
Analysis of Chromatin Accessibility and DNA Methylation to Reveal the Functions of Epigenetic Modifications in Cyprinus carpio Gonads

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
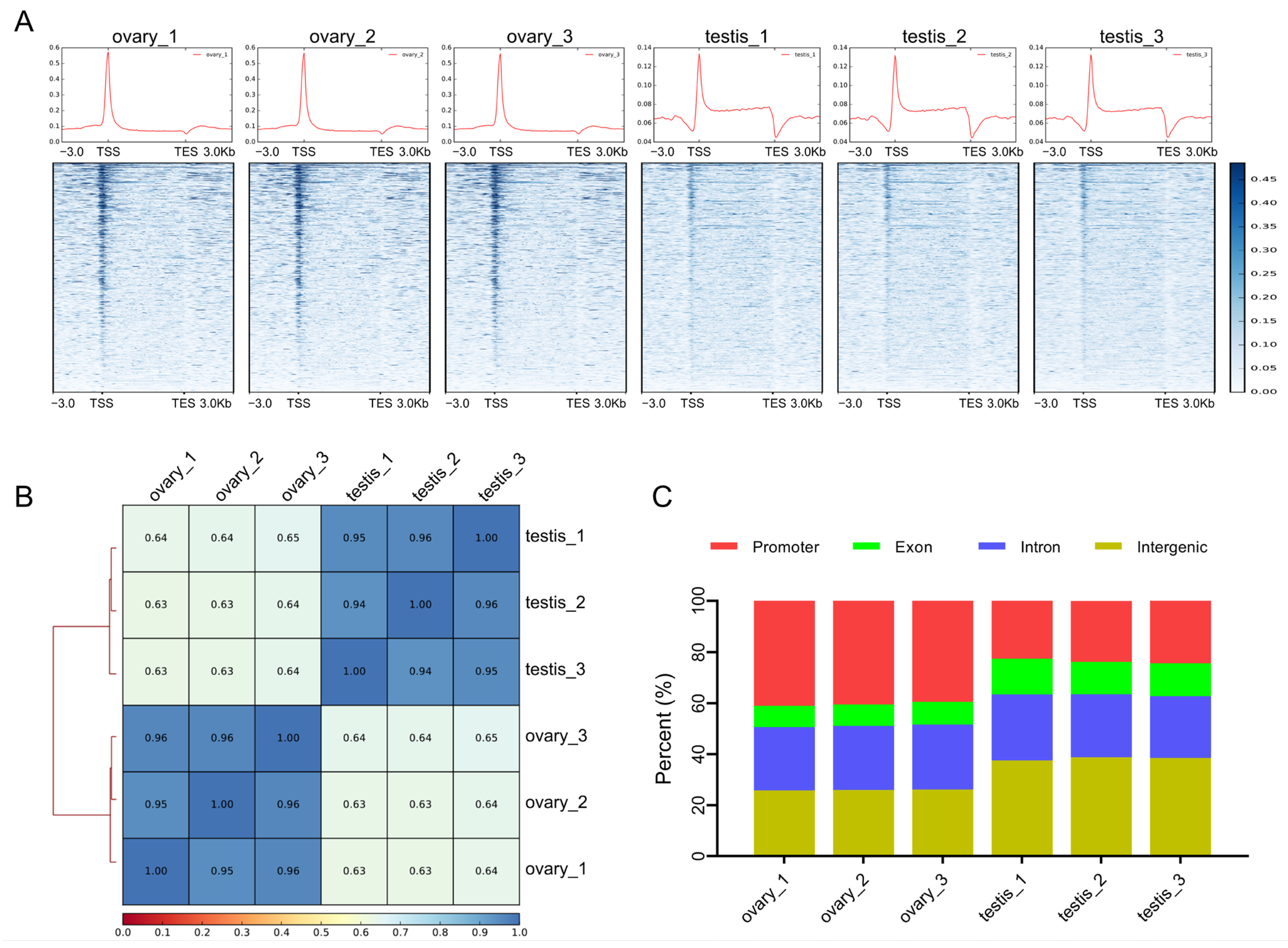
2.1. Profiles of Chromatin Accessibility in Ovaries and Testes
2.2. Differential Peaks between Ovaries and Testes
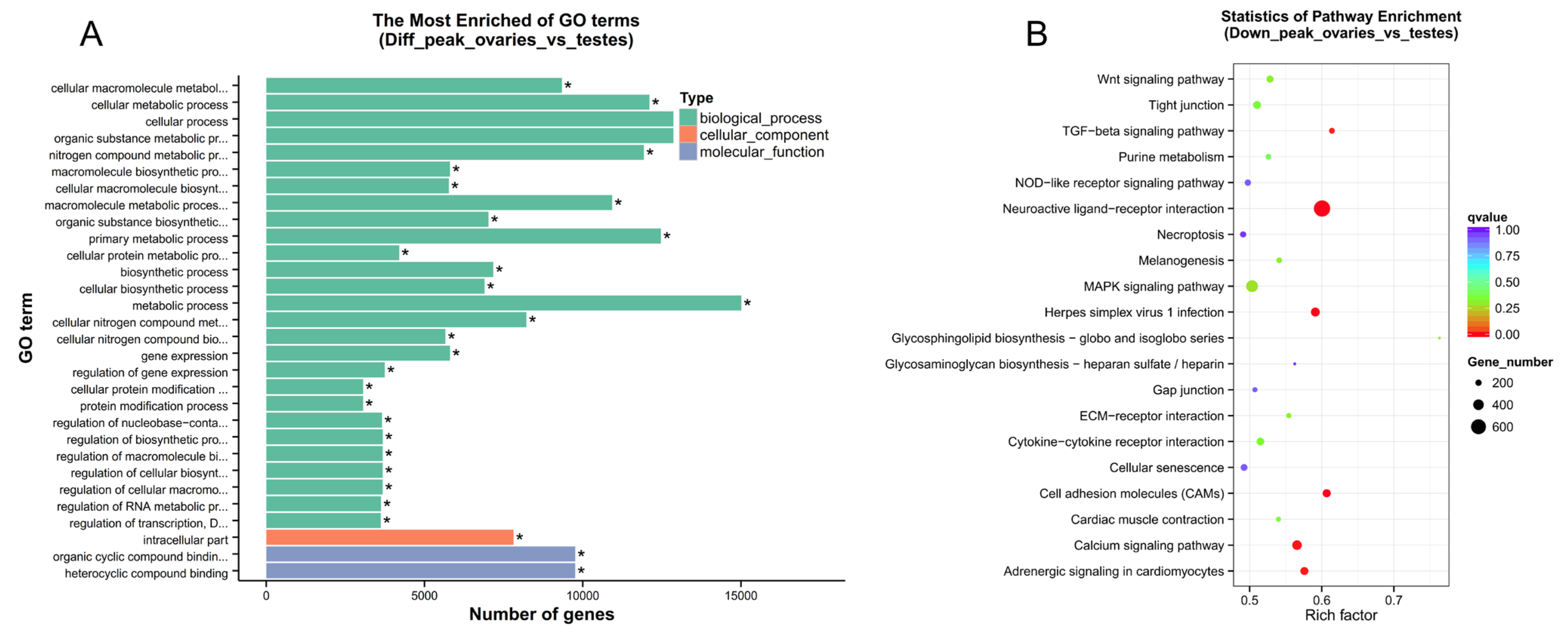
2.3. Functional Annotation of Chromatin Accessibility in Common Carp Gonads
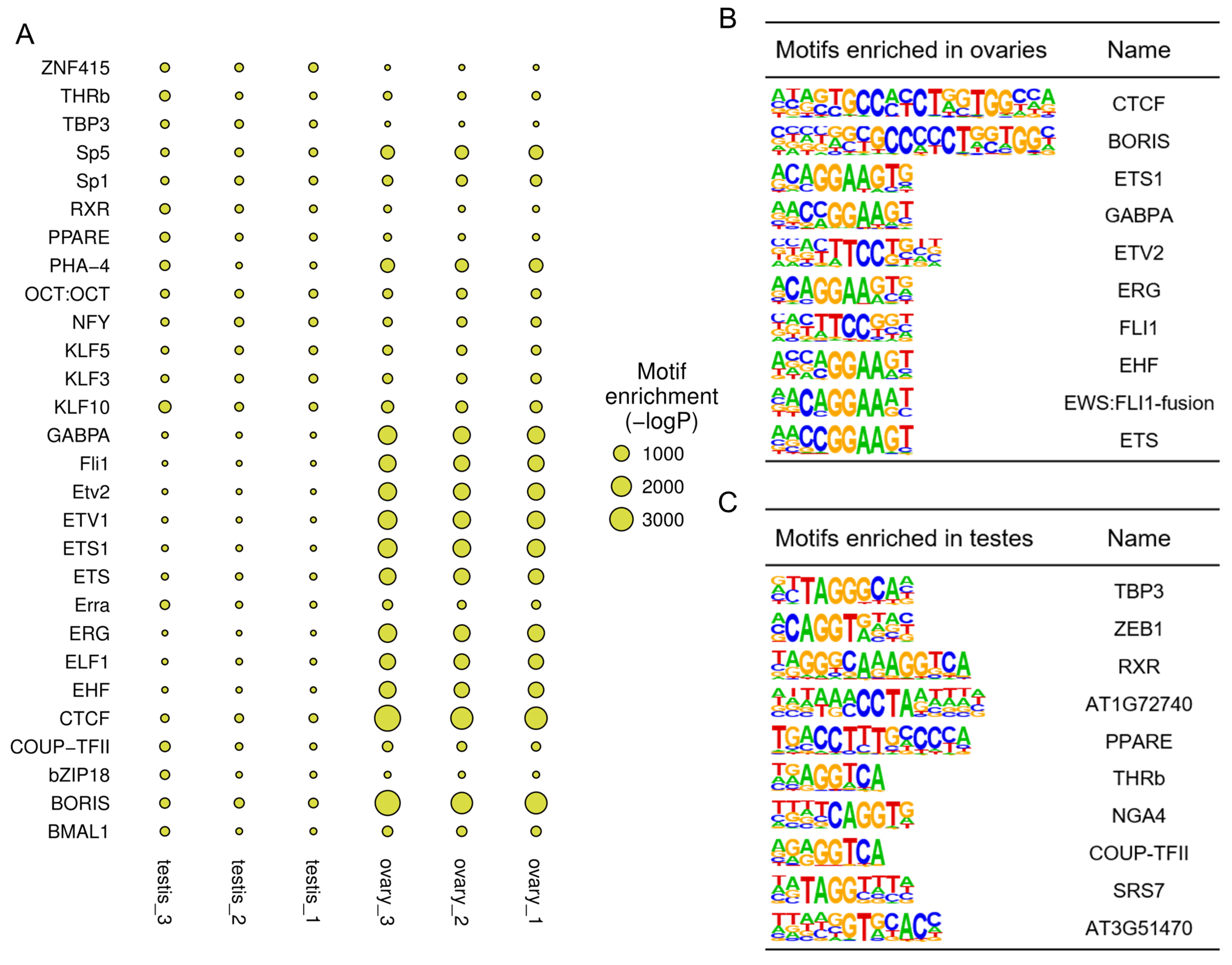
2.4. Motif Enrichment Analysis
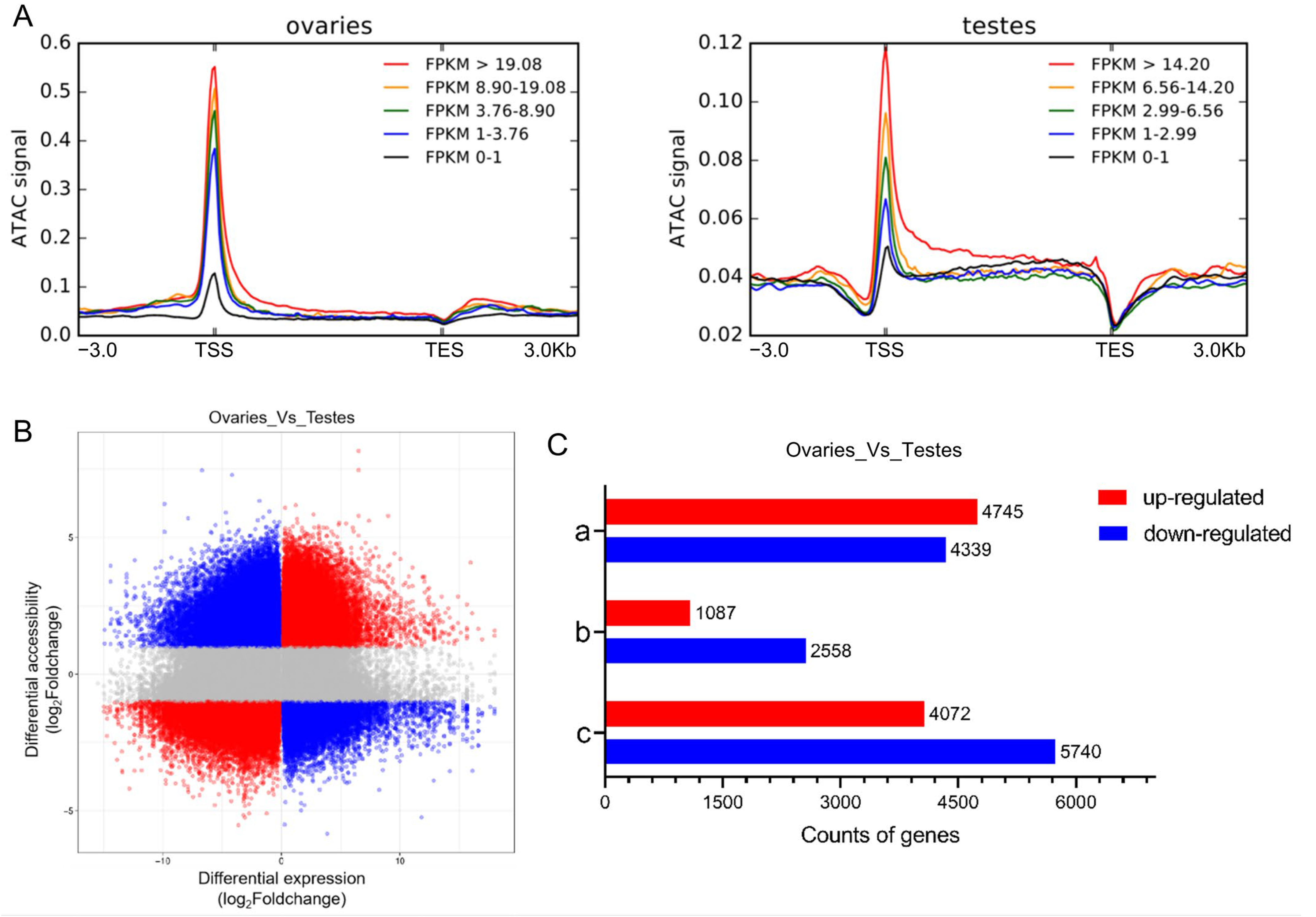
2.5. Relationship between Chromatin Accessibility and Gene Expression
2.6. Summary of Bisulfite Sequencing Data
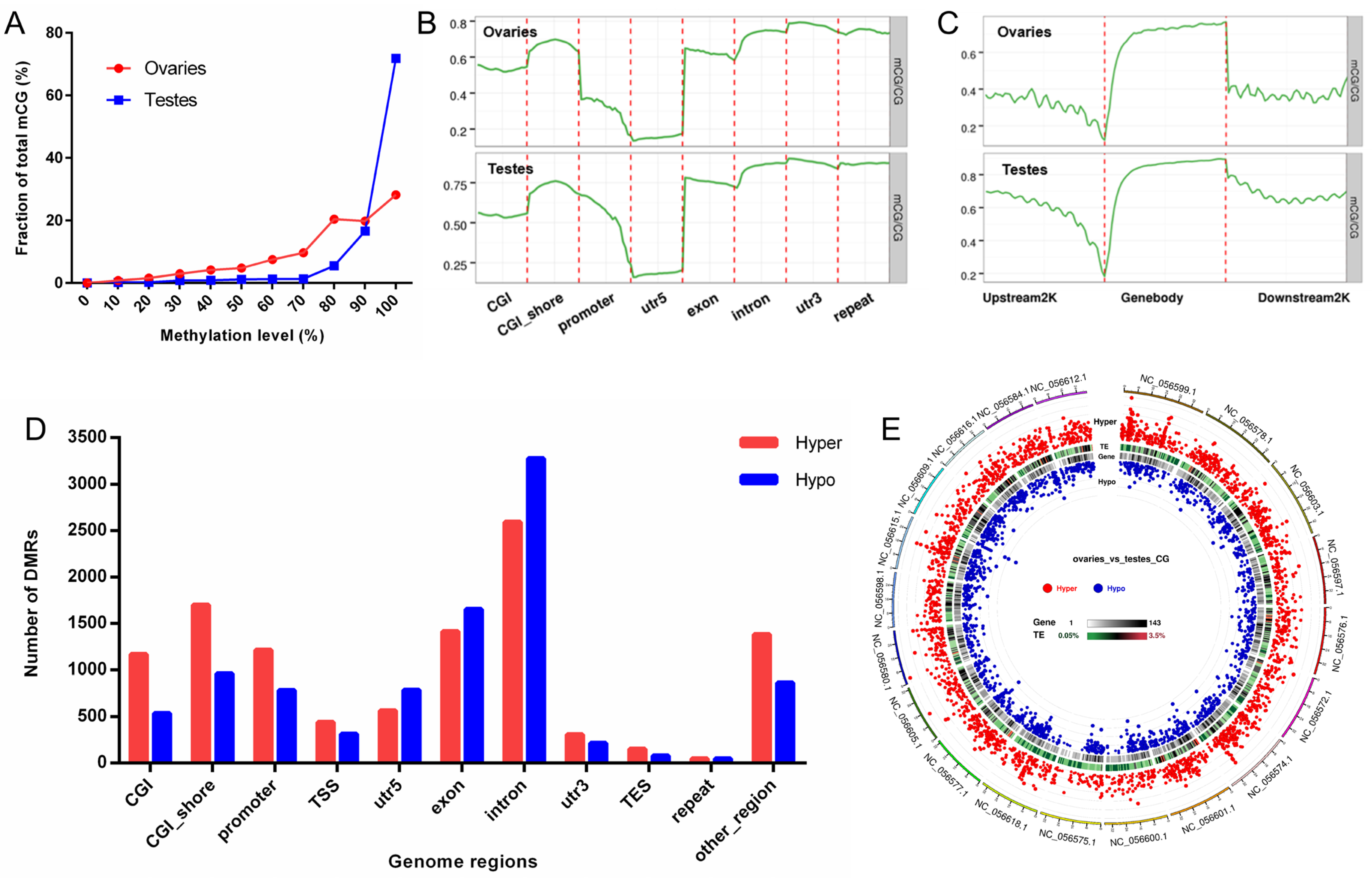
2.7. DNA Methylation Profiles of Ovaries and Testes
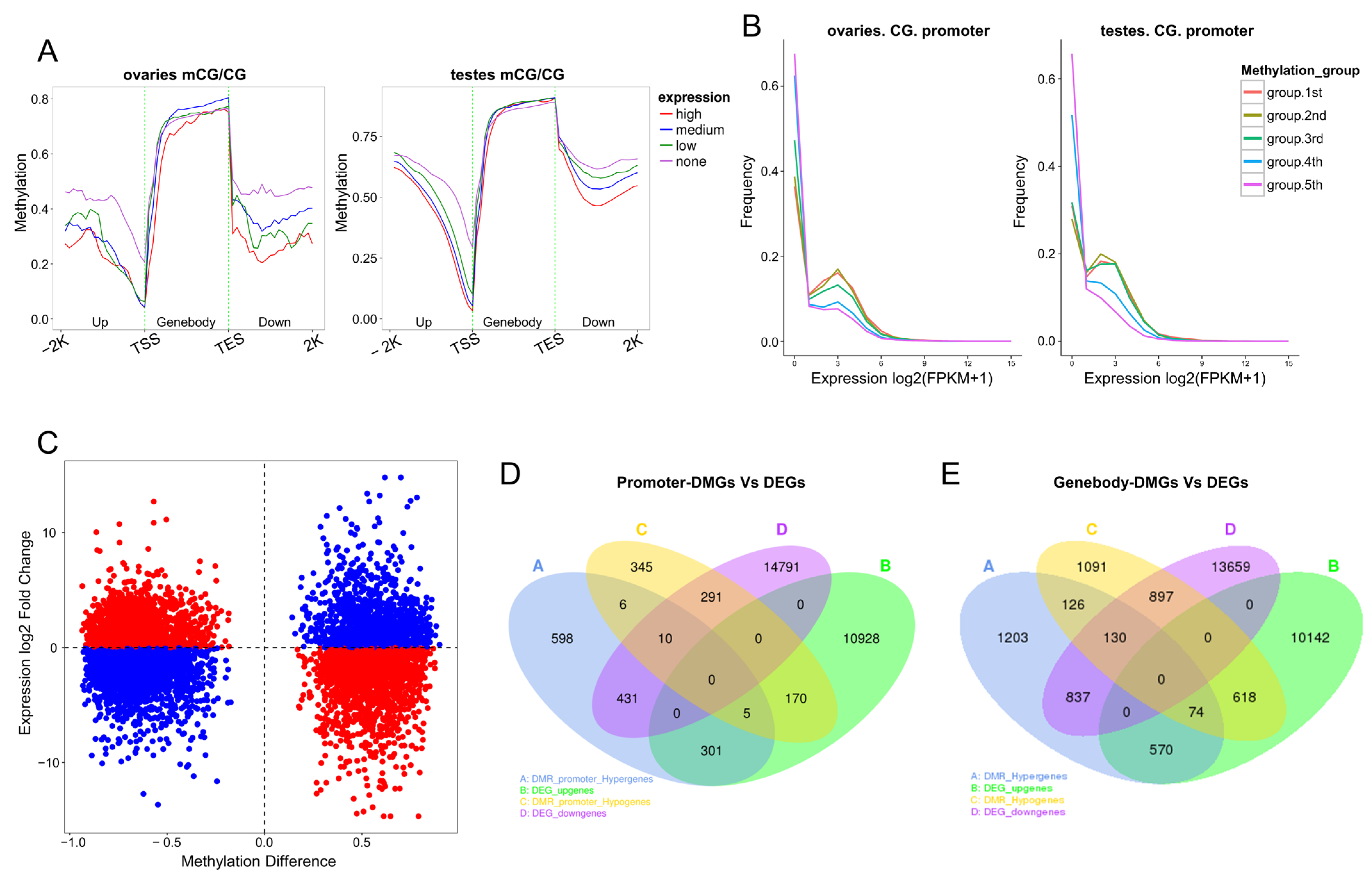
2.8. Overall Relationship between DNA Methylation and Gene Expression
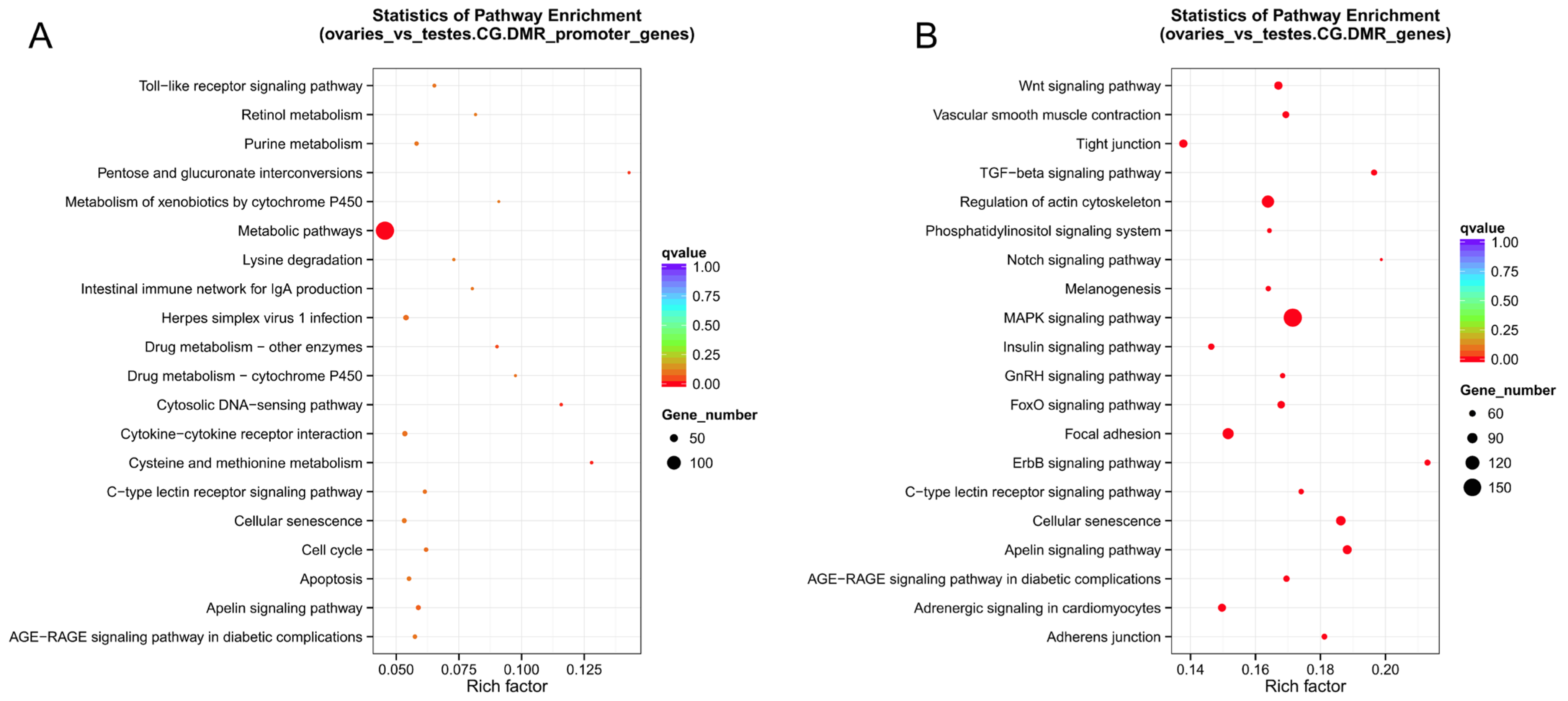
2.9. Combined Analysis of DMGs and DEGs
3. Discussion
3.1. Chromatin Accessibility Profiles in Common Carp Gonads
3.2. Differential DNA Methylation between Ovaries and Testes
4. Materials and Methods
4.1. Sample Collection
4.2. ATAC Library Preparation and Sequencing
4.3. ATAC-Seq Reads Alignment
4.4. Peak Calling and Motif Analysis
4.5. Differential Peaks Analysis and Annotation
4.6. Bisulfite Library Construction and Sequencing
4.7. Bisulfite Sequencing Reads Mapping
4.8. Methylation Level Estimation and Differential Analysis
4.9. Gene Functional Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bachtrog, D.; Mank, J.E.; Peichel, C.L.; Kirkpatrick, M.; Otto, S.P.; Ashman, T.L.; Hahn, M.W.; Kitano, J.; Mayrose, I.; Ming, R.; et al. Sex determination: Why so many ways of doing it? PLoS Biol. 2014, 12, e1001899. [Google Scholar] [CrossRef] [PubMed]
- Holleley, C.E.; O‘Meally, D.; Sarre, S.D.; Marshall Graves, J.A.; Ezaz, T.; Matsubara, K.; Azad, B.; Zhang, X.; Georges, A. Sex reversal triggers the rapid transition from genetic to temperature-dependent sex. Nature 2015, 523, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, L.; Wang, D. Roles of estrogens in fish sexual plasticity and sex differentiation. Gen. Comp. Endocrinol. 2019, 277, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Aharon, D.; Marlow, F.L. Sexual determination in zebrafish. Cell. Mol. Life Sci. 2021, 79, 8. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran, P.; Jaafar, F.; Kar, S.; Sudhakumari, C.; Senthilkumaran, B.; Parhar, I.S. Sex determination and differentiation in teleost: Roles of genetics, environment, and brain. Biology 2021, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Viñas, A.M.; Sánchez, L.; Díaz, N.; Ribas, L.; Piferrer, F. Genetic architecture of sex determination in fish: Applications to sex ratio control in aquaculture. Front. Genet. 2014, 5, 340. [Google Scholar] [CrossRef] [PubMed]
- Budd, A.; Banh, Q.; Domingos, J.; Jerry, D. Sex control in fish: Approaches, challenges and opportunities for aquaculture. J. Mar. Sci. Eng. 2015, 3, 329–355. [Google Scholar] [CrossRef]
- Ge, C.; Ye, J.; Weber, C.; Sun, W.; Zhang, H.; Zhou, Y.; Cai, C.; Qian, G.; Capel, B. The histone demethylase KDM6B regulates temperature-dependent sex determination in a turtle species. Science 2018, 360, 645–648. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Tafessu, A.; O’Hara, R.; Martire, S.; Dube, A.L.; Saha, P.; Gant, V.U.; Banaszynski, L.A. H3.3 contributes to chromatin accessibility and transcription factor binding at promoter-proximal regulatory elements in embryonic stem cells. Genome Biol. 2023, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Recalde, O.; Goikoetxea, A.; Hore, T.A.; Todd, E.V.; Gemmell, N.J. The genetics and epigenetics of sex change in fish. Annu. Rev. Anim. Biosci. 2020, 8, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhuang, Z.; Sun, Y.; Chen, Q.; Zheng, X.; Liang, Y.; Mahboob, S.; Wang, Q. Changes in DNA methylation during epigenetic-associated sex reversal under low temperature in Takifugu rubripes. PLoS ONE 2019, 14, e0221641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Liu, Z.; Zhang, L.; Zhang, W. Epigenetic modifications during sex change repress gonadotropin stimulation of cyp19a1a in a teleost ricefield eel (Monopterus albus). Endocrinology 2013, 154, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.C.; Li, H.W.; Huang, C.H.; Lin, H.J.; Lin, C.J.; Chang, C.F. The testis is a primary factor that contributes to epigenetic modifications in the ovaries of the protandrous Black Porgy, Acanthopagrus schlegelii. Biol. Reprod. 2016, 94, 132. [Google Scholar] [CrossRef]
- Wen, A.Y.; You, F.; Sun, P.; Li, J.; Xu, D.D.; Wu, Z.H.; Ma, D.Y.; Zhang, P.J. CpG methylation of dmrt1 and cyp19a promoters in relation to their sexual dimorphic expression in the Japanese flounder Paralichthys olivaceus. J. Fish Biol. 2014, 84, 193–205. [Google Scholar] [CrossRef]
- Si, Y.; Ding, Y.; He, F.; Wen, H.; Li, J.; Zhao, J.; Huang, Z. DNA methylation level of cyp19a1a and Foxl2 gene related to their expression patterns and reproduction traits during ovary development stages of Japanese flounder (Paralichthys olivaceus). Gene 2016, 575, 321–330. [Google Scholar] [CrossRef]
- Todd, E.V.; Ortega-Recalde, O.; Liu, H.; Lamm, M.S.; Rutherford, K.M.; Cross, H.; Black, M.A.; Kardailsky, O.; Graves, J.A.M.; Hore, T.A.; et al. Stress, novel sex genes, and epigenetic reprogramming orchestrate socially controlled sex change. Sci. Adv. 2019, 5, eaaw7006. [Google Scholar] [CrossRef] [PubMed]
- Shabir, U.; Dar, J.S.; Bhat, A.H.; Nazir, R.; Ameen, F. The hidden world of fish fungal pathogens: Molecular identification and phylogenetic analysis in common carp, Cyprinus carpio. Arch. Microbiol. 2023, 205, 311. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, L.; Zhou, Z.; Xiao, J.; Chen, B.; Huang, P.; Li, C.; Xue, Y.; Liu, R.; Bai, Y.; et al. Comparative transcriptome analysis of early sexual differentiation in the male and female gonads of common carp (Cyprinus carpio). Aquaculture 2023, 563, 738984. [Google Scholar] [CrossRef]
- Feng, X.; Yu, X.; Fu, B.; Wang, X.; Liu, H.; Pang, M.; Tong, J. A high-resolution genetic linkage map and QTL fine mapping for growth-related traits and sex in the Yangtze River common carp (Cyprinus carpio haematopterus). BMC Genom. 2018, 19, 230. [Google Scholar] [CrossRef] [PubMed]
- Kocour, M.; Linhart, O.; Gela, D.; Rodina, M. Growth performance of all-female and mixed-sex common carp Cyprinus Carpio L. populations in the central Europe climatic conditions. J. World Aquac. Soc. 2005, 36, 103–113. [Google Scholar] [CrossRef]
- Chen, L.; Xu, J.; Sun, X.; Xu, P. Research advances and future perspectives of genomics and genetic improvement in allotetraploid common carp. Rev. Aquac. 2021, 14, 957–978. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, S.; Chen, J.; Chen, K.; Ma, W.; Wu, X.; Luo, H.; Li, Y.; Zhu, Z.; Hu, W. Timing of gonadal development and dimorphic expression of sex-related genes in gonads during early sex differentiation in the Yellow River carp. Aquaculture 2020, 518, 734825. [Google Scholar] [CrossRef]
- Chen, G.; Huang, J.; Jia, J.; Lou, Q.; Shi, C.; Yasheng, M.; Zhao, Y.; Yuan, Q.; Tang, K.; Liu, X.; et al. The food safety assessment of all-female common carp (Cyprinus carpio) (cyp17a1+/−; XX genotype) generated using genome editing technology. Food Chem. Toxicol. 2023, 181, 114103. [Google Scholar] [CrossRef]
- Chen, J.J.; Xia, X.H.; Wang, L.F.; Jia, Y.F.; Nan, P.; Li, L.; Chang, Z.J. Identification and comparison of gonadal transcripts of testis and ovary of adult common carp Cyprinus carpio using suppression subtractive hybridization. Theriogenology 2015, 83, 1416–1427. [Google Scholar] [CrossRef]
- Anitha, A.; Gupta, Y.R.; Deepa, S.; Ningappa, M.; Rajanna, K.B.; Senthilkumaran, B. Gonadal transcriptome analysis of the common carp, Cyprinus carpio: Identification of differentially expressed genes and SSRs. Gen. Comp. Endocrinol. 2019, 279, 67–77. [Google Scholar] [CrossRef]
- Li, J.T.; Wang, Q.; Huang Yang, M.D.; Li, Q.-S.; Cui, M.S.; Dong, Z.J.; Wang, H.W.; Yu, J.H.; Zhao, Y.J.; Yang, C.R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Lareau, C.A.; Ramirez, R.N.; Rose, S.A.; Maier, B.; Wroblewska, A.; Desland, F.; Chudnovskiy, A.; Mortha, A.; Dominguez, C.; et al. The cis-regulatory atlas of the mouse immune system. Cell 2019, 176, 897–912.e20. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Wang, Q.; Zhang, J.; Zhao, R.; Cao, Y.; Yu, S.; Wang, K.; Chen, Y.; Ma, Z.; Sun, X.; et al. Differential expression of miRNAs, lncRNAs, and circRNAs between ovaries and testes in common carp (Cyprinus carpio). Cells 2023, 12, 2631. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Y.; Zhong, C.; Guo, Y.; Wei, T.; Li, S.; Lin, H.; Liu, X. Integration of ATAC-seq and RNA-seq unravels chromatin accessibility during sex reversal in Orange-Spotted Grouper (Epinephelus coioides). Int. J. Mol. Sci. 2020, 21, 2800. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Lian, Z.; Xia, X.; Tian, H.; Li, Z. Integrated chromatin accessibility and DNA methylation analysis to reveal the critical epigenetic modification and regulatory mechanism in gonadal differentiation of the sequentially hermaphroditic fish, Monopterus albus. Biol. Sex Differ. 2022, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- He, H.H.; Meyer, C.A.; Shin, H.; Bailey, S.T.; Wei, G.; Wang, Q.; Zhang, Y.; Xu, K.; Ni, M.; Lupien, M.; et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010, 42, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, W.; Hu, S.; Wang, X.; Zhang, Y. Inherited DNA methylation primes the establishment of accessible chromatin during genome activation. Genome Res. 2018, 28, 998–1007. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Yang, H.; Zhang, T.; Naruse, K.; Tu, Q. Dynamic transcriptional and chromatin accessibility landscape of medaka embryogenesis. Genome Res. 2020, 30, 924–937. [Google Scholar] [CrossRef]
- Xu, P.; Meng, A. Maternal factors regulate embryonic development in zebrafish. Sci. Sin. Vitae 2014, 44, 954–964. [Google Scholar] [CrossRef]
- Navarro-Martín, L.; Viñas, J.; Ribas, L.; Díaz, N.; Gutiérrez, A.; Di Croce, L.; Piferrer, F. DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European Sea Bass. PLoS Genet. 2011, 7, e1002447. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Cairns, B.R. Genome-wide DNA methylation profiling in zebrafish. In The Zebrafish-Genetics, Genomics, and Transcriptomics; Methods in Cell Biology; Academic Press: Salt Lake City, UT, USA, 2016; pp. 345–359. [Google Scholar] [CrossRef]
- He, L.; Liang, X.; Wang, Q.; Yang, C.; Li, Y.; Liao, L.; Zhu, Z.; Wang, Y. Genome-wide DNA methylation reveals potential epigenetic mechanism of age-dependent viral susceptibility in grass carp. Immun. Ageing 2022, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Tenugu, S.; Pranoty, A.; Mamta, S.-K.; Senthilkumaran, B. Development and organisation of gonadal steroidogenesis in bony fishes. Aquac. Fish. 2021, 6, 223–246. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef] [PubMed]
- Nanavaty, V.; Abrash, E.W.; Hong, C.; Park, S.; Fink, E.E.; Li, Z.; Sweet, T.J.; Bhasin, J.M.; Singuri, S.; Lee, B.H.; et al. DNA methylation regulates alternative polyadenylation via CTCF and the cohesin complex. Mol. Cell 2020, 78, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Kossack, M.E.; High, S.K.; Hopton, R.E.; Yan, Y.-l.; Postlethwait, J.H.; Draper, B.W. Female sex development and reproductive duct formation depend on Wnt4a in zebrafish. Genetics 2019, 211, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Zohar, Y.; Muñoz-Cueto, J.A.; Elizur, A.; Kah, O. Neuroendocrinology of reproduction in teleost fish. Gen. Comp. Endocrinol. 2010, 165, 438–455. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Wu, H.; Xu, T.; Feng, H.; Chen, L.; Li, B.; Yao, B.; Qin, Z.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, M.; Wang, Q.; Zhao, R.; Cao, Y.; Zhang, J.; Sun, X.; Yu, S.; Wang, K.; Chen, Y.; Zhang, Y.; et al. Analysis of Chromatin Accessibility and DNA Methylation to Reveal the Functions of Epigenetic Modifications in Cyprinus carpio Gonads. Int. J. Mol. Sci. 2024, 25, 321. https://doi.org/10.3390/ijms25010321
Hou M, Wang Q, Zhao R, Cao Y, Zhang J, Sun X, Yu S, Wang K, Chen Y, Zhang Y, et al. Analysis of Chromatin Accessibility and DNA Methylation to Reveal the Functions of Epigenetic Modifications in Cyprinus carpio Gonads. International Journal of Molecular Sciences. 2024; 25(1):321. https://doi.org/10.3390/ijms25010321
Chicago/Turabian StyleHou, Mingxi, Qi Wang, Ran Zhao, Yiming Cao, Jin Zhang, Xiaoqing Sun, Shuangting Yu, Kaikuo Wang, Yingjie Chen, Yan Zhang, and et al. 2024. "Analysis of Chromatin Accessibility and DNA Methylation to Reveal the Functions of Epigenetic Modifications in Cyprinus carpio Gonads" International Journal of Molecular Sciences 25, no. 1: 321. https://doi.org/10.3390/ijms25010321
APA StyleHou, M., Wang, Q., Zhao, R., Cao, Y., Zhang, J., Sun, X., Yu, S., Wang, K., Chen, Y., Zhang, Y., & Li, J. (2024). Analysis of Chromatin Accessibility and DNA Methylation to Reveal the Functions of Epigenetic Modifications in Cyprinus carpio Gonads. International Journal of Molecular Sciences, 25(1), 321. https://doi.org/10.3390/ijms25010321