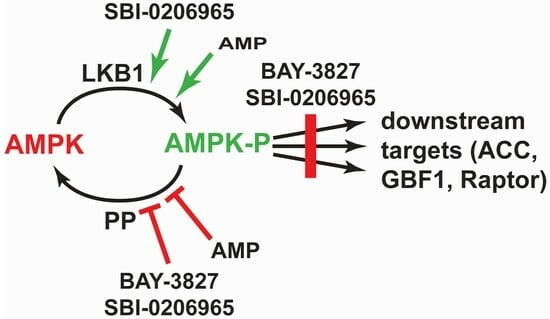
:
1. Introduction
2. Results
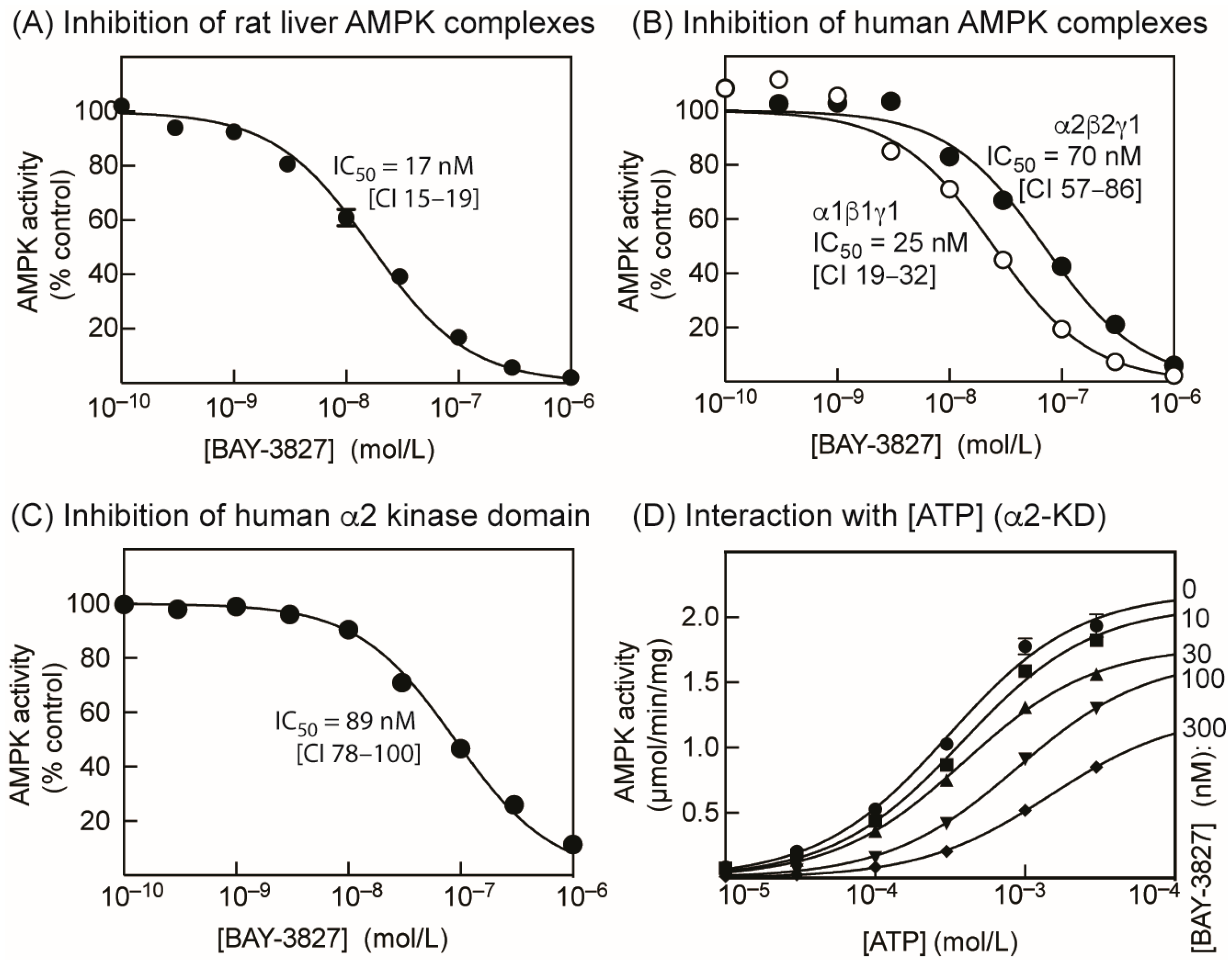
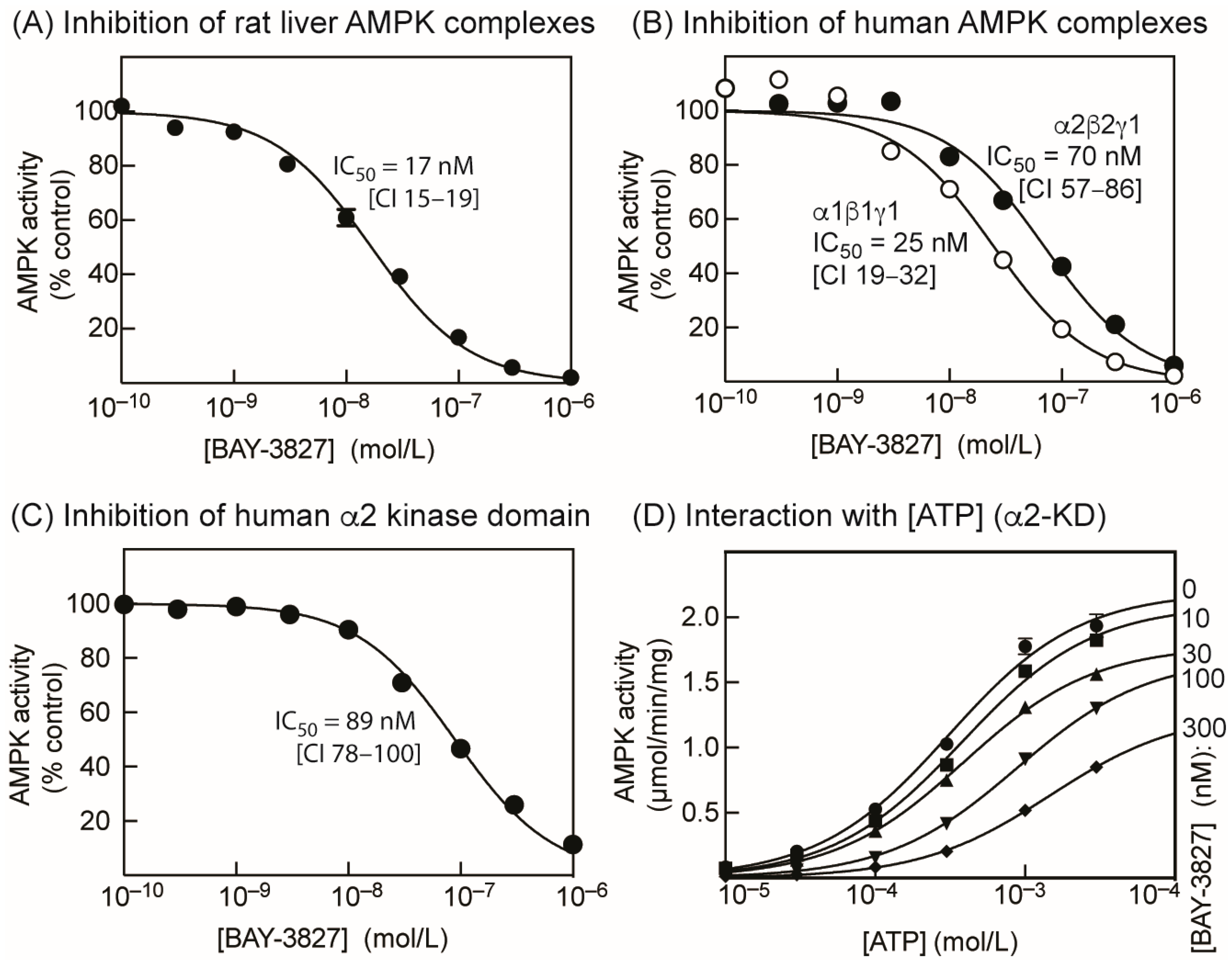
2.1. BAY-3827 Is a Mixed Type Inhibitor of AMPK
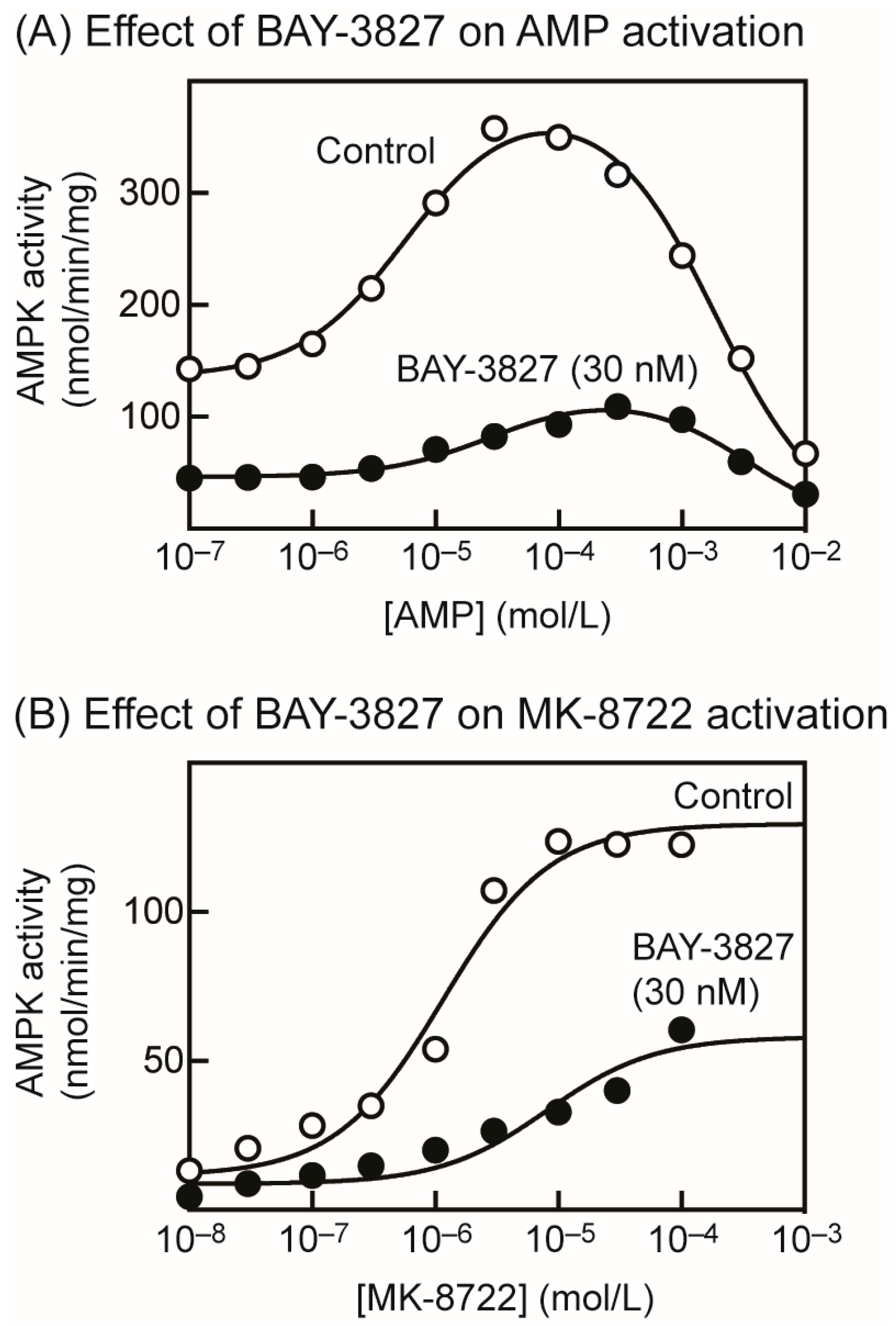
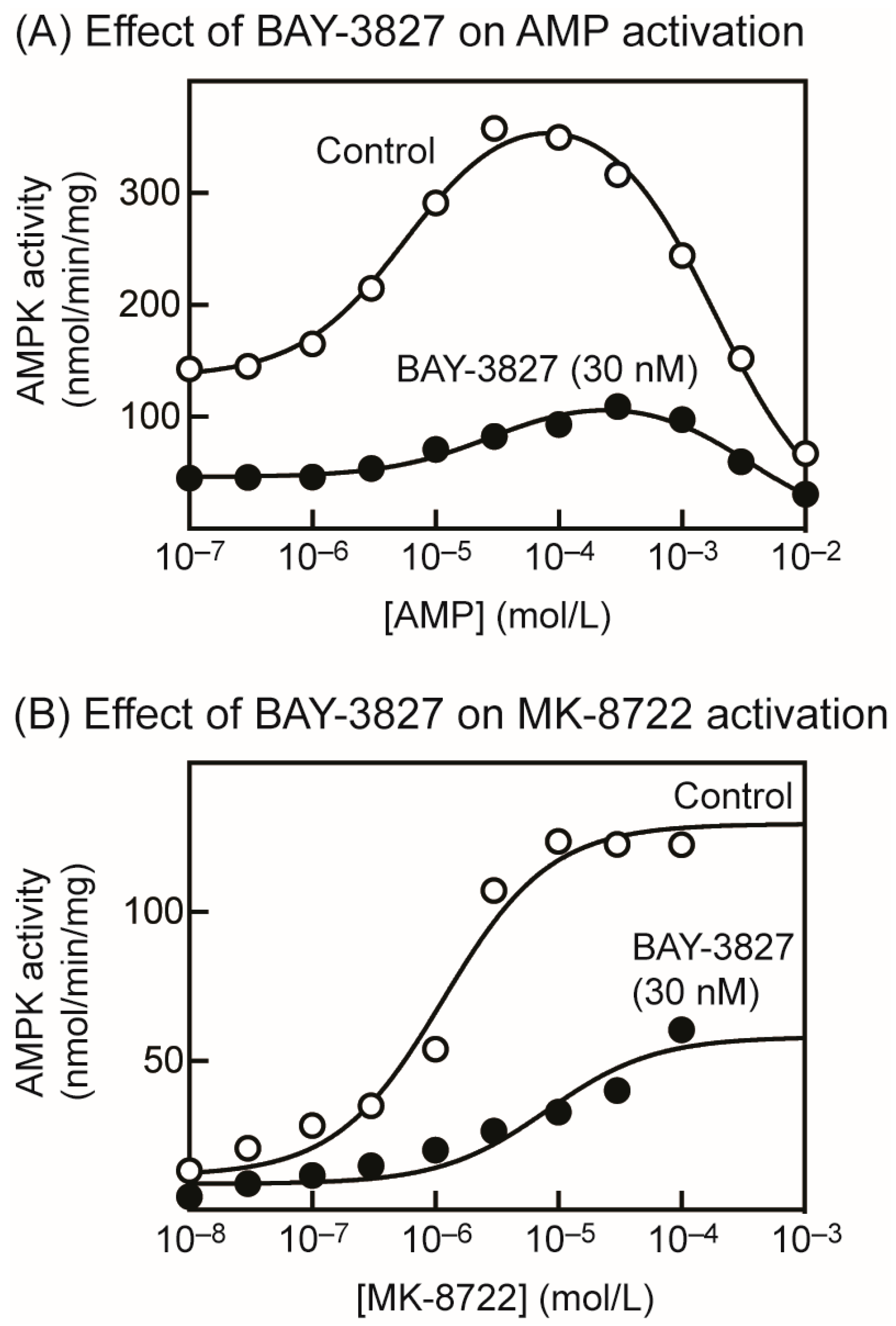
2.2. BAY-3827 Interacts with Activation by AMP and MK-8722
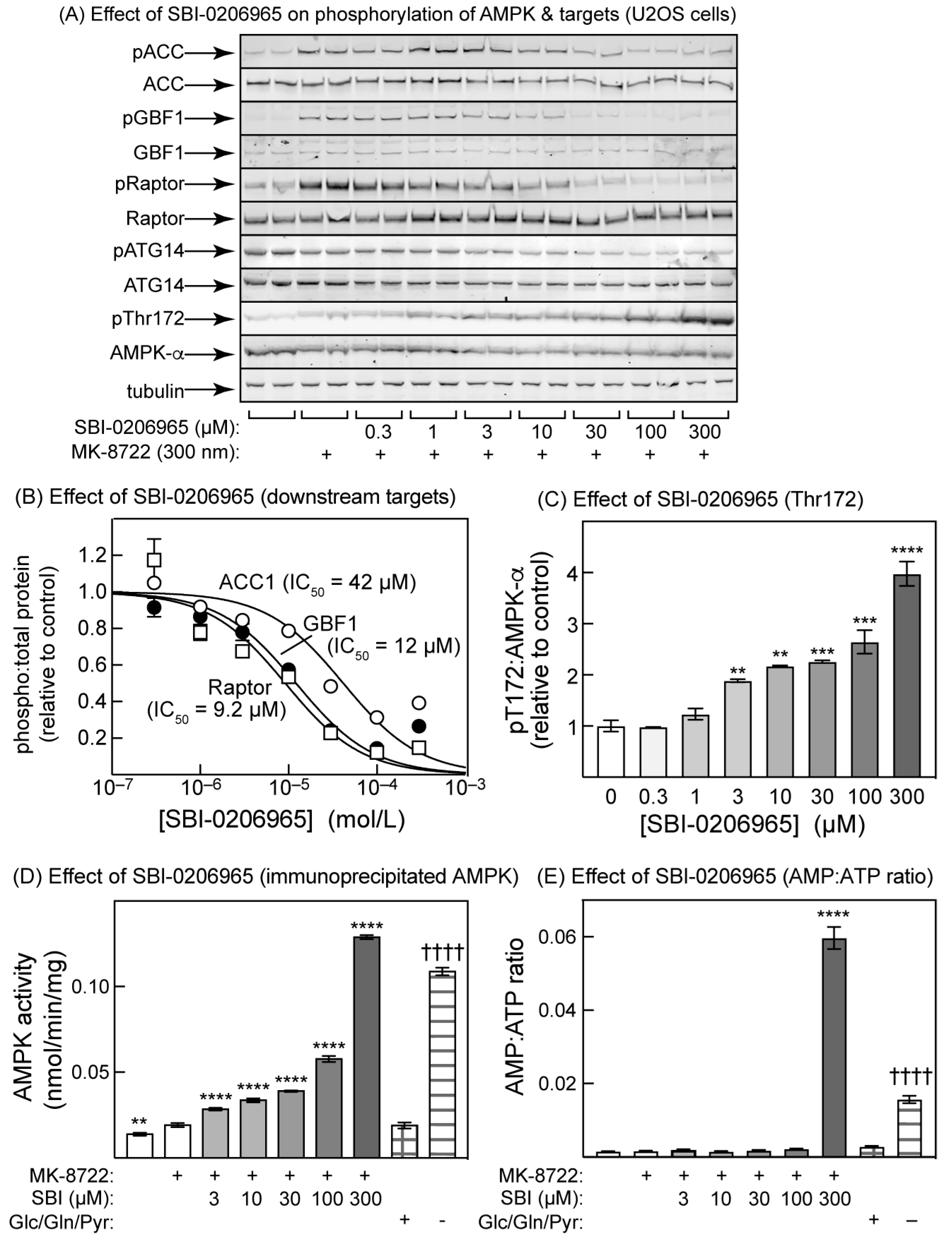
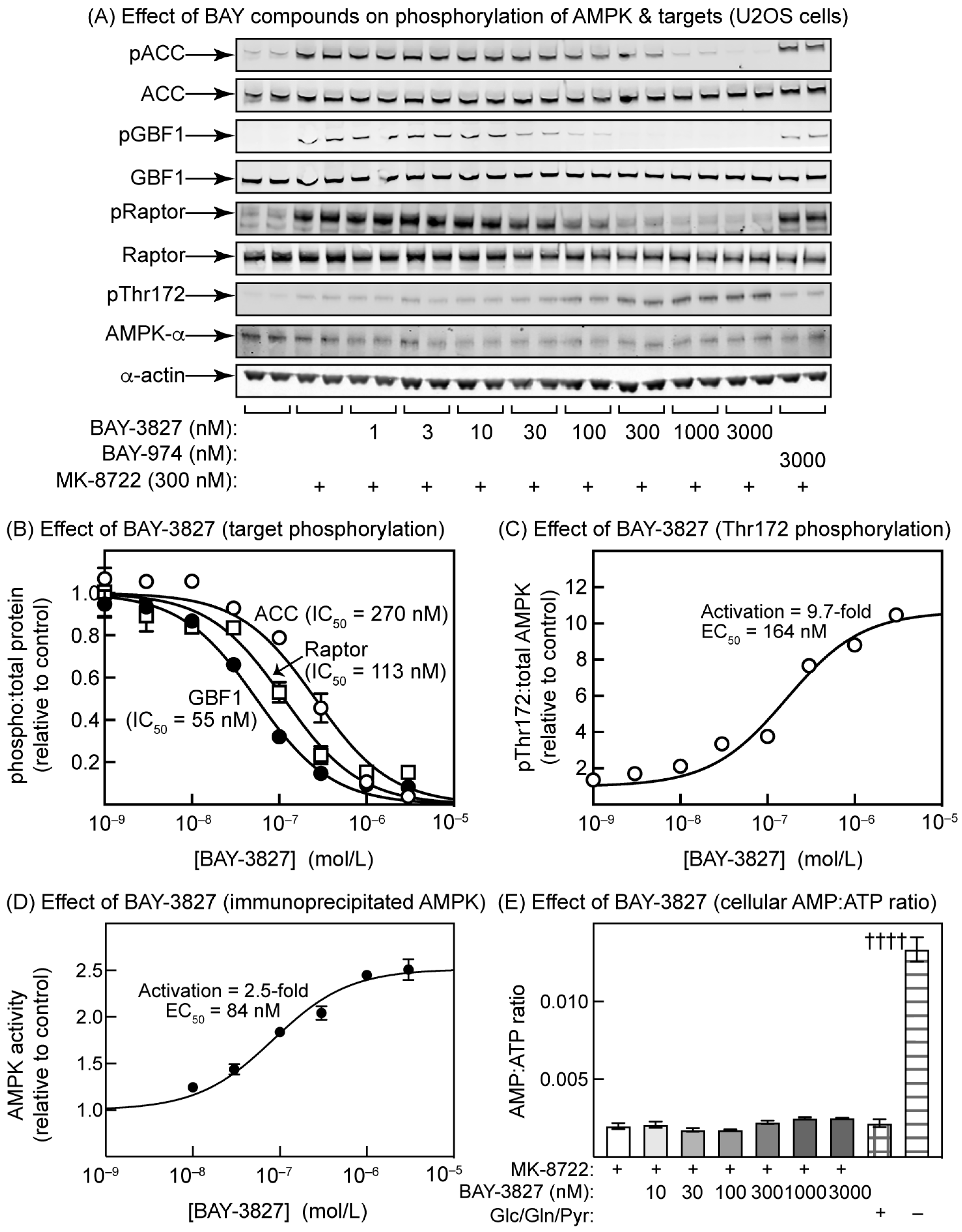
2.3. Effects of BAY-3827 on Phosphorylation of AMPK Targets in U2OS Cells
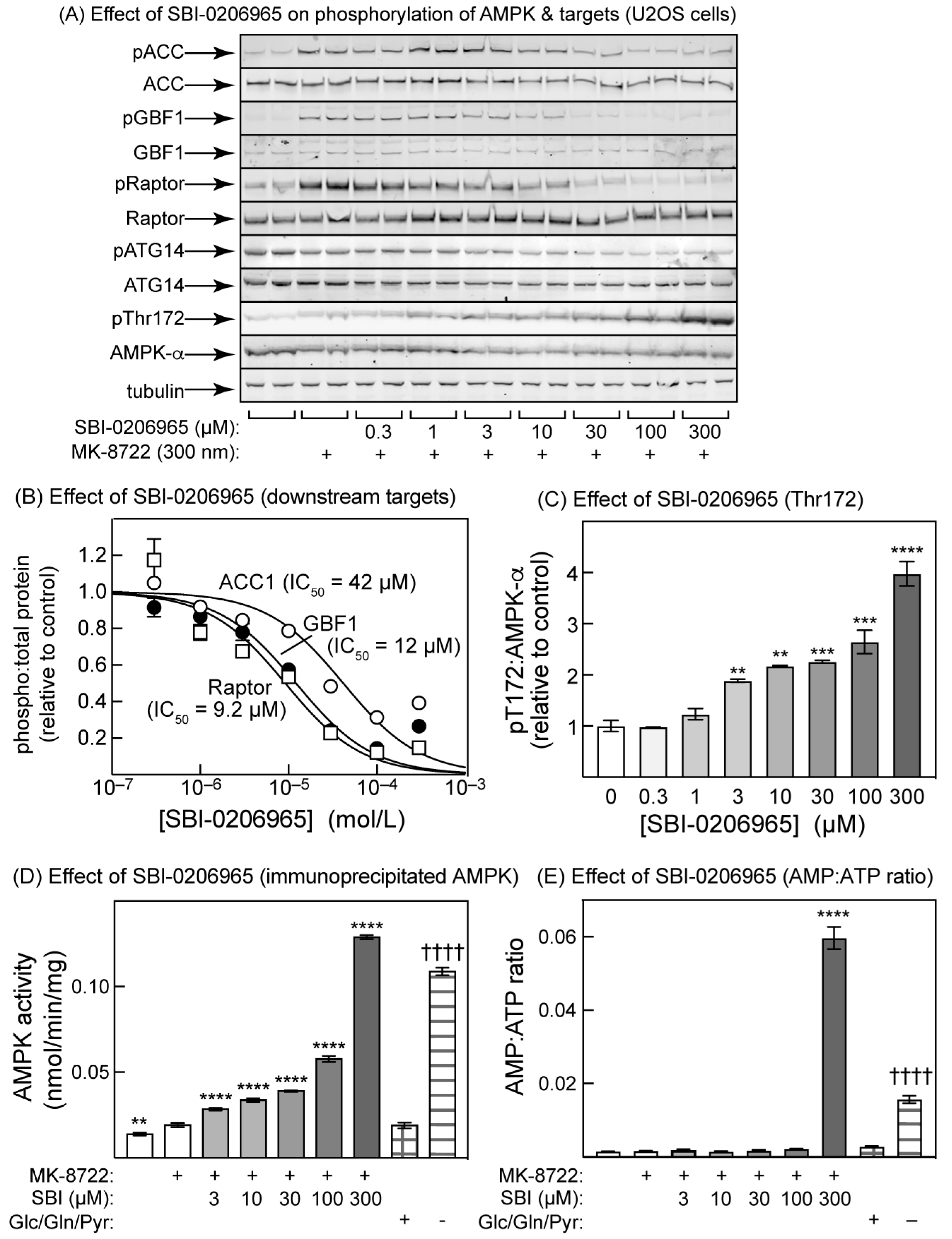
2.4. SBI-0206965 Also Promotes Thr172 Phosphorylation in U2OS Cells
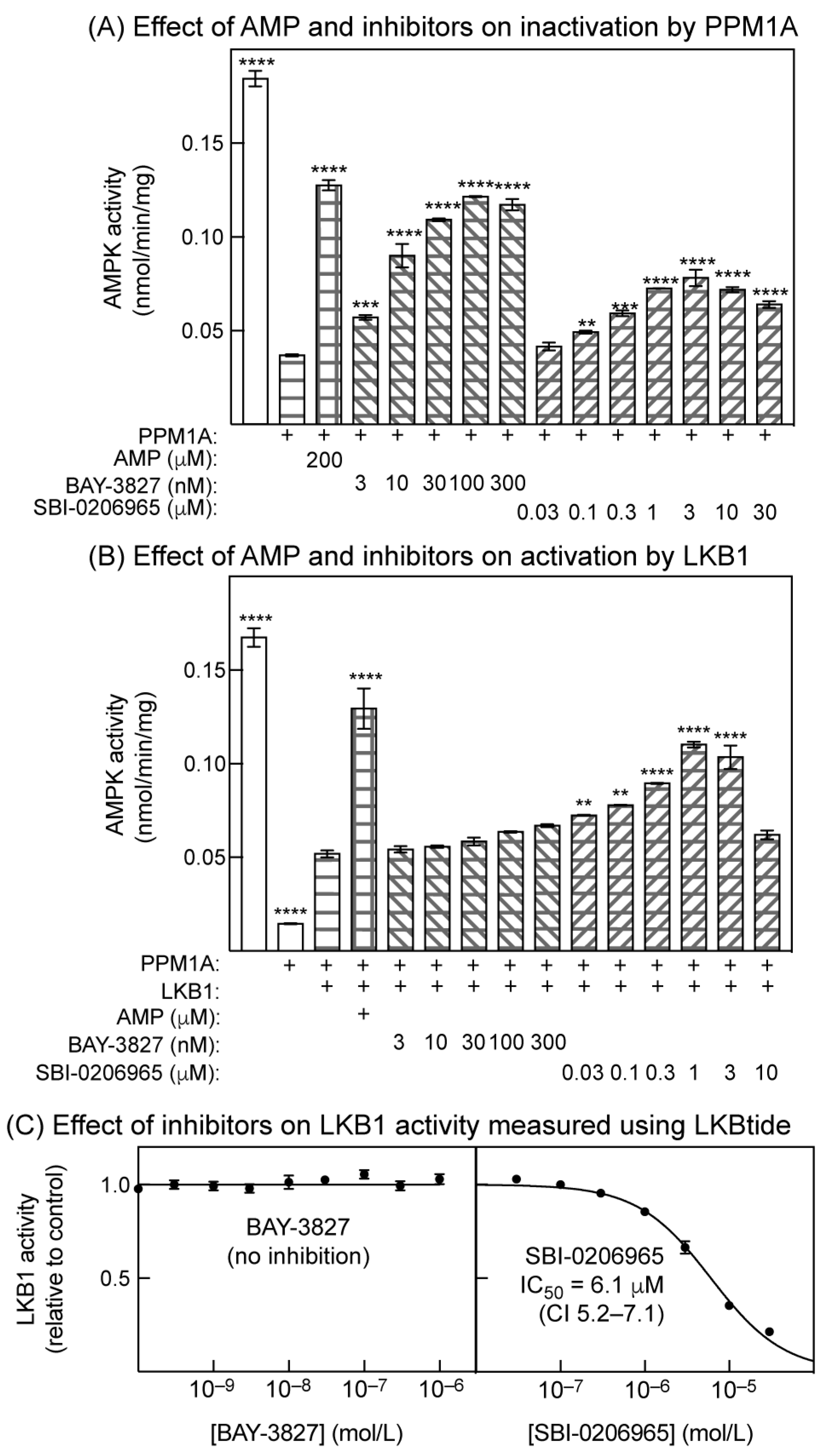
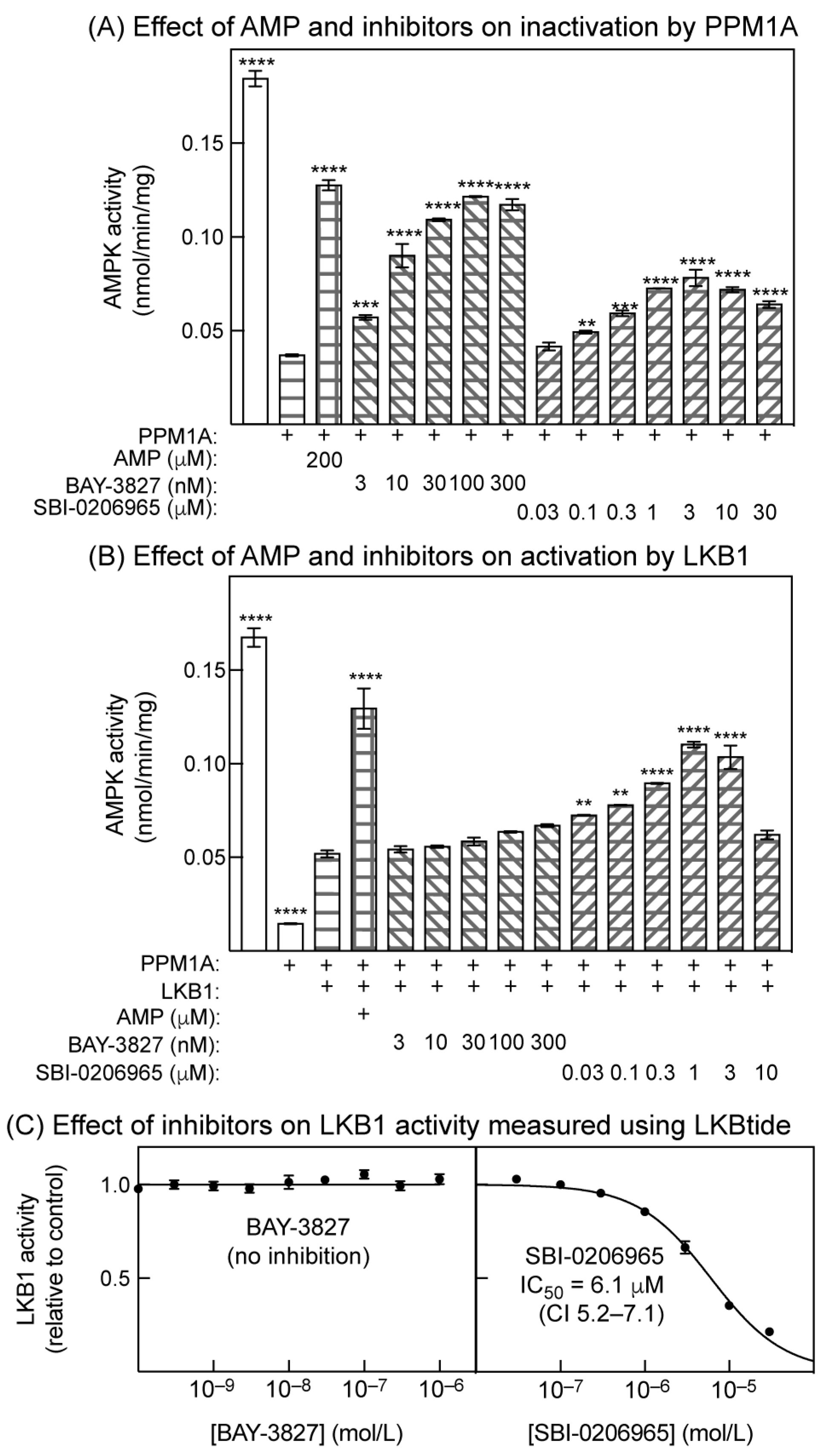
2.5. Effects of Inhibitors on Inactivation by Protein Phosphatase and Activation by LKB1 in Cell-Free Assays
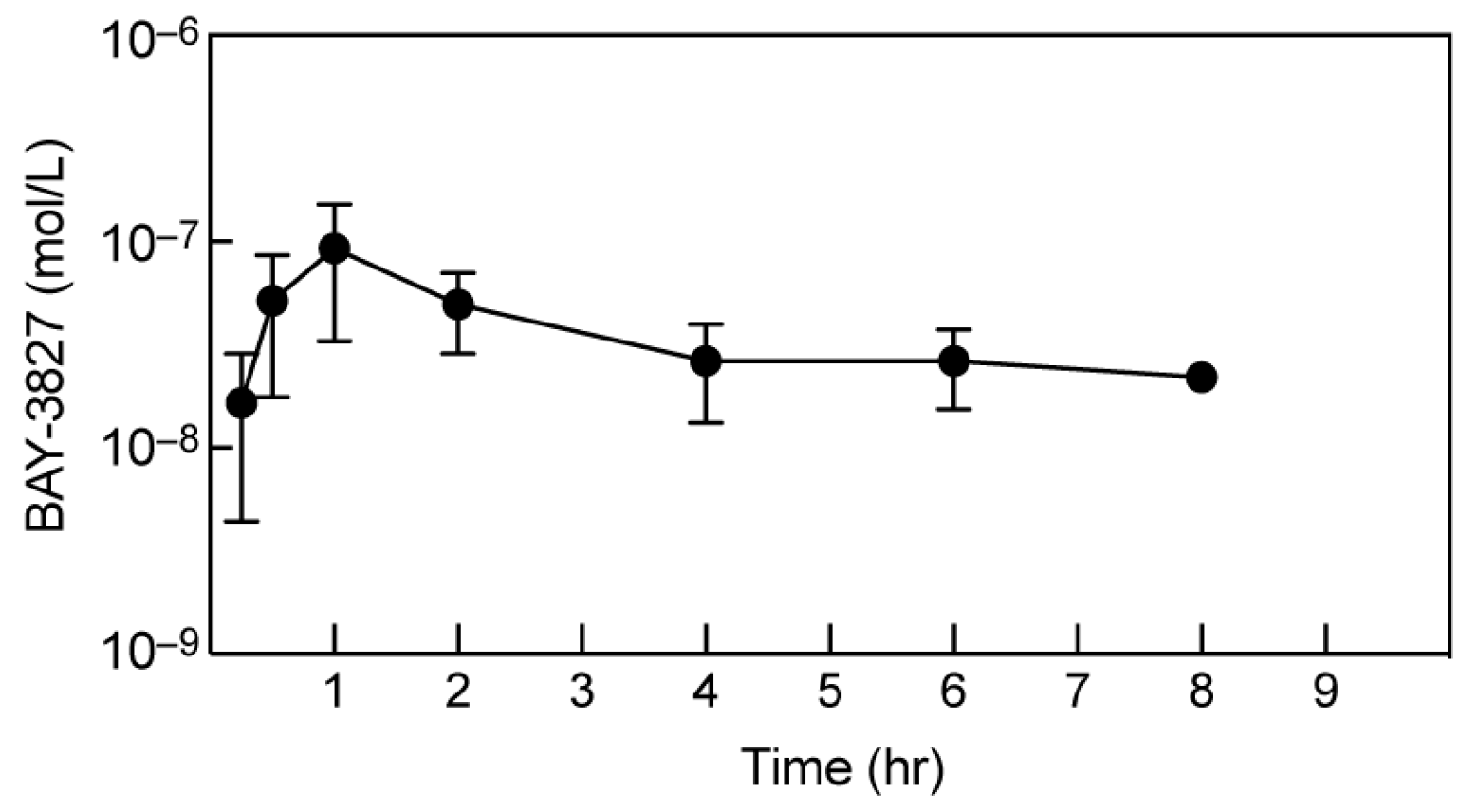
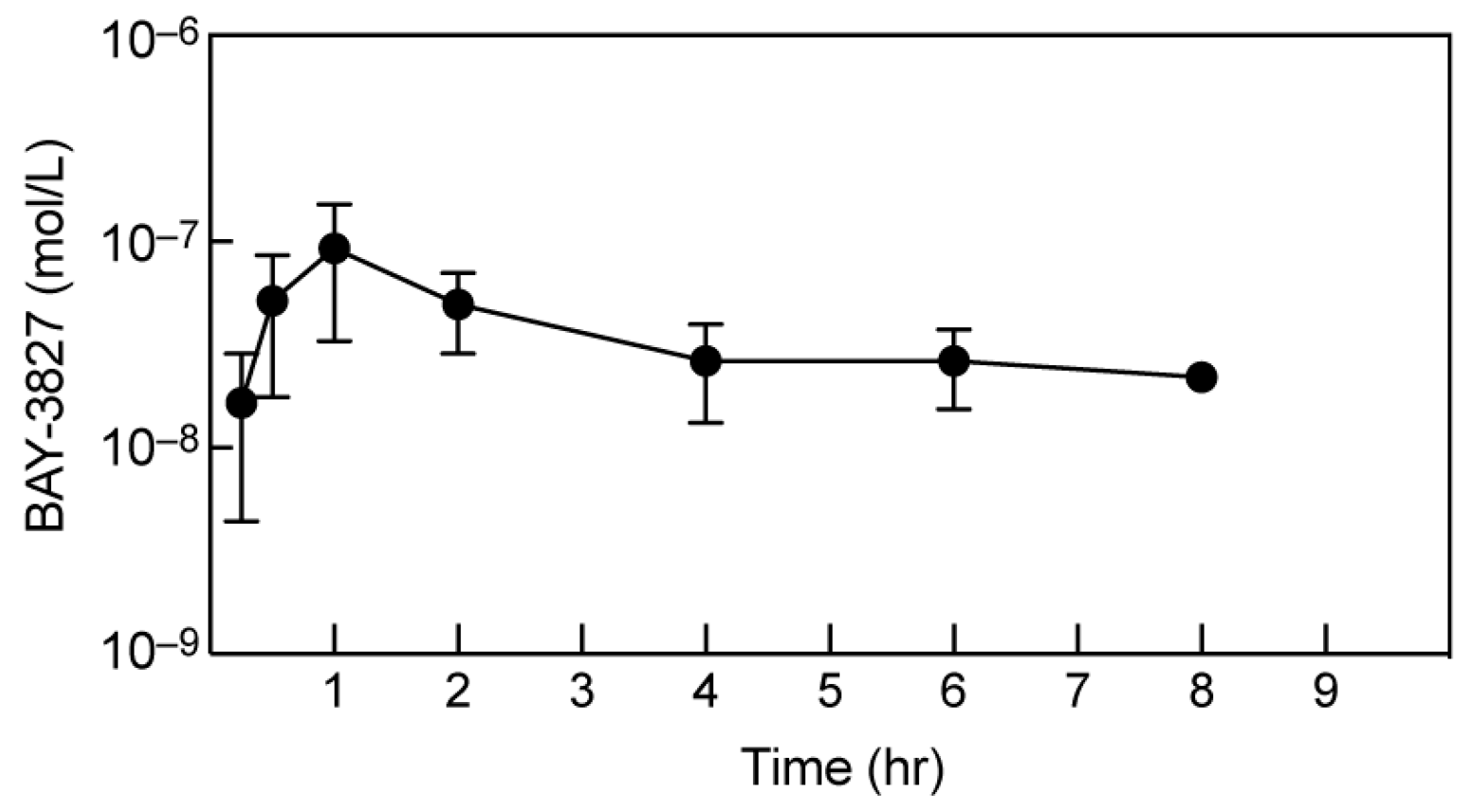
2.6. BAY-3827 Bio-Availability Is Low following Oral Administration in Mice
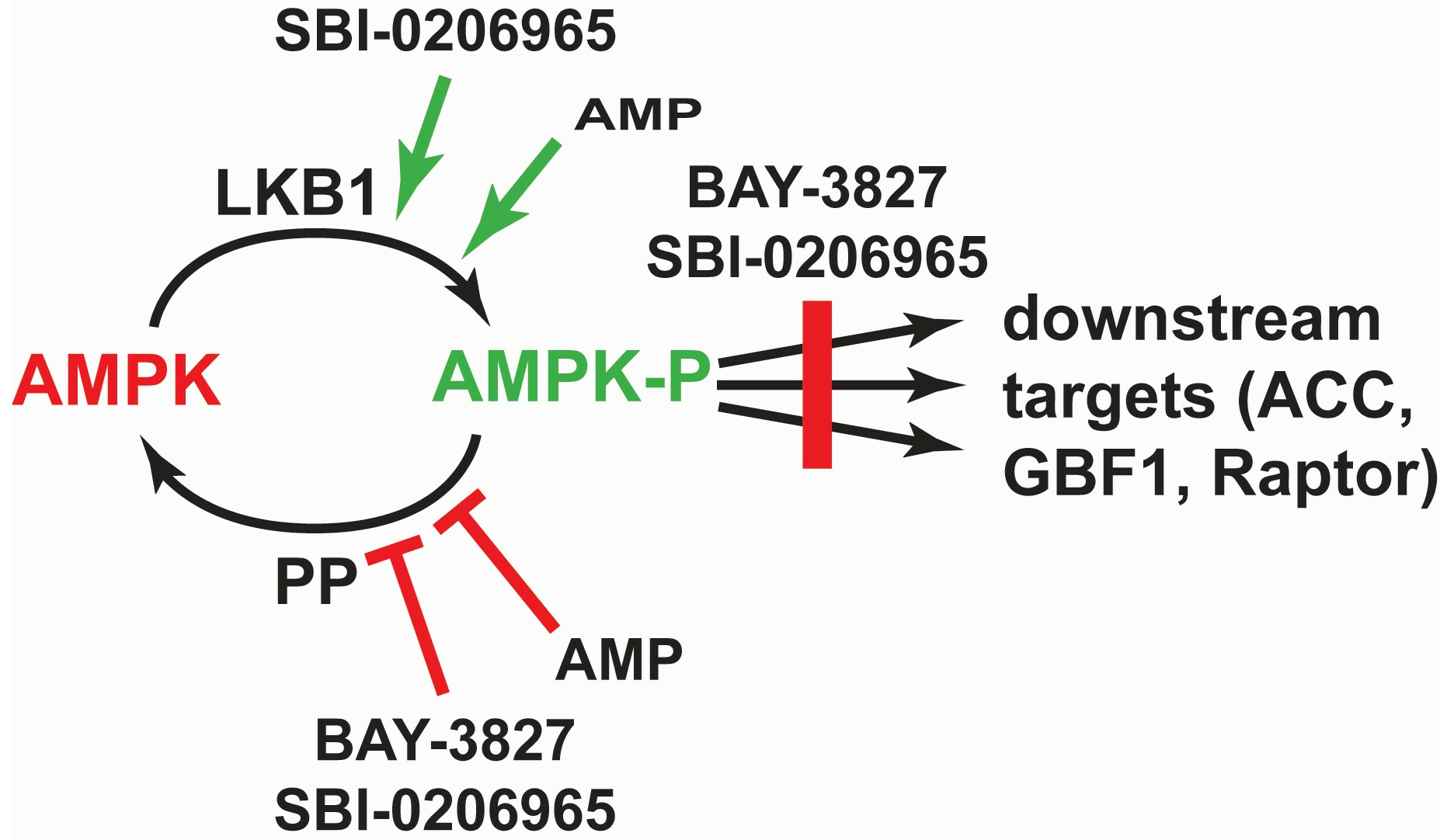
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies and Other Probes
4.3. Chemicals, Peptides, and Recombinant Proteins
4.4. Cell Lysis
4.5. Kinase Assays
4.6. Phosphorylation and Activation of Bacterially Expressed AMPK
4.7. Assays of AMPK Dephosphorylation by PP2Cα (PPM1A)
4.8. Assays of AMPK Phosphorylation by the LKB1 Complex
4.9. Assays of LKB1 Using LKBtide and PPM1A Using P-Nitrophenyl Phosphate
4.10. Pharmacokinetic Study in Mice
4.11. Analysis of Blood Levels of BAY-3827
4.12. Preparation of Internal Standards
4.13. Preparation of BAY-3827 Solutions
4.14. Sample Preparation
4.15. LC:MS Sample Analysis
4.16. Other Analytical Procedures
4.17. Curve Fitting
4.18. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yan, Y.; Novick, S.J.; Kovach, A.; Goswami, D.; Ke, J.; Tan, M.H.E.; Wang, L.; Li, X.; de Waal, P.W.; et al. Deconvoluting AMP-activated protein kinase (AMPK) adenine nucleotide binding and sensing. J. Biol. Chem. 2017, 292, 12653–12666. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Mukherjee, S.; Harikumar, K.G.; Strutzenberg, T.S.; Zhou, X.E.; Suino-Powell, K.; Xu, T.H.; Sheldon, R.D.; Lamp, J.; Brunzelle, J.S.; et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science 2021, 373, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Riek, U.; Scholz, R.; Konarev, P.; Rufer, A.; Suter, M.; Nazabal, A.; Ringler, P.; Chami, M.; Muller, S.A.; Neumann, D.; et al. Structural properties of AMP-activated protein kinase. Dimerization, molecular shape, and changes upon ligand binding. J. Biol. Chem. 2008, 283, 18331–18343. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef]
- Oakhill, J.S.; Chen, Z.P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef]
- Yeh, L.A.; Lee, K.H.; Kim, K.H. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J. Biol. Chem. 1980, 255, 2308–2314. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Hardie, D.G. The AMP-activated protein kinase, a metabolic master switch: Possible roles in Type 2 diabetes. Am. J. Physiol. 1999, 277, E1–E10. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Trotter, D.G.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017, 25, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Alkhouri, N.; Harrison, S.A.; Fouqueray, P.; Moller, D.E.; Hallakou-Bozec, S.; Bolze, S.; Grouin, J.M.; Jeannin Megnien, S.; Dubourg, J.; et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): A randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol. Hepatol. 2021, 6, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Langendorf, C.G.; Kemp, B.E. Choreography of AMPK activation. Cell Res. 2015, 25, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK beta1 isoforms. Nat. Metab. 2020, 2, 873–881. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 2019, 27, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Monteverde, T.; Muthalagu, N.; Port, J.; Murphy, D.J. Evidence of cancer promoting roles for AMPK and related kinases. FEBS J. 2015, 282, 4658–4671. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- International Centre for Kinase Profiling. Kinase Profiling Inhibitor Database. Available online: https://www.kinase-screen.mrc.ac.uk/kinase-inhibitors (accessed on 14 November 2023).
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Dite, T.A.; Langendorf, C.G.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.W.; Ling, N.X.Y.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef]
- Ahwazi, D.; Neopane, K.; Markby, G.R.; Kopietz, F.; Ovens, A.J.; Dall, M.; Hassing, A.S.; Grasle, P.; Alshuweishi, Y.; Treebak, J.T.; et al. Investigation of the specificity and mechanism of action of the ULK1/AMPK inhibitor SBI-0206965. Biochem. J. 2021, 478, 2977–2997. [Google Scholar] [CrossRef]
- Lemos, C.; Schulze, V.K.; Baumgart, S.J.; Nevedomskaya, E.; Heinrich, T.; Lefranc, J.; Bader, B.; Christ, C.D.; Briem, H.; Kuhnke, L.P.; et al. The potent AMPK inhibitor BAY-3827 shows strong efficacy in androgen-dependent prostate cancer models. Cell Oncol. 2021, 44, 581–594. [Google Scholar] [CrossRef]
- Lemos, C.; Schulze, A. Overview for BAY-3827, an Inhibitor of PRKAA1, RPS6KA1. Available online: https://www.sgc-ffm.uni-frankfurt.de/chemProbes#!specificprobeoverview/BAY-3827 (accessed on 14 November 2023).
- Storer, A.C.; Cornish-Bowden, A. Concentration of MgATP2- and other ions in solution. Calculation of the true concentrations of species present in mixtures of associating ions. Biochem. J. 1976, 159, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Ling, N.; Issa, S.M.; Dite, T.A.; O’Brien, M.T.; Chen, Z.P.; Galic, S.; Langendorf, C.G.; Steinberg, G.R.; Kemp, B.E.; et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 2014, 21, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Oshiro, N.; Yoshino, K.; Nakashima, A.; Eguchi, S.; Takahashi, M.; Ono, Y.; Kikkawa, U.; Yonezawa, K. AMP-activated protein kinase phosphorylates Golgi-specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus. J. Biol. Chem. 2008, 283, 4430–4438. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Li, P.; Nguyen, J.; Cardoza, K.; Zeng, H.; Mu, D.X.; Wu, J.T.; Gan, L.S.; Lee, F.W. Effective dosing regimen of 1-aminobenzotriazole for inhibition of antipyrine clearance in guinea pigs and mice using serial sampling. Drug Metab. Dispos. 2004, 32, 1092–1095. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef]
- Scott, J.W.; van Denderen, B.J.; Jorgensen, S.B.; Honeyman, J.E.; Steinberg, G.R.; Oakhill, J.S.; Iseli, T.J.; Koay, A.; Gooley, P.R.; Stapleton, D.; et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem. Biol. 2008, 15, 1220–1230. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; Hawley, S.A.; Auciello, F.R.; Gowans, G.J.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanisms of paradoxical activation of AMPK by the kinase inhibitors SU6656 and sorafenib. Cell Chem. Biol. 2017, 24, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekarappa, D.G.; McCartney, R.R.; Schmidt, M.C. Ligand binding to the AMP-activated protein kinase active site mediates protection of the activation loop from dephosphorylation. J. Biol. Chem. 2013, 288, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Caligaris, M.; Nicastro, R.; Hu, Z.; Tripodi, F.; Hummel, J.E.; Pillet, B.; Deprez, M.A.; Winderickx, J.; Rospert, S.; Coccetti, P.; et al. Snf1/AMPK fine-tunes TORC1 signaling in response to glucose starvation. eLife 2023, 12, e84319. [Google Scholar] [CrossRef] [PubMed]
- Desai, J.M.; Karve, A.S.; Gudelsky, G.A.; Gawali, M.V.; Seibel, W.; Sallans, L.; DasGupta, B.; Desai, P.B. Brain pharmacokinetics and metabolism of the AMP-activated protein kinase selective inhibitor SBI-0206965, an investigational agent for the treatment of glioblastoma. Investig. New Drugs 2022, 40, 944–952. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Woods, A.; Salt, I.; Scott, J.; Hardie, D.G.; Carling, D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996, 397, 347–351. [Google Scholar] [CrossRef]
- Anand, R.; Apgar, J.M.; Biftu, T.; Chen, P.; Chu, L.; Colandrea, V.J.; Dong, G.; Dropinski, D.F.; Feng, D.; Hicks, J.D.; et al. Novel Cyclic Azabenzimidazole Derivatives Useful as Anti-Diabetic Agents. WO/2012/116145 23, 30 August 2012. [Google Scholar]
- Neumann, D.; Woods, A.; Carling, D.; Wallimann, T.; Schlattner, U. Mammalian AMP-activated protein kinase: Functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr. Purif. 2003, 30, 230–237. [Google Scholar] [CrossRef]
- Zeqiraj, E.; Filippi, B.M.; Deak, M.; Alessi, D.R.; van Aalten, D.M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 2009, 326, 1707–1711. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Fyffe, F.A.; Russell, F.M.; Gowans, G.J.; Hardie, D.G. Intact cell assays to monitor AMPK and determine the contribution of the AMP-binding or ADaM sites to activation. Methods Mol. Biol. 2018, 1732, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Fyffe, F.A.; Hawley, S.A.; Gray, A.; Hardie, D.G. Cell-free assays to measure effects of regulatory ligands on AMPK. Methods Mol. Biol. 2018, 1732, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; Rafferty, J.N.; Dallas, M.L.; Ogunbayo, O.; Ikematsu, N.; McClafferty, H.; Tian, L.; Widmer, H.; Rowe, I.C.; Wyatt, C.N.; et al. Selective expression in carotid body Type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J. Biol. Chem. 2011, 286, 11929–11936. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | BAY-3827 (10 nM) | BAY-3827 (30 nM) | BAY-3827 (100 nM) | BAY-3827 (300 nM) | |
|---|---|---|---|---|---|
| Apparent Km (µM) | 309 | 382 | 391 | 887 | 1480 |
| CI Km (µM) | 253–378 | 326–446 | 357–429 | 825–954 | 1224–1807 |
| Apparent Vmax (µmol/min/mg) | 2.19 | 2.09 | 1.78 | 1.69 | 1.27 |
| CI Vmax (µmol/min/mg) | 2.07–2.32 | 2.00–2.20 | 1.73–1.83 | 1.65–1.74 | 1.22–1.81 |
| Compound | Parent (m/z) | Daughter (m/z) | Cone Voltage (V) | Collision Energy (V) |
|---|---|---|---|---|
| BAY-3827 | 469.2 | 132.6 | 12 | 30 |
| Donepezil | 380.2 | 90.9 | 60 | 33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawley, S.A.; Russell, F.M.; Ross, F.A.; Hardie, D.G. BAY-3827 and SBI-0206965: Potent AMPK Inhibitors That Paradoxically Increase Thr172 Phosphorylation. Int. J. Mol. Sci. 2024, 25, 453. https://doi.org/10.3390/ijms25010453
Hawley SA, Russell FM, Ross FA, Hardie DG. BAY-3827 and SBI-0206965: Potent AMPK Inhibitors That Paradoxically Increase Thr172 Phosphorylation. International Journal of Molecular Sciences. 2024; 25(1):453. https://doi.org/10.3390/ijms25010453
Chicago/Turabian StyleHawley, Simon A., Fiona M. Russell, Fiona A. Ross, and D. Grahame Hardie. 2024. "BAY-3827 and SBI-0206965: Potent AMPK Inhibitors That Paradoxically Increase Thr172 Phosphorylation" International Journal of Molecular Sciences 25, no. 1: 453. https://doi.org/10.3390/ijms25010453
APA StyleHawley, S. A., Russell, F. M., Ross, F. A., & Hardie, D. G. (2024). BAY-3827 and SBI-0206965: Potent AMPK Inhibitors That Paradoxically Increase Thr172 Phosphorylation. International Journal of Molecular Sciences, 25(1), 453. https://doi.org/10.3390/ijms25010453