

SMC5 Plays Independent Roles in Congenital Heart Disease and Neurodevelopmental Disability

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identifying SMC5 as a Candidate Genetic Etiology for NDD and CHD
2.2. smc5 KO Reduces Cardiac Ventricle Size in X. tropicalis
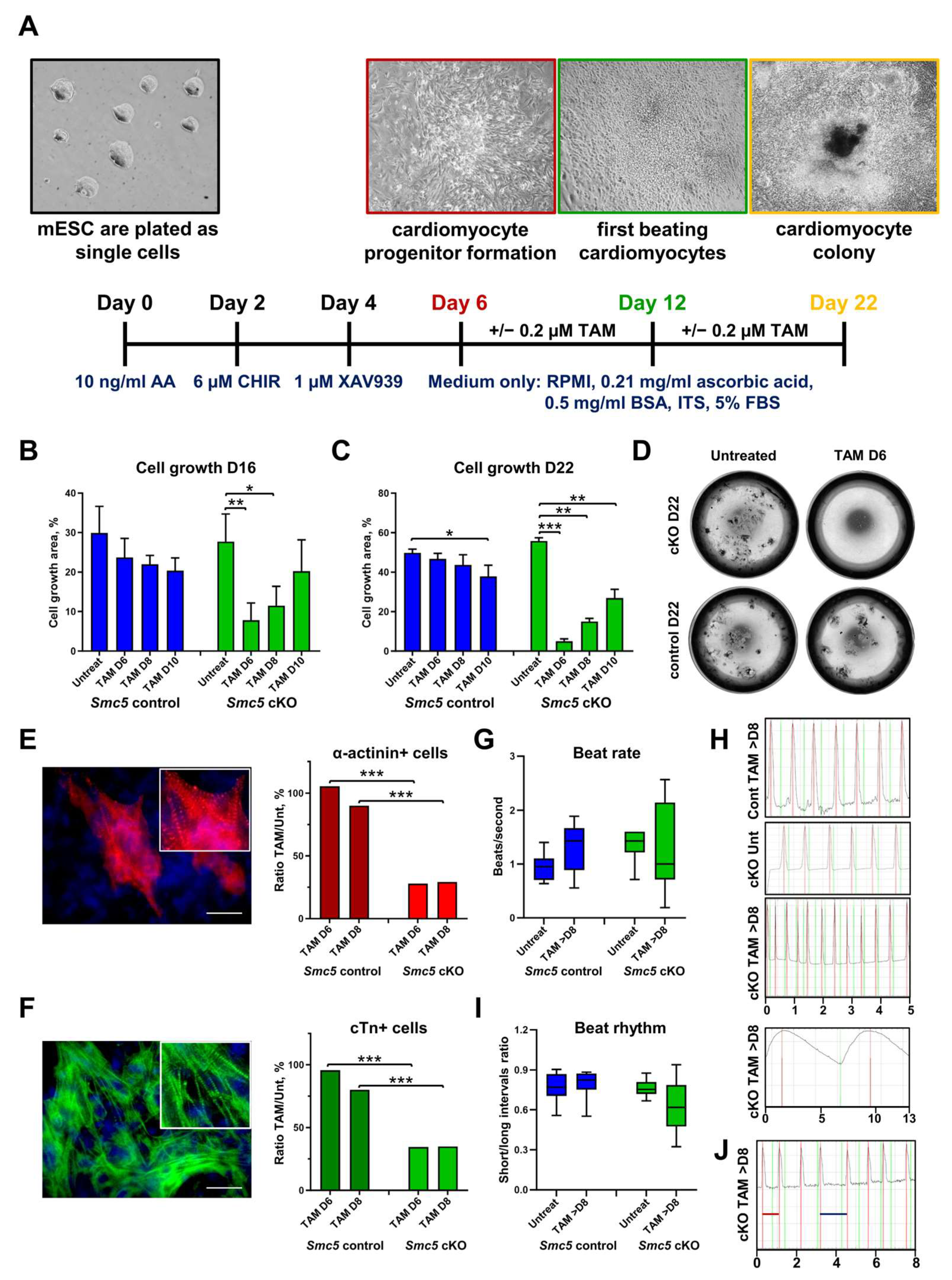
2.3. SMC5 Is Required for Mouse Cardiac Development and Function
2.4. Smc5 Is Required for Proper Brain Size in X. tropicalis
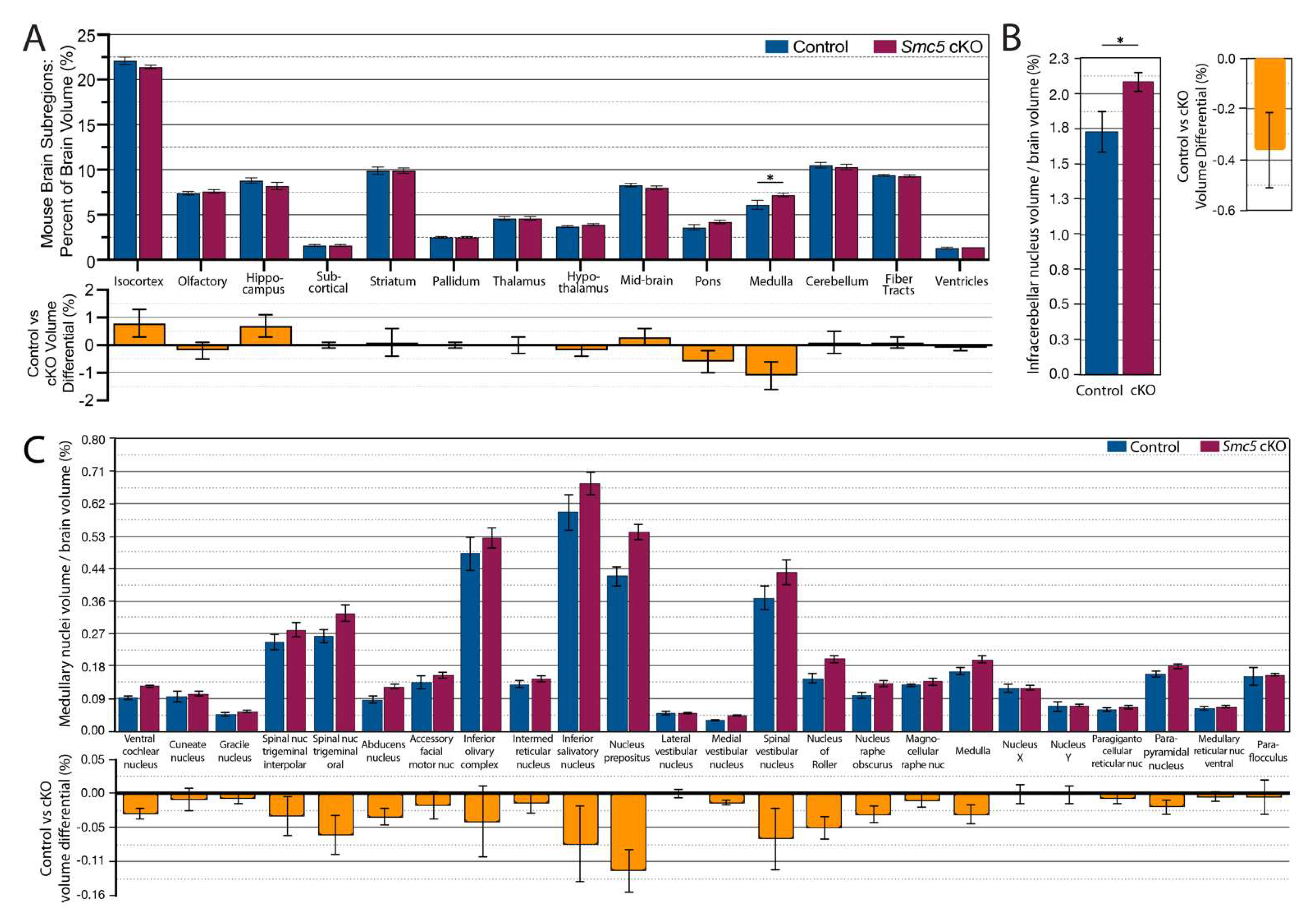
2.5. Reduced Brain Size in Smc5 cKO Mice Occurs Independent of CHD
2.6. Smc5 cKO Does Not Significantly Impact Brain Vascular Volume or Spatial Distribution
2.7. Smc5 cKO Disrupts Functional Connectivity in the Mouse Brain
3. Discussion
3.1. Cardiac Phenotype Associated with SMC5 Variants Informs the Pathophysiology of HLHS
3.2. SMC5 Is Required for Cardiac Development
3.3. SMC5 Is Required for Development of Proper Brain Structures and Function
3.4. Variants in SMC5 Are Associated with Atelis Syndrome
3.5. SMC5 Plays an Independent Role in NDD in the Setting of CHD
4. Materials and Methods
4.1. Editorial Compliance and Ethical Considerations
4.2. Patient Recruitment and Whole Exome Sequencing
4.3. Echocardiography
4.4. X. tropicalis Husbandry and Habitat
4.5. X. tropicalis Genome Editing and Genotyping
4.6. Optical Coherence Tomography of X. tropicalis Heart and Brain
4.7. Whole Mount In Situ Hybridization of X. tropicalis Embryos
4.8. mESC Differentiation into Cardiomyocytes and Their Characterization
4.9. Mouse Husbandry and Genotyping
4.10. MRI of Mouse Brain
4.11. Obtaining fMRI of Mouse Brains
4.12. Analysis of Mouse MRI Data
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CHD | congenital heart disease |
| NDD | neurodevelopmental disability; cKO: conditional knockout |
| HLHS | hypoplastic left heart syndrome |
| LOF | loss of function |
| FC | functional connectivity |
| BOLD | blood oxygen level dependent |
| fMRI | functional magnetic resonance imaging |
| OCT | optical coherence tomography |
| SMC | structural maintenance of chromosomes |
| SLF | SMC5/6 localization factor |
| mESC | mouse embryonic stem cell |
References
- Van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef]
- Talner, C.N. Report of the New England Regional Infant Cardiac Program, by Donald C. Fyler, MD, Pediatrics, 1980; 65(suppl): 375–461. Pediatrics 1998, 102, 258–259. [Google Scholar] [CrossRef]
- Egbe, A.; Lee, S.; Ho, D.; Uppu, S.; Srivastava, S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 2014, 7, 86–91. [Google Scholar] [CrossRef]
- Naef, N.; Liamlahi, R.; Beck, I.; Bernet, V.; Dave, H.; Knirsch, W.; Latal, B. Neurodevelopmental Profiles of Children with Congenital Heart Disease at School Age. J. Pediatr. 2017, 188, 75–81. [Google Scholar] [CrossRef]
- Verrall, C.E.; Blue, G.M.; Loughran-Fowlds, A.; Kasparian, N.; Gecz, J.; Walker, K.; Dunwoodie, S.L.; Cordina, R.; Sholler, G.; Badawi, N.; et al. ‘Big issues’ in neurodevelopment for children and adults with congenital heart disease. Open Heart 2019, 6, e000998. [Google Scholar] [CrossRef]
- Petit, C.J.; Rome, J.J.; Wernovsky, G.; Mason, S.E.; Shera, D.M.; Nicolson, S.C.; Montenegro, L.M.; Tabbutt, S.; Zimmerman, R.A.; Licht, D.J. Preoperative brain injury in transposition of the great arteries is associated with oxygenation and time to surgery, not balloon atrial septostomy. Circulation 2009, 119, 709–716. [Google Scholar] [CrossRef]
- Mebius, M.J.; van der Laan, M.E.; Verhagen, E.A.; Roofthooft, M.T.; Bos, A.F.; Kooi, E.M. Cerebral oxygen saturation during the first 72h after birth in infants diagnosed prenatally with congenital heart disease. Early Hum. Dev. 2016, 103, 199–203. [Google Scholar] [CrossRef]
- Morton, P.D.; Korotcova, L.; Lewis, B.K.; Bhuvanendran, S.; Ramachandra, S.D.; Zurakowski, D.; Zhang, J.; Mori, S.; Frank, J.A.; Jonas, R.A.; et al. Abnormal neurogenesis and cortical growth in congenital heart disease. Sci. Transl. Med. 2017, 9, eaah7029. [Google Scholar] [CrossRef]
- von Rhein, M.; Buchmann, A.; Hagmann, C.; Dave, H.; Bernet, V.; Scheer, I.; Knirsch, W.; Latal, B.; Heart and Brain Research Group. Severe Congenital Heart Defects Are Associated with Global Reduction of Neonatal Brain Volumes. J. Pediatr. 2015, 167, 1259–1263.e1. [Google Scholar] [CrossRef]
- Sun, L.; Macgowan, C.K.; Sled, J.G.; Yoo, S.-J.; Manlhiot, C.; Porayette, P.; Grosse-Wortmann, L.; Jaeggi, E.; McCrindle, B.W.; Kingdom, J.; et al. Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation 2015, 131, 1313–1323. [Google Scholar] [CrossRef]
- Donofrio, M.T.; Duplessis, A.J.; Limperopoulos, C. Impact of congenital heart disease on fetal brain development and injury. Curr. Opin. Pediatr. 2011, 23, 502–511. [Google Scholar] [CrossRef]
- Laraja, K.; Sadhwani, A.; Tworetzky, W.; Marshall, A.C.; Gauvreau, K.; Freud, L.; Hass, C.; Dunbar-Masterson, C.; Ware, J.; Lafranchi, T.; et al. Neurodevelopmental Outcome in Children after Fetal Cardiac Intervention for Aortic Stenosis with Evolving Hypoplastic Left Heart Syndrome. J. Pediatr. 2017, 184, 130–136.e4. [Google Scholar] [CrossRef]
- Peyvandi, S.; Xu, D.; Barkovich, A.J.; Gano, D.; Chau, V.; Reddy, V.M.; Selvanathan, T.; Guo, T.; Gaynor, J.W.; Seed, M.; et al. Declining incidence of postoperative neonatal brain injury in congenital heart disease. J. Am. Coll. Cardiol. 2023, 81, 253–266. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Blue, G.M.; Kirk, E.P.; Giannoulatou, E.; Sholler, G.F.; Dunwoodie, S.L.; Harvey, R.P.; Winlaw, D.S. Advances in the genetics of congenital heart disease: A clinician’s guide. J. Am. Coll. Cardiol. 2017, 69, 859–870. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef]
- Ji, W.; Ferdman, D.; Copel, J.; Scheinost, D.; Shabanova, V.; Brueckner, M.; Khokha, M.K.; Ment, L.R. De novo damaging variants associated with congenital heart diseases contribute to the connectome. Sci. Rep. 2020, 10, 7046. [Google Scholar] [CrossRef]
- Nishiyama, T. Cohesion and cohesin-dependent chromatin organization. Curr. Opin. Cell Biol. 2019, 58, 8–14. [Google Scholar] [CrossRef]
- Hirano, T. Condensins: Universal organizers of chromosomes with diverse functions. Genes. Dev. 2012, 26, 1659–1678. [Google Scholar] [CrossRef]
- Atkins, A.; Xu, M.J.; Li, M.; Rogers, N.P.; Pryzhkova, M.V.; Jordan, P.W. SMC5/6 is required for replication fork stability and faithful chromosome segregation during neurogenesis. Elife 2020, 9, e61171. [Google Scholar] [CrossRef]
- Rossi, F.; Helbling-Leclerc, A.; Kawasumi, R.; Jegadesan, N.K.; Xu, X.; Devulder, P.; Abe, T.; Takata, M.; Xu, D.; Rosselli, F.; et al. SMC5/6 acts jointly with Fanconi anemia factors to support DNA repair and genome stability. EMBO Rep. 2020, 21, e48222. [Google Scholar] [CrossRef]
- Grange, L.J.; Reynolds, J.J.; Ullah, F.; Isidor, B.; Shearer, R.F.; Latypova, X.; Baxley, R.M.; Oliver, A.W.; Ganesh, A.; Cooke, S.L.; et al. Pathogenic variants in SLF2 and SMC5 cause segmented chromosomes and mosaic variegated hyperploidy. Nat. Commun. 2022, 13, 6664. [Google Scholar] [CrossRef]
- Hwang, G.; Sun, F.; O’Brien, M.; Eppig, J.J.; Handel, M.A.; Jordan, P.W. SMC5/6 is required for the formation of segregation-competent bivalent chromosomes during meiosis I in mouse oocytes. Development 2017, 144, 1648–1660. [Google Scholar] [CrossRef]
- Jacome, A.; Gutierrez-Martinez, P.; Schiavoni, F.; Tenaglia, E.; Martinez, P.; Rodríguez-Acebes, S.; Lecona, E.; Murga, M.; Méndez, J.; Blasco, M.A.; et al. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 2015, 34, 2604–2619. [Google Scholar] [CrossRef]
- Salman, R.; More, S.R.; Ferreira Botelho, M.P.; Ketwaroo, P.M.; Masand, P.M.; Molossi, S.; Jadhav, S.P. Role of gated cardiac computed tomographic angiography in the evaluation of postsurgical complications after stage I Norwood procedure and its implications on management: A comparative study with two-dimensional echocardiography. Pediatr. Radiol. 2021, 51, 1185–1191. [Google Scholar] [CrossRef]
- Woods, R.K.; Kindel, S.; Mitchell, M.E.; Hraska, V.; Niebler, R.A. Evolving understanding of total artificial heart support of young infants and children. J. Thorac. Cardiovasc. Surg. 2020, 159, 1075–1082. [Google Scholar] [CrossRef]
- Zhu, F.; Bo, H.; Liu, G.; Li, R.; Liu, Z.; Fan, L. SPANXN2 functions a cell migration inhibitor in testicular germ cell tumor cells. PeerJ 2020, 8, e9358. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Kim, M.-S.; Fleres, B.; Lovett, J.; Anfinson, M.; Samudrala, S.S.K.; Kelly, L.J.; Teigen, L.E.; Cavanaugh, M.; Marquez, M.; Geurts, A.M.; et al. Contractility of Induced Pluripotent Stem Cell-Cardiomyocytes With an MYH6 Head Domain Variant Associated With Hypoplastic Left Heart Syndrome. Front. Cell Dev. Biol. 2020, 8, 440. [Google Scholar] [CrossRef]
- Pryzhkova, M.V.; Jordan, P.W. Conditional mutation of Smc5 in mouse embryonic stem cells perturbs condensin localization and mitotic progression. J. Cell Sci. 2016, 129, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.; Verver, D.E.; Handel, M.A.; Hamer, G.; Jordan, P.W. Depletion of SMC5/6 sensitizes male germ cells to DNA damage. Mol. Biol. Cell 2018, 29, 3003–3016. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, H.; Pryzhkova, M.V.; Jordan, P.W. Conditional mutation of SMC5 in mouse embryonic fibroblasts. Methods Mol. Biol. 2019, 2004, 35–46. [Google Scholar] [CrossRef]
- Burridge, P.W.; Holmström, A.; Wu, J.C. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr. Protoc. Hum. Genet. 2015, 87, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hao, J.; Hong, C.C. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/β-catenin signaling. ACS Chem. Biol. 2011, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Limperopoulos, C.; Tworetzky, W.; McElhinney, D.B.; Newburger, J.W.; Brown, D.W.; Robertson, R.L.; Guizard, N.; McGrath, E.; Geva, J.; Annese, D.; et al. Brain volume and metabolism in fetuses with congenital heart disease: Evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation 2010, 121, 26–33. [Google Scholar] [CrossRef]
- Ochi, S.; Manabe, S.; Kikkawa, T.; Osumi, N. Thirty Years’ History since the Discovery of Pax6: From Central Nervous System Development to Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 6115. [Google Scholar] [CrossRef]
- Exner, C.R.T.; Willsey, H.R. Xenopus leads the way: Frogs as a pioneering model to understand the human brain. Genesis 2021, 59, e23405. [Google Scholar] [CrossRef]
- Odanaka, Y.; Ashida, A.; Nemoto, S.; Hamanaka, K.; Matsumoto, N. Severe cardiac defect in Cornelia de Lange syndrome from a novel SMC1A variant. Pediatr. Int. 2022, 64, e15031. [Google Scholar] [CrossRef]
- Chinen, Y.; Nakamura, S.; Kaneshi, T.; Nakayashiro, M.; Yanagi, K.; Kaname, T.; Naritomi, K.; Nakanishi, K. A novel nonsense SMC1A mutation in a patient with intractable epilepsy and cardiac malformation. Hum. Gen. Variation 2019, 6, 23. [Google Scholar] [CrossRef]
- Martin, C.-A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef]
- Khan, T.N.; Khan, K.; Sadeghpour, A.; Reynolds, H.; Perilla, Y.; McDonald, M.T.; Gallentine, W.B.; Baig, S.M.; Task Force for Neonatal Genomics; Davis, E.E.; et al. Mutations in NCAPG2 Cause a Severe Neurodevelopmental Syndrome that Expands the Phenotypic Spectrum of Condensinopathies. Am. J. Hum. Genet. 2019, 104, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Hofmann, D.; Kaloulis, K.; Bishop, J.M.; Trumpp, A. Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 2006, 44, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Giusti, S.A.; Vercelli, C.A.; Vogl, A.M.; Kolarz, A.W.; Pino, N.S.; Deussing, J.M.; Refojo, D. Behavioral phenotyping of Nestin-Cre mice: Implications for genetic mouse models of psychiatric disorders. J. Psychiatr. Res. 2014, 55, 87–95. [Google Scholar] [CrossRef]
- Wang, Q.; Ding, S.-L.; Li, Y.; Royall, J.; Feng, D.; Lesnar, P.; Graddis, N.; Naeemi, M.; Facer, B.; Ho, A.; et al. The allen mouse brain common coordinate framework: A 3D reference atlas. Cell 2020, 181, 936–953.e20. [Google Scholar] [CrossRef] [PubMed]
- Mlczoch, E.; Brugger, P.; Ulm, B.; Novak, A.; Frantal, S.; Prayer, D.; Salzer-Muhar, U. Structural congenital brain disease in congenital heart disease: Results from a fetal MRI program. Eur. J. Paediatr. Neurol. 2013, 17, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.; Book, W.M.; Nelson, T.J.; Xu, C. Hypoplastic left heart syndrome: From bedside to bench and back. J. Mol. Cell Cardiol. 2019, 135, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Gebbia, M.; Ferrero, G.B.; Pilia, G.; Bassi, M.T.; Aylsworth, A.; Penman-Splitt, M.; Bird, L.M.; Bamforth, J.S.; Burn, J.; Schlessinger, D.; et al. X-linked situs abnormalities result from mutations in ZIC3. Nat. Genet. 1997, 17, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef]
- Huang, M.; Akerberg, A.A.; Zhang, X.; Yoon, H.; Joshi, S.; Hallinan, C.; Nguyen, C.; Pu, W.T.; Haigis, M.C.; Burns, C.G.; et al. Intrinsic myocardial defects underlie an Rbfox-deficient zebrafish model of hypoplastic left heart syndrome. Nat. Commun. 2022, 13, 5877. [Google Scholar] [CrossRef]
- Payne, F.; Colnaghi, R.; Rocha, N.; Seth, A.; Harris, J.; Carpenter, G.; Bottomley, W.E.; Wheeler, E.; Wong, S.; Saudek, V.; et al. Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J. Clin. Investig. 2014, 124, 4028–4038. [Google Scholar] [CrossRef]
- Zhu, W.; Shi, Y.; Zhang, C.; Peng, Y.; Wan, Y.; Xu, Y.; Liu, X.; Han, B.; Zhao, S.; Kuang, Y.; et al. In-frame deletion of SMC5 related with the phenotype of primordial dwarfism, chromosomal instability and insulin resistance. Clin. Transl. Med. 2023, 13, e1007. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Klein, S.D.; Mulatinho, M.V.; Senaratne, T.N.; Singh, K.; UCLA Clinical Genomics Center; Nguyen, D.C.; Gallant, N.M.; Strom, S.P.; Ghahremani, S.; et al. De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. A 2017, 173, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange syndrome-overlapping phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Paez, L.M.; Tanaka, H.; Bando, M.; Takahashi, M.; Nozaki, N.; Nakato, R.; Shirahige, K.; Hirota, T. Smc5/6-mediated regulation of replication progression contributes to chromosome assembly during mitosis in human cells. Mol. Biol. Cell 2014, 25, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Irmisch, A.; Ampatzidou, E.; Mizuno, K.; O’Connell, M.J.; Murray, J.M. Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J. 2009, 28, 144–155. [Google Scholar] [CrossRef]
- Ampatzidou, E.; Irmisch, A.; O’Connell, M.J.; Murray, J.M. Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol. 2006, 26, 9387–9401. [Google Scholar] [CrossRef]
- Menolfi, D.; Delamarre, A.; Lengronne, A.; Pasero, P.; Branzei, D. Essential Roles of the Smc5/6 Complex in Replication through Natural Pausing Sites and Endogenous DNA Damage Tolerance. Mol. Cell 2015, 60, 835–846. [Google Scholar] [CrossRef]
- Etheridge, T.J.; Villahermosa, D.; Campillo-Funollet, E.; Herbert, A.D.; Irmisch, A.; Watson, A.T.; Dang, H.Q.; Osborne, M.A.; Oliver, A.W.; Carr, A.M.; et al. Live-cell single-molecule tracking highlights requirements for stable Smc5/6 chromatin association in vivo. Elife 2021, 10, e68579. [Google Scholar] [CrossRef]
- Yong-Gonzales, V.; Hang, L.E.; Castellucci, F.; Branzei, D.; Zhao, X. The Smc5-Smc6 complex regulates recombination at centromeric regions and affects kinetochore protein sumoylation during normal growth. PLoS ONE 2012, 7, e51540. [Google Scholar] [CrossRef]
- Gómez, R.; Jordan, P.W.; Viera, A.; Alsheimer, M.; Fukuda, T.; Jessberger, R.; Llano, E.; Pendás, A.M.; Handel, M.A.; Suja, J.A. Dynamic localization of SMC5/6 complex proteins during mammalian meiosis and mitosis suggests functions in distinct chromosome processes. J. Cell Sci. 2013, 126, 4239–4252. [Google Scholar] [CrossRef]
- Venegas, A.B.; Natsume, T.; Kanemaki, M.; Hickson, I.D. Inducible Degradation of the Human SMC5/6 Complex Reveals an Essential Role Only during Interphase. Cell Rep. 2020, 31, 107533. [Google Scholar] [CrossRef] [PubMed]
- Moradi-Fard, S.; Sarthi, J.; Tittel-Elmer, M.; Lalonde, M.; Cusanelli, E.; Chartrand, P.; Cobb, J.A. Smc5/6 Is a Telomere-Associated Complex that Regulates Sir4 Binding and TPE. PLoS Genet. 2016, 12, e1006268. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.; Cordero, G.; Kawamura, R.; Sverzhinsky, A.; Sarker, M.; Roy, S.; Malo, C.; Pascal, J.M.; Marko, J.F.; D’Amours, D. The Smc5/6 Core Complex Is a Structure-Specific DNA Binding and Compacting Machine. Mol. Cell 2020, 80, 1025–1038.e5. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Escribano, P.; Hormeño, S.; Madariaga-Marcos, J.; Solé-Soler, R.; O’Reilly, F.J.; Morris, K.; Aicart-Ramos, C.; Aramayo, R.; Montoya, A.; Kramer, H.; et al. Purified smc5/6 complex exhibits DNA substrate recognition and compaction. Mol. Cell 2020, 80, 1039–1054.e6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, S.; Ser, Z.; Sanyal, T.; Choi, K.; Wan, B.; Kuang, H.; Sali, A.; Kentsis, A.; Patel, D.J.; et al. Integrative analysis reveals unique structural and functional features of the Smc5/6 complex. Proc. Natl. Acad. Sci. USA 2021, 118, e2026844118. [Google Scholar] [CrossRef] [PubMed]
- Yatskevich, S.; Rhodes, J.; Nasmyth, K. Organization of chromosomal DNA by SMC complexes. Annu. Rev. Genet. 2019, 53, 445–482. [Google Scholar] [CrossRef]
- Dowen, J.M.; Young, R.A. SMC complexes link gene expression and genome architecture. Curr. Opin. Genet. Dev. 2014, 25, 131–137. [Google Scholar] [CrossRef]
- Pradhan, B.; Barth, R.; Kim, E.; Davidson, I.F.; Bauer, B.; van Laar, T.; Yang, W.; Ryu, J.-K.; van der Torre, J.; Peters, J.-M.; et al. SMC complexes can traverse physical roadblocks bigger than their ring size. Cell Rep. 2022, 41, 111491. [Google Scholar] [CrossRef]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef]
- Moudgil, R.; Samra, G.; Ko, K.A.; Vu, H.T.; Thomas, T.N.; Luo, W.; Chang, J.; Reddy, A.K.; Fujiwara, K.; Abe, J.-I. Topoisomerase 2B decrease results in diastolic dysfunction via p53 and akt: A novel pathway. Front Cardiovasc. Med. 2020, 7, 594123. [Google Scholar] [CrossRef]
- Madabhushi, R. The roles of DNA topoisomerase iiβ in transcription. Int. J. Mol. Sci. 2018, 19, 1917. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Wilson, M.D. Untangling the roles of TOP2A and TOP2B in transcription and cancer. Sci. Adv. 2022, 8, eadd4920. [Google Scholar] [CrossRef]
- McKinnon, P.J. Topoisomerases and the regulation of neural function. Nat. Rev. Neurosci. 2016, 17, 673–679. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Burger, L.; Nikoletopoulou, V.; Deogracias, R.; Thakurela, S.; Wirbelauer, C.; Kaut, J.; Terranova, R.; Hoerner, L.; Mielke, C.; et al. Target genes of Topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. USA 2012, 109, E934–E943. [Google Scholar] [CrossRef]
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The first thirty years. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef]
- Cowell, I.G.; Casement, J.W.; Austin, C.A. To break or not to break: The role of TOP2B in transcription. Int. J. Mol. Sci. 2023, 24, 14806. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Saito, S.; Adachi, N.; Yano, K.-I. Dynamic behavior of DNA topoisomerase IIβ in response to DNA double-strand breaks. Sci. Rep. 2018, 8, 10344. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Hiromoto, Y.; Higaki, T.; Yano, K.-I. Disease-associated H58Y mutation affects the nuclear dynamics of human DNA topoisomerase IIβ. Sci. Rep. 2022, 12, 20627. [Google Scholar] [CrossRef]
- Hiraide, T.; Watanabe, S.; Matsubayashi, T.; Yanagi, K.; Nakashima, M.; Ogata, T.; Saitsu, H. A de novo TOP2B variant associated with global developmental delay and autism spectrum disorder. Mol. Genet. Genomic Med. 2020, 8, e1145. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.E.; Zheng, Y.; Speijer, D.; Hoebe, R.; Dekker, H.L.; Repping, S.; Stap, J.; Hamer, G. Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress. Int. J. Mol. Sci. 2016, 17, 1782. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Alveal, C.; Outwin, E.A.; Trempolec, N.; Dziadkowiec, D.; Murray, J.M.; O’Connell, M.J. SMC complexes and topoisomerase II work together so that sister chromatids can work apart. Cell Cycle 2010, 9, 2065–2070. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lauridsen, M.H.; Uldbjerg, N.; Henriksen, T.B.; Petersen, O.B.; Stausbøl-Grøn, B.; Matthiesen, N.B.; Peters, D.A.; Ringgaard, S.; Hjortdal, V.E. Cerebral oxygenation measurements by magnetic resonance imaging in fetuses with and without heart defects. Circ. Cardiovasc. Imaging 2017, 10, e006459. [Google Scholar] [CrossRef] [PubMed]
- De Asis-Cruz, J.; Donofrio, M.T.; Vezina, G.; Limperopoulos, C. Aberrant brain functional connectivity in newborns with congenital heart disease before cardiac surgery. Neuroimage Clin. 2018, 17, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Ma, X.-H.; Liang, J.-W. Application of voxel-based morphometric method to detect brain changes in children with non-cyanotic congenital heart disease. World J. Radiol. 2020, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Masoller, N.; Sanz-Cortés, M.; Crispi, F.; Gómez, O.; Bennasar, M.; Egaña-Ugrinovic, G.; Bargalló, N.; Martínez, J.M.; Gratacós, E. Severity of Fetal Brain Abnormalities in Congenital Heart Disease in Relation to the Main Expected Pattern of in utero Brain Blood Supply. Fetal Diagn. Ther. 2016, 39, 269–278. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Boston Children’s Hospital Z-Score Calculator. Available online: https://zscore.chboston.org/ (accessed on 16 October 2023).
- Lane, M.; Khokha, M.K. Obtaining Xenopus tropicalis Embryos by In Vitro Fertilization. Cold Spring Harb. Protoc. 2022, 2022, Pdb.prot106351. [Google Scholar] [CrossRef]
- Lane, M.; Mis, E.K.; Khokha, M.K. Microinjection of Xenopus tropicalis Embryos. Cold Spring Harb. Protoc. 2022, 2022, Pdb.prot107644. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef]
- Deniz, E.; Mis, E.K.; Lane, M.; Khokha, M.K. Xenopus tadpole craniocardiac imaging using optical coherence tomography. Cold Spring Harb. Protoc. 2022, 2022, Pdb.prot105676. [Google Scholar] [CrossRef]
- Khokha, M.K.; Chung, C.; Bustamante, E.L.; Gaw, L.W.K.; Trott, K.A.; Yeh, J.; Lim, N.; Lin, J.C.Y.; Taverner, N.; Amaya, E.; et al. Techniques and probes for the study of Xenopus tropicalis development. Dev. Dyn. 2002, 225, 499–510. [Google Scholar] [CrossRef]
- Pryzhkova, M.V.; Aria, I.; Cheng, Q.; Harris, G.M.; Zan, X.; Gharib, M.; Jabbarzadeh, E. Carbon nanotube-based substrates for modulation of human pluripotent stem cell fate. Biomaterials 2014, 35, 5098–5109. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Grune, T.; Ott, C.; Häseli, S.; Höhn, A.; Jung, T. The “MYOCYTER”—Convert cellular and cardiac contractions into numbers with ImageJ. Sci. Rep. 2019, 9, 15112. [Google Scholar] [CrossRef]
- Noble, S.; Spann, M.N.; Tokoglu, F.; Shen, X.; Constable, R.T.; Scheinost, D. Influences on the Test-Retest Reliability of Functional Connectivity MRI and its Relationship with Behavioral Utility. Cereb. Cortex 2017, 27, 5415–5429. [Google Scholar] [CrossRef]
- Papademetris, X.; Jackowski, M.P.; Rajeevan, N.; DiStasio, M.; Okuda, H.; Constable, R.T.; Staib, L.H. BioImage Suite: An integrated medical image analysis suite: An update. Insight J. 2006, 2006, 209. [Google Scholar] [CrossRef]
- Papademetris, X.; Jackowski, A.P.; Schultz, R.T.; Staib, L.H.; Duncan, J.S. Computing 3D Non-rigid Brain Registration Using Extended Robust Point Matching for Composite Multisubject fMRI Analysis. In Medical Image Computing and Computer-Assisted Intervention—MICCAI 2003; Lecture Notes in Computer Science; Ellis, R.E., Peters, T.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2879, pp. 788–795. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, M.P.; Pryzhkova, M.V.; Lake, E.M.R.; Mandino, F.; Shen, X.; Karnik, R.; Atkins, A.; Xu, M.J.; Ji, W.; Konstantino, M.; et al. SMC5 Plays Independent Roles in Congenital Heart Disease and Neurodevelopmental Disability. Int. J. Mol. Sci. 2024, 25, 430. https://doi.org/10.3390/ijms25010430
O’Brien MP, Pryzhkova MV, Lake EMR, Mandino F, Shen X, Karnik R, Atkins A, Xu MJ, Ji W, Konstantino M, et al. SMC5 Plays Independent Roles in Congenital Heart Disease and Neurodevelopmental Disability. International Journal of Molecular Sciences. 2024; 25(1):430. https://doi.org/10.3390/ijms25010430
Chicago/Turabian StyleO’Brien, Matthew P., Marina V. Pryzhkova, Evelyn M. R. Lake, Francesca Mandino, Xilin Shen, Ruchika Karnik, Alisa Atkins, Michelle J. Xu, Weizhen Ji, Monica Konstantino, and et al. 2024. "SMC5 Plays Independent Roles in Congenital Heart Disease and Neurodevelopmental Disability" International Journal of Molecular Sciences 25, no. 1: 430. https://doi.org/10.3390/ijms25010430
APA StyleO’Brien, M. P., Pryzhkova, M. V., Lake, E. M. R., Mandino, F., Shen, X., Karnik, R., Atkins, A., Xu, M. J., Ji, W., Konstantino, M., Brueckner, M., Ment, L. R., Khokha, M. K., & Jordan, P. W. (2024). SMC5 Plays Independent Roles in Congenital Heart Disease and Neurodevelopmental Disability. International Journal of Molecular Sciences, 25(1), 430. https://doi.org/10.3390/ijms25010430