

Intracellular Trafficking of Size-Tuned Nanoparticles for Drug Delivery





Abstract
:
1. Introduction
2. Results
2.1. NP Synthesis and Characterization
2.2. Rh123 and Dox-Release Profile
2.3. Cytotoxicity Assay
2.4. Intracellular Trafficking
3. Discussion
4. Methods and Materials
4.1. NPs Synthesis
4.2. NPs Characterization
4.2.1. Size Distribution and Zeta-Potential Determinations
4.2.2. NPs Fluorescence and Rh123 Release Profile
4.2.3. Drug Content and Release Profile
4.3. Biological Assays
4.3.1. Cell Seeding
4.3.2. Cell Metabolic Activity
4.3.3. Intracellular Trafficking Analyses
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gajbhiye, K.R.; Salve, R.; Narwade, M.; Sheikh, A.; Kesharwani, P.; Gajbhiye, V. Lipid Polymer Hybrid Nanoparticles: A Custom-Tailored next-Generation Approach for Cancer Therapeutics. Mol. Cancer 2023, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N. Poly(Glycerol)-Based Biomedical Nanodevices Constructed by Functional Programming on Inorganic Nanoparticles for Cancer Nanomedicine. Acc. Chem. Res. 2023, 56, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Violatto, M.B.; Casarin, E.; Talamini, L.; Russo, L.; Baldan, S.; Tondello, C.; Messmer, M.; Hintermann, E.; Rossi, A.; Passoni, A.; et al. Dexamethasone Conjugation to Biodegradable Avidin-Nucleic-Acid-Nano-Assemblies Promotes Selective Liver Targeting and Improves Therapeutic Efficacy in an Autoimmune Hepatitis Murine Model. ACS Nano 2019, 13, 4410–4423. [Google Scholar] [CrossRef] [PubMed]
- Morelli, L.; Gimondi, S.; Sevieri, M.; Salvioni, L.; Guizzetti, M.; Colzani, B.; Palugan, L.; Foppoli, A.; Talamini, L.; Morosi, L.; et al. Monitoring the Fate of Orally Administered PLGA Nanoformulation for Local Delivery of Therapeutic Drugs. Pharmaceutics 2019, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Mendanha, D.; Vieira de Castro, J.; Ferreira, H.; Neves, N.M. Biomimetic and Cell-Based Nanocarriers—New Strategies for Brain Tumor Targeting. J. Control. Release 2021, 337, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Mendanha, D.; Vieira de Castro, J.; Casanova, M.R.; Gimondi, S.; Ferreira, H.; Neves, N.M. Macrophage Cell Membrane Infused Biomimetic Liposomes for Glioblastoma Targeted Therapy. Nanomedicine 2023, 49, 102663. [Google Scholar] [CrossRef] [PubMed]
- McNiven, M.A.; Ridley, A.J. Focus on Membrane Dynamics. Trends Cell Biol. 2006, 16, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Gimondi, S.; Vieira de Castro, J.; Reis, R.L.; Ferreira, H.; Neves, N.M. On the Size-Dependent Internalization of Sub-Hundred Polymeric Nanoparticles. Colloids Surf. B Biointerfaces 2023, 225, 113245. [Google Scholar] [CrossRef]
- Gimondi, S.; Ferreira, H.; Reis, R.L.; Neves, N.M. Size-Dependent Polymeric Nanoparticle Distribution in a Static versus Dynamic Microfluidic Blood Vessel Model: Implications for Nanoparticle-Based Drug Delivery. ACS Appl. Nano Mater. 2023, 6, 7364–7374. [Google Scholar] [CrossRef]
- Lostao, A.; Lim, K.; Pallarés, M.C.; Ptak, A.; Marcuello, C. Recent Advances in Sensing the Inter-Biomolecular Interactions at the Nanoscale—A Comprehensive Review of AFM-Based Force Spectroscopy. Int. J. Biol. Macromol. 2023, 238, 124089. [Google Scholar] [CrossRef]
- Wang, S.-H.; Lee, C.-W.; Chiou, A.; Wei, P.-K. Size-Dependent Endocytosis of Gold Nanoparticles Studied by Three-Dimensional Mapping of Plasmonic Scattering Images. J. Nanobiotechnol. 2010, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kröger, M.; Liu, W.K. Endocytosis of PEGylated Nanoparticles Accompanied by Structural and Free Energy Changes of the Grafted Polyethylene Glycol. Biomaterials 2014, 35, 8467–8478. [Google Scholar] [CrossRef] [PubMed]
- Toscano, F.; Torres-Arias, M. Nanoparticles Cellular Uptake, Trafficking, Activation, Toxicity and In Vitro Evaluation. Curr. Res. Immunol. 2023, 4, 100073. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.; Alkawareek, M.; Dreaden, E.; Brown, D.; Alkilany, A.; Farokhzad, O.; Mahmoudi, M. Cellular Uptake of Nanoparticles: Journey inside the Cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.J.; Burgers, T.C.Q.; Vlijm, R.; Roos, W.H.; Åberg, C. Rapid Internalization of Nanoparticles by Human Cells at the Single Particle Level. ACS Nano 2023, 17, 16517–16529. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.-G.; Skotland, T.; Sandvig, K. Endocytosis and Intracellular Transport of Nanoparticles: Present Knowledge and Need for Future Studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Kjeken, R.; Egeberg, M.; Habermann, A.; Kuehnel, M.; Peyron, P.; Floetenmeyer, M.; Walther, P.; Jahraus, A.; Defacque, H.; Kuznetsov, S.A.; et al. Fusion between Phagosomes, Early and Late Endosomes: A Role for Actin in Fusion between Late, but Not Early Endocytic Organelles. Mol. Biol. Cell 2004, 15, 345–358. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding Biophysicochemical Interactions at the Nano–Bio Interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Bannunah, A.M.; Vllasaliu, D.; Lord, J.; Stolnik, S. Mechanisms of Nanoparticle Internalization and Transport Across an Intestinal Epithelial Cell Model: Effect of Size and Surface Charge. Mol. Pharm. 2014, 11, 4363–4373. [Google Scholar] [CrossRef]
- Xue, J.; Guan, Z.; Zhu, X.; Lin, J.; Cai, C.; Jin, X.; Li, Y.; Ye, Z.; Zhang, W.; Jiang, X. Cellular Internalization of Polypeptide-Based Nanoparticles: Effects of Size, Shape and Surface Morphology. Biomater. Sci. 2018, 6, 3251–3261. [Google Scholar] [CrossRef]
- Petithory, T.; Pieuchot, L.; Josien, L.; Ponche, A.; Anselme, K.; Vonna, L. Size-Dependent Internalization Efficiency of Macrophages from Adsorbed Nanoparticle-Based Monolayers. Nanomaterials 2021, 11, 1963. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Rai, B. Effect of Size and Surface Charge of Gold Nanoparticles on Their Skin Permeability: A Molecular Dynamics Study. Sci. Rep. 2017, 7, 45292. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Y.; Yuan, H.; Gao, H.; Zhang, S. Role of Nanoparticle Geometry in Endocytosis: Laying down to Stand Up. Nano Lett. 2013, 13, 4546–4550. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Zhang, X. Cooperative Effect in Receptor-Mediated Endocytosis of Multiple Nanoparticles. ACS Nano 2012, 6, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, C.F.; Ahmed, R.; Mataji-Kojouri, A.; Soto, F.; Wang, J.; Liu, S.; Stoyanova, T.; Marques, A.P.; Reis, R.L.; Demirci, U. Engineering Polysaccharide-Based Hydrogel Photonic Constructs: From Multiscale Detection to the Biofabrication of Living Optical Fibers. Adv. Mater. 2021, 33, 2105361. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, C.F.; Gasperini, L.; Marques, A.P.; Reis, R.L. 3D Flow-Focusing Microfluidic Biofabrication: One-Chip-Fits-All Hydrogel Fiber Architectures. Appl. Mater. Today 2021, 23, 101013. [Google Scholar] [CrossRef]
- Guimarães, C.F.; Gasperini, L.; Ribeiro, R.S.; Carvalho, A.F.; Marques, A.P.; Reis, R.L. High-Throughput Fabrication of Cell-Laden 3D Biomaterial Gradients. Mater. Horiz. 2020, 7, 2414–2421. [Google Scholar] [CrossRef]
- Liu, Z.; Fontana, F.; Python, A.; Hirvonen, J.T.; Santos, H.A. Microfluidics for Production of Particles: Mechanism, Methodology, and Applications. Small 2020, 16, 1904673. [Google Scholar] [CrossRef]
- Gimondi, S.; Ferreira, H.; Reis, R.L.; Neves, N.M. Microfluidic Devices: A Tool for Nanoparticle Synthesis and Performance Evaluation. ACS Nano 2023, 17, 14205–14228. [Google Scholar] [CrossRef]
- Gimondi, S.; Guimarães, C.F.; Vieira, S.F.; Gonçalves, V.M.F.; Tiritan, M.E.; Reis, R.L.; Ferreira, H.; Neves, N.M. Microfluidic Mixing System for Precise PLGA-PEG Nanoparticles Size Control. Nanomed. Nanotechnol. Biol. Med. 2021, 40, 102482. [Google Scholar] [CrossRef]
- Gimondi, S.; Reis, R.L.; Ferreira, H.; Neves, N.M. Microfluidic-Driven Mixing of High Molecular Weight Polymeric Complexes for Precise Nanoparticle Downsizing. Nanomedicine 2022, 43, 102560. [Google Scholar] [CrossRef] [PubMed]
- Mendanha, D.; Gimondi, S.; Costa, B.M.; Ferreira, H.; Neves, N.M. Microfluidic-Derived Docosahexaenoic Acid Liposomes for Glioblastoma Therapy. Nanomed. Nanotechnol. Biol. Med. 2023, 53, 102704. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, Q.; Gao, J.; Chen, L.; Vasanthan, T. Preparation and Characterization of Nanoparticles from Field Pea Starch by Batch versus Continuous Nanoprecipitation Techniques. Food Hydrocoll. 2022, 122, 107098. [Google Scholar] [CrossRef]
- Mou, J.; Wang, C.; Zhao, H.; Xiong, C.; Ren, Y.; Wang, J.; Jiang, D.; Zheng, Z. Centrifugal Microfluidic Synthesis of Nickel Sesquioxide Nanoparticles. Micromachines 2023, 14, 1741. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, A.; Bansal, I.; Jaiswar, A.; Roy, N.; Verma, D. A Simple Cost-Effective Microfluidic Platform for Rapid Synthesis of Diverse Metal Nanoparticles: A Novel Approach towards Fighting SARS-CoV-2. Mater. Today Proc. 2023, 80, 1852–1857. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, T.; Li, Y.; Huang, G.; White, M.A.; Gao, J. Investigation of Endosome and Lysosome Biology by Ultra pH-Sensitive Nanoprobes. Adv. Drug Deliv. Rev. 2017, 113, 87–96. [Google Scholar] [CrossRef]
- Dong, H.; Sun, L.-D.; Yan, C.-H. Upconversion Emission Studies of Single Particles. Nano Today 2020, 35, 100956. [Google Scholar] [CrossRef]
- Weiss, S. Shattering the Diffraction Limit of Light: A Revolution in Fluorescence Microscopy? Proc. Natl. Acad. Sci. USA 2000, 97, 8747–8749. [Google Scholar] [CrossRef]
- Strauch, M.; Lüdke, A.; Münch, D.; Laudes, T.; Galizia, C.G.; Martinelli, E.; Lavra, L.; Paolesse, R.; Ulivieri, A.; Catini, A.; et al. More than Apples and Oranges--Detecting Cancer with a Fruit Fly’s Antenna. Sci. Rep. 2014, 4, 3576. [Google Scholar] [CrossRef]
- Wosikowski, K.; Schuurhuis, D.; Kops, G.J.; Saceda, M.; Bates, S.E. Altered Gene Expression in Drug-Resistant Human Breast Cancer Cells. Clin. Cancer Res. 1997, 3, 2405–2414. [Google Scholar] [PubMed]
- Partikel, K.; Korte, R.; Stein, N.C.; Mulac, D.; Herrmann, F.C.; Humpf, H.-U.; Langer, K. Effect of Nanoparticle Size and PEGylation on the Protein Corona of PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2019, 141, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Li, N.; Zhang, W.; Zhang, W. Exploring the Impact of PEGylation on the Cell-Nanomicelle Interactions by AFM-Based Single-Molecule Force Spectroscopy and Force Tracing. Acta Biomater. 2023, 157, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle Colloidal Stability in Cell Culture Media and Impact on Cellular Interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef] [PubMed]
- Ayala, V.; Herrera, A.P.; Latorre-Esteves, M.; Torres-Lugo, M.; Rinaldi, C. Effect of Surface Charge on the Colloidal Stability and in Vitro Uptake of Carboxymethyl Dextran-Coated Iron Oxide Nanoparticles. J. Nanopart. Res. 2013, 15, 1874. [Google Scholar] [CrossRef] [PubMed]
- Ben-Akiva, E.; Hickey, J.W.; Meyer, R.A.; Isser, A.; Shannon, S.R.; Livingston, N.K.; Rhodes, K.R.; Kosmides, A.K.; Warren, T.R.; Tzeng, S.Y.; et al. Shape Matters: Biodegradable Anisotropic Nanoparticle Artificial Antigen Presenting Cells for Cancer Immunotherapy. Acta Biomater. 2023, 160, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Walsh, M.L.; Chen, L.B. Localization of Mitochondria in Living Cells with Rhodamine 123. Proc. Natl. Acad. Sci. USA 1980, 77, 990–994. [Google Scholar] [CrossRef]
- Juan, G.; Cavazzoni, M.; Sáez, G.T.; O’Connor, J.-E. A Fast Kinetic Method for Assessing Mitochondrial Membrane Potential in Isolated Hepatocytes with Rhodamine 123 and Flow Cytometry. Cytometry 1994, 15, 335–342. [Google Scholar] [CrossRef]
- Huang, M.; Camara, A.K.S.; Stowe, D.F.; Qi, F.; Beard, D.A. Mitochondrial Inner Membrane Electrophysiology Assessed by Rhodamine-123 Transport and Fluorescence. Ann. Biomed. Eng. 2007, 35, 1276–1285. [Google Scholar] [CrossRef]
- Zorova, L.D.; Demchenko, E.A.; Korshunova, G.A.; Tashlitsky, V.N.; Zorov, S.D.; Andrianova, N.V.; Popkov, V.A.; Babenko, V.A.; Pevzner, I.B.; Silachev, D.N.; et al. Is the Mitochondrial Membrane Potential (∆Ψ) Correctly Assessed? Intracellular and Intramitochondrial Modifications of the ∆Ψ Probe, Rhodamine 123. Int. J. Mol. Sci. 2022, 23, 482. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Zolnik, B.S.; Burgess, D.J. Effect of Acidic pH on PLGA Microsphere Degradation and Release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.K.; Reineke, J.J. Quantitative Detection of PLGA Nanoparticle Degradation in Tissues Following Intravenous Administration. Mol. Pharm. 2013, 10, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Tong, H.H.Y.; Chow, S.F. In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics 2020, 12, 732. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Della Sala, A.; Tocchetti, C.G.; Porporato, P.E.; Ghigo, A. Metabolic Aspects of Anthracycline Cardiotoxicity. Curr. Treat. Options Oncol. 2021, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-Induced Cardiotoxicity: An Update on the Molecular Mechanism and Novel Therapeutic Strategies for Effective Management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, S.B.; de Oliveira Silva, J.; Garcia-Fossa, F.; Leite, E.A.; Malachias, A.; Pound-Lana, G.; Mosqueira, V.C.F.; Oliveira, M.C.; de Barros, A.L.B.; de Jesus, M.B. Mechanistic Insights into the Intracellular Release of Doxorubicin from pH-Sensitive Liposomes. Biomed. Pharmacother. 2021, 134, 110952. [Google Scholar] [CrossRef]
- Javan, N.; Khadem Ansari, M.H.; Dadashpour, M.; Khojastehfard, M.; Bastami, M.; Rahmati-Yamchi, M.; Zarghami, N. Synergistic Antiproliferative Effects of Co-Nanoencapsulated Curcumin and Chrysin on MDA-MB-231 Breast Cancer Cells through Upregulating miR-132 and miR-502c. Nutr. Cancer 2019, 71, 1201–1213. [Google Scholar] [CrossRef]
- Liu, X.; Huang, N.; Li, H.; Jin, Q.; Ji, J. Surface and Size Effects on Cell Interaction of Gold Nanoparticles with Both Phagocytic and Nonphagocytic Cells. Langmuir 2013, 29, 9138–9148. [Google Scholar] [CrossRef]
- Yuan, T.; Gao, L.; Zhan, W.; Dini, D. Effect of Particle Size and Surface Charge on Nanoparticles Diffusion in the Brain White Matter. Pharm. Res. 2022, 39, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Shah, S.; Thomas, A.; Ou-Yang, H.D.; Liu, Y. The Influence of Size, Shape and Vessel Geometry on Nanoparticle Distribution. Microfluid. Nanofluid. 2013, 14, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of Nanoparticle Size, Shape and Surface Chemistry in Oral Drug Delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Guzmán, M.; Molpeceres, J.; Aberturas, M.R. Freeze-Drying of Polycaprolactone and Poly(D,L-Lactic-Glycolic) Nanoparticles Induce Minor Particle Size Changes Affecting the Oral Pharmacokinetics of Loaded Drugs. Eur. J. Pharm. Biopharm. 2000, 50, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Shi, B.; Pei, Y.-Y.; Hong, M.-H.; Wu, J.; Chen, H.-Z. In Vivo Tumor Targeting of Tumor Necrosis Factor-Alpha-Loaded Stealth Nanoparticles: Effect of MePEG Molecular Weight and Particle Size. Eur. J. Pharm. Sci. 2006, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic Pathways and Endosomal Trafficking: A Primer. Wien Med Wochenschr 2016, 166, 196–204. [Google Scholar] [CrossRef]
- Rainero, E.; Norman, J.C. Late Endosomal and Lysosomal Trafficking during Integrin-mediated Cell Migration and Invasion: Cell Matrix Receptors Are Trafficked through the Late Endosomal Pathway in a Way That Dictates How Cells Migrate. Bioessays 2013, 35, 523–532. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome Maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Zhukova, V.; Osipova, N.; Semyonkin, A.; Malinovskaya, J.; Melnikov, P.; Valikhov, M.; Porozov, Y.; Solovev, Y.; Kuliaev, P.; Zhang, E.; et al. Fluorescently Labeled PLGA Nanoparticles for Visualization In Vitro and In Vivo: The Importance of Dye Properties. Pharmaceutics 2021, 13, 1145. [Google Scholar] [CrossRef]
- Zhang, E.; Zhukova, V.; Semyonkin, A.; Osipova, N.; Malinovskaya, Y.; Maksimenko, O.; Chernikov, V.; Sokolov, M.; Grigartzik, L.; Sabel, B.A.; et al. Release Kinetics of Fluorescent Dyes from PLGA Nanoparticles in Retinal Blood Vessels: In Vivo Monitoring and Ex Vivo Localization. Eur. J. Pharm. Biopharm. 2020, 150, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Fang, L.; Ling, J.; Ding, C.Z.; Kang, B.; Huang, C.Z. Nanotoxicity of Silver Nanoparticles to Red Blood Cells: Size Dependent Adsorption, Uptake, and Hemolytic Activity. Chem. Res. Toxicol. 2015, 28, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Li, X. Optimal Size of Gold Nanoparticles for Surface-Enhanced Raman Spectroscopy under Different Conditions. J. Nanomater. 2013, 2013, e790323. [Google Scholar] [CrossRef]
- Prabha, S.; Arya, G.; Chandra, R.; Ahmed, B.; Nimesh, S. Effect of Size on Biological Properties of Nanoparticles Employed in Gene Delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 83–91. [Google Scholar] [CrossRef]
- Meng, X.; Li, X. Size Limit and Energy Analysis of Nanoparticles during Wrapping Process by Membrane. Nanomaterials 2018, 8, 899. [Google Scholar] [CrossRef]
- Fröhlich, E.; Meindl, C.; Roblegg, E.; Ebner, B.; Absenger, M.; Pieber, T.R. Action of Polystyrene Nanoparticles of Different Sizes on Lysosomal Function and Integrity. Part. Fibre Toxicol. 2012, 9, 26. [Google Scholar] [CrossRef]
- Wu, M.; Guo, H.; Liu, L.; Liu, Y.; Xie, L. Size-Dependent Cellular Uptake and Localization Profiles of Silver Nanoparticles. Int. J. Nanomed. 2019, 14, 4247–4259. [Google Scholar] [CrossRef]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in Speed, Utility and Usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef]
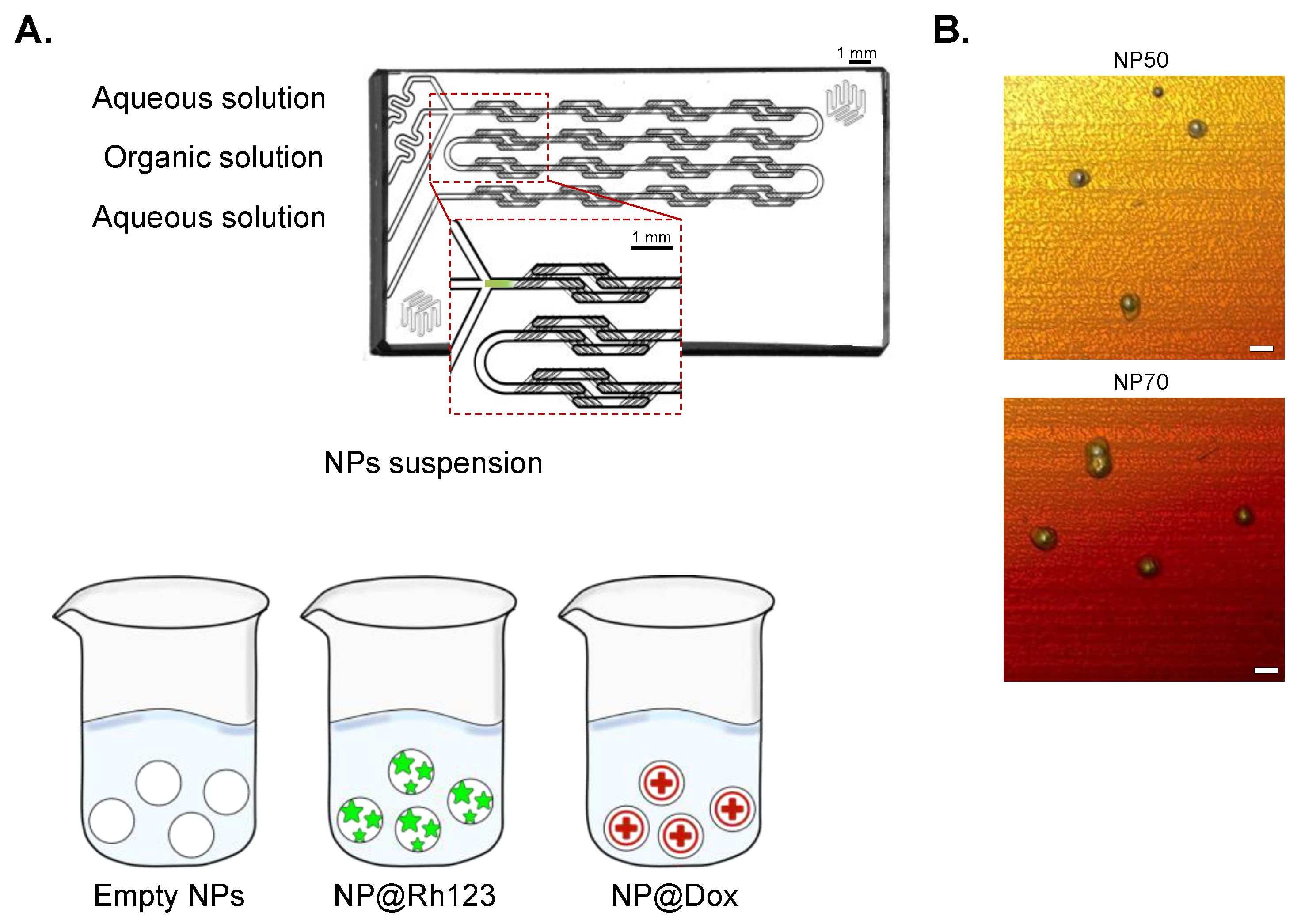

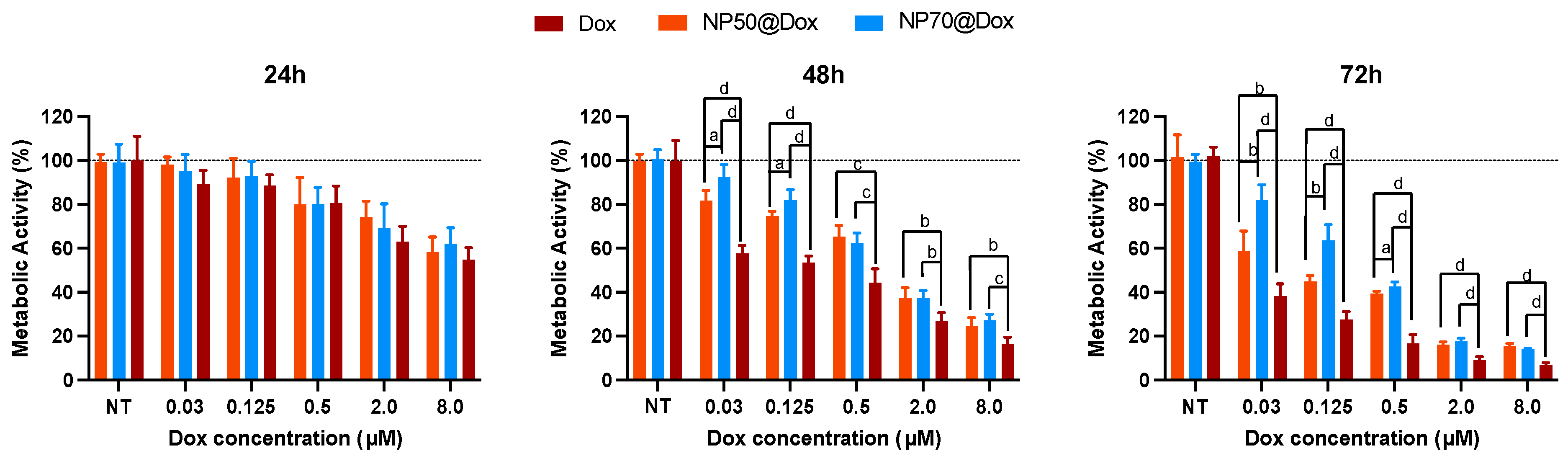
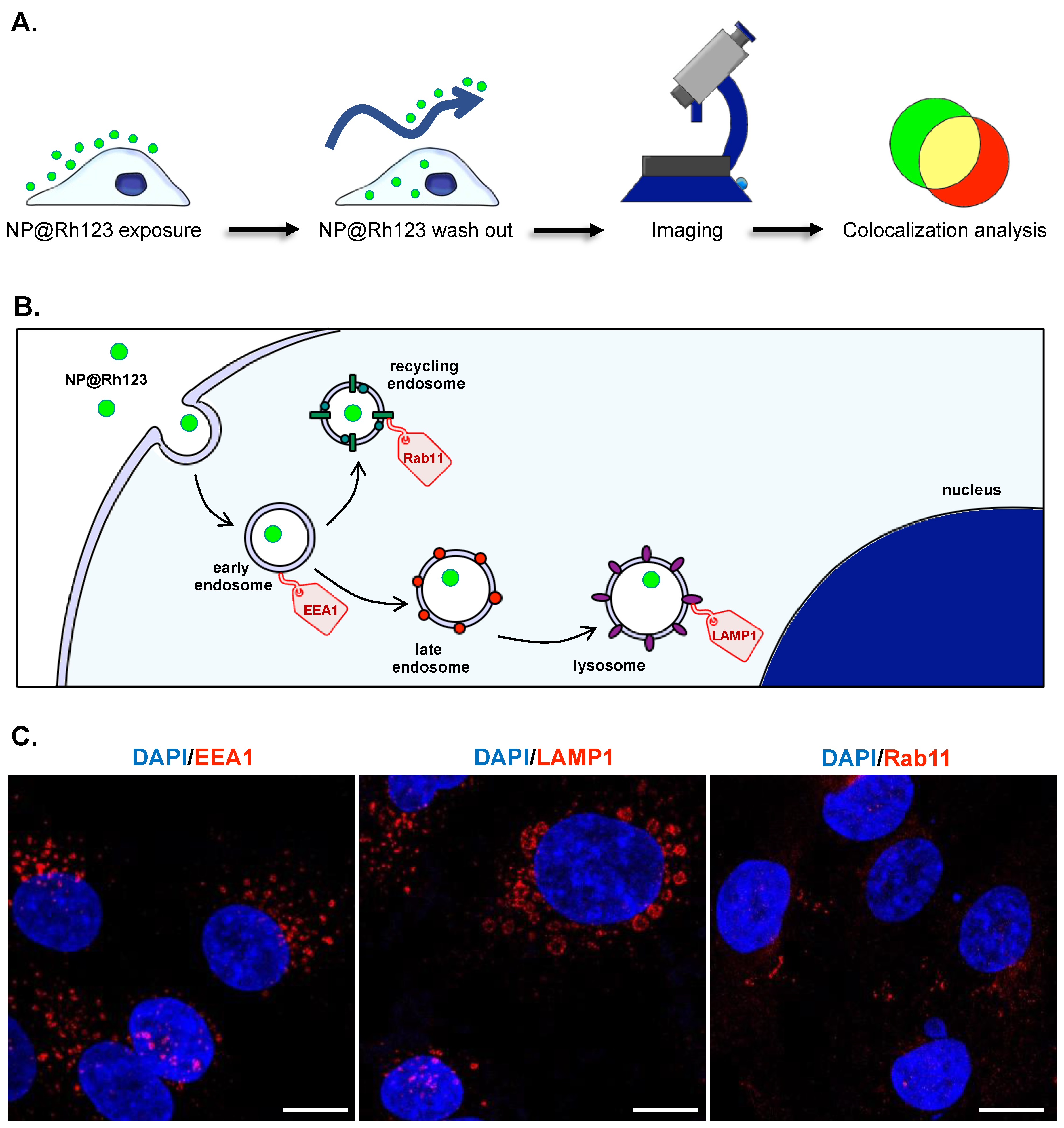



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) | PDI | ζ-Potential (mV) | E.E (%) |
|---|---|---|---|---|
| NP-50 | 50.1 ± 1.0 | 0.13 ± 0.01 | −20.1 ± 1.3 | --- |
| NP-70 | 69.9 ± 1.5 | 0.12 ± 0.01 | −22.2 ± 1.0 | --- |
| NP50@Rh123 | 51.4 ± 0.6 | 0.11 ± 0.01 | −18.1 ± 1.0 | 17.6 ± 1.5 |
| NP70@Rh123 | 72.6 ± 2.9 | 0.13 ± 0.04 | −16.2 ± 2.6 | 14.6 ± 1.0 |
| NP50@Dox | 55.2 ± 1.7 | 0.12 ± 0.02 | −19.9 ± 2.1 | 18.1 ± 0.6 |
| NP70@Dox | 78.1 ± 2.0 | 0.11 ± 0.01 | −20.5 ± 1.8 | 21.1 ± 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimondi, S.; Ferreira, H.; Reis, R.L.; Neves, N.M. Intracellular Trafficking of Size-Tuned Nanoparticles for Drug Delivery. Int. J. Mol. Sci. 2024, 25, 312. https://doi.org/10.3390/ijms25010312
Gimondi S, Ferreira H, Reis RL, Neves NM. Intracellular Trafficking of Size-Tuned Nanoparticles for Drug Delivery. International Journal of Molecular Sciences. 2024; 25(1):312. https://doi.org/10.3390/ijms25010312
Chicago/Turabian StyleGimondi, Sara, Helena Ferreira, Rui L. Reis, and Nuno M. Neves. 2024. "Intracellular Trafficking of Size-Tuned Nanoparticles for Drug Delivery" International Journal of Molecular Sciences 25, no. 1: 312. https://doi.org/10.3390/ijms25010312
APA StyleGimondi, S., Ferreira, H., Reis, R. L., & Neves, N. M. (2024). Intracellular Trafficking of Size-Tuned Nanoparticles for Drug Delivery. International Journal of Molecular Sciences, 25(1), 312. https://doi.org/10.3390/ijms25010312