
Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents?

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
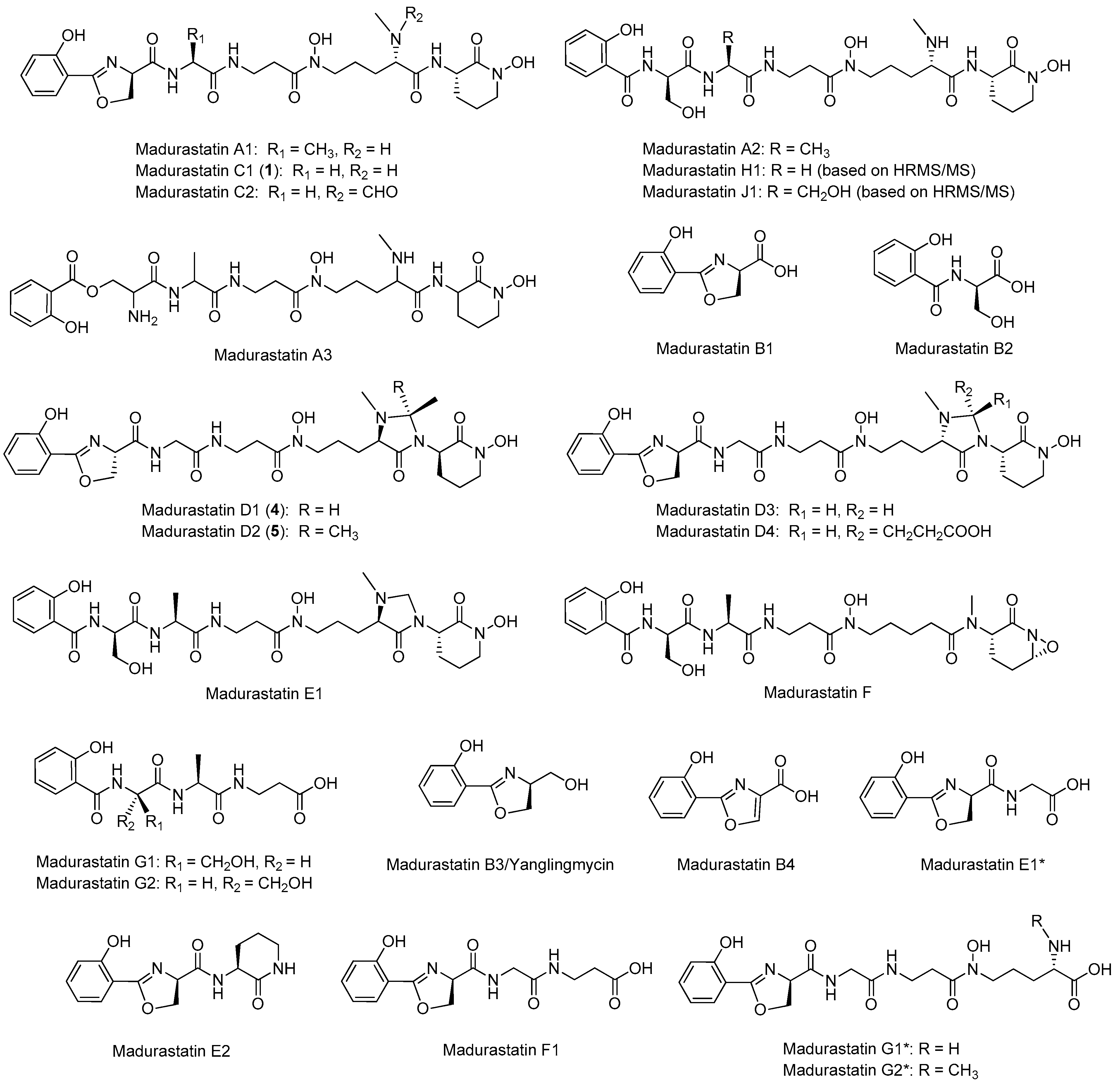
2.1. LC-UV-HRMS Analysis of the Extract from Actinomadura sp. CA-135719 and Identification of Components
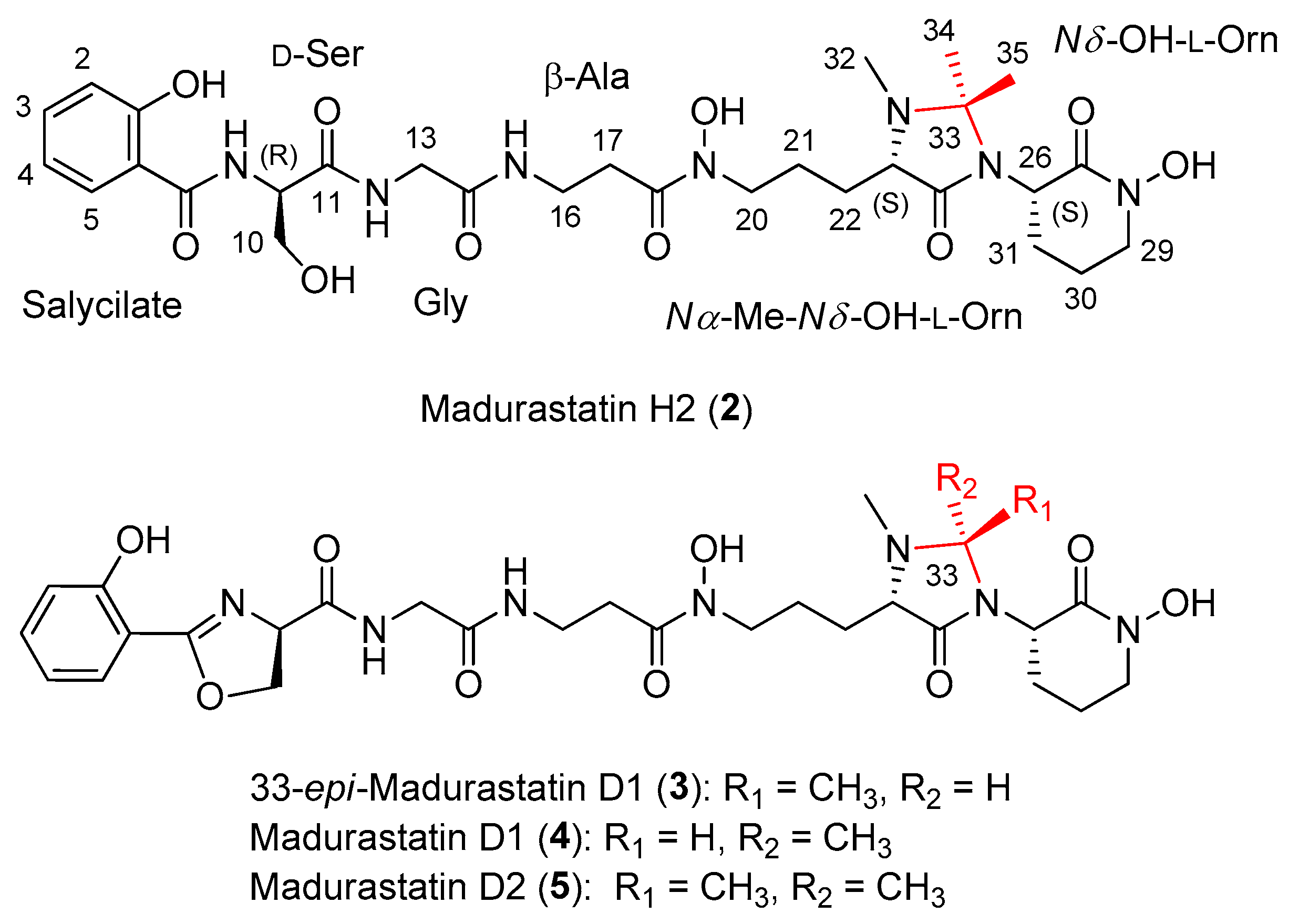
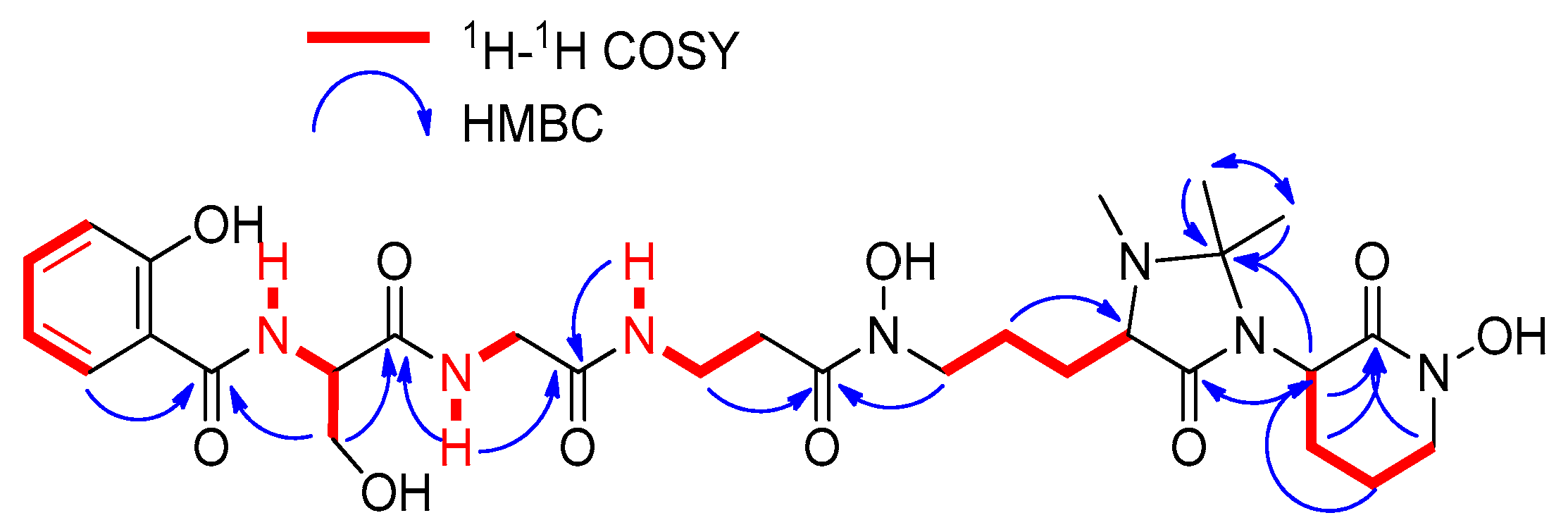
2.2. Extraction, Isolation, and Structural Elucidation
2.3. In Silico Analysis of the Madurastatin Biosynthetic Gene Cluster
2.4. Effect of the Solvent Used in the Extraction of Madurastatins
2.5. Obtainment of Madurastatin D2 from Madurastatin C1 Using Acetone
2.6. Antiparasitic Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Taxonomic Identification of the Producing Microorganism
3.3. Fermentation of the Producing Microorganism
3.4. Extraction and Isolation
3.5. Marfey’s Analysis of Compounds 1–4
3.6. Genome Sequencing and Biosynthetic Gene Clusters Prediction
3.7. Extraction of CA-135719 Cultures
3.8. Madurastatin C1 Reaction with Acetone
3.9. Antiparasitic Activity
3.9.1. Antiparasitic Activity on T. cruzi
3.9.2. Antiparasitic Activity on L. donovani
3.9.3. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haradaa, K.-I.; Tomita, K.; Fujii, K.; Masuda, K.; Mikami, Y.; Yazawa, K.; Komaki, H. Isolation and structural characterization of siderophores, madurastatins, produced by a pathogenic Actinomadura madurae. J. Antibiot. 2004, 57, 125–135. [Google Scholar] [CrossRef]
- Mazzei, E.; Iorio, M.; Maffioli, S.I.; Sosio, M.; Donadio, S. Characterization of madurastatin C1, a novel siderophore from Actinomadura sp. J. Antibiot. 2012, 65, 267–269. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; He, H.; Ma, R.; Ji, Z.; Wei, Q.; Dai, H.; Zhang, L.; Song, F. Madurastatin B3, a rare aziridine derivative from actinomycete Nocardiopsis sp. LS150010 with potent anti-tuberculosis activity. J. Ind. Microbiol. Biotechnol. 2017, 44, 589–594. [Google Scholar] [CrossRef]
- Yan, J.X.; Chevrette, M.G.; Braun, D.R.; Harper, M.K.; Currie, C.R.; Bugni, T.S. Madurastatin D1 and D2, oxazoline containing siderophores isolated from an Actinomadura sp. Org. Lett. 2019, 21, 6275–6279. [Google Scholar] [CrossRef]
- Lee, S.R.; Schalk, F.; Schwitalla, J.W.; Guo, H.; Yu, J.S.; Song, M.; Jung, W.H.; de Beer, Z.W.; Beemelmanns, C.; Kim, K.H. GNPS**-Guided discovery of madurastatin siderophores from the termite-associated Actinomadura sp. RB99. Chem. Eur. J. 2022, 28, e202200612. [Google Scholar] [CrossRef]
- Bill, M.K.; Kleiner, Y.; Flügel, J.L.; Kurz, M.; Spohn, M.; Marner, M.; Mihajlovic, S.; Vilcinskas, A.; Schäberle, T.F.; Hammann, P.E.; et al. Isolation, characterization, and bioactivity profiling of oxazoline containing madurastatin siderophores from Actinomadura sp. J. Antibiot. 2022, 75, 576–582. [Google Scholar] [CrossRef]
- Zhang, J.W.; Zhang, W.J.; Wei, S.P.; Wu, W.J. A New dihydrooxazole antibiotic from the fermentation broth of Streptomyces djakartensis. Heterocycles 2014, 89, 1656–1661. [Google Scholar]
- Tyler, A.R.; Mosaei, H.; Morton, S.; Waddell, P.G.; Wills, C.; McFarlane, W.; Gray, J.; Goodfellow, M.; Errington, J.; Allenby, N.; et al. Structural reassignment and absolute stereochemistry of madurastatin C1 (MBJ-0034) and the related aziridine siderophores: Madurastatins A1, B1, and MBJ-0035. J. Nat. Prod. 2017, 80, 1558–1562. [Google Scholar] [CrossRef]
- No, J.H. Visceral Leishmaniasis: Revisiting Current Treatments and Approaches for Future Discoveries. Acta Trop. 2016, 155, 113–123. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas Disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Marfey, P. Determination of D-amino acids. II Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Ayon, N.J.; Sharma, A.D.; Gutheil, W.G. LC-MS/MS-based separation and quantification of Marfey’s reagent derivatized proteinogenic amino acid DL-stereoisomers. J. Am. Soc. Mass Spectrom. 2019, 30, 448–458. [Google Scholar] [CrossRef]
- Liao, R.; Gao, Y.; Chen, M.; Li, L.; Hu, X. A Ubiquitous but overlooked side reaction in dimethyl labeling of peptides. Anal. Chem. 2018, 90, 13533–13540. [Google Scholar] [CrossRef]
- Mostardeiro, M.A.; Ilha, V.; Dahmer, J.; Caro, M.S.B.; Dalcol, I.I.; da Silva, U.F.; Morel, A.F. Cyclopeptide alkaloids: Stereochemistry and synthesis of the precursors of discarines C and D and myrianthine A. J. Nat. Prod. 2013, 76, 1343–1350. [Google Scholar] [CrossRef]
- Tschesche, R.; Elgamal, M.; Eckhardt, G. Alkaloide aus Rhamnaceen, XXVIII. Nummularin-G, -H und -K, weitere peptidalkaloide aus Ziziphus nummularia. Chem. Berichte 1977, 110, 2649–2655. [Google Scholar] [CrossRef]
- Tschesche, R.; Shah, A.H.; Eckhardt, G. Sativanine-a and sativanine-b, two new cyclopeptide alkaloids from the bark of Zizyphus sativa. Phytochemistry 1979, 18, 702–704. [Google Scholar] [CrossRef]
- Lomchoey, N.; Panseeta, P.; Boonsri, P.; Apiratikul, N.; Prabpai, S.; Kongsaeree, P.; Suksamrarn, S. New bioactive cyclopeptide alkaloids with rare terminal unit from the root bark of Ziziphus cambodiana. RSC Adv. 2018, 8, 18204–18215. [Google Scholar] [CrossRef]
- Heck, A.J.R.; Bonnici, P.J.; Breukink, E.; Morris, D.; Wills, M. Modification and inhibition of vancomycin group antibiotics by formaldehyde and acetaldehyde. Chem. Eur. J. 2001, 7, 910–916. [Google Scholar] [CrossRef]
- Koppel, C.; Tenczer, J.; Peixoto-Menezes, K.M. Formation of formaldehyde adducts from various drugs by use of methanol in a toxicological screening procedure with gas chromatography-mass spectrometry. J. Chromatogr. 1991, 563, 73–81. [Google Scholar] [CrossRef]
- Bak, A.; Fich, M.; Larsen, B.D.; Frokjaer, S.; Friis, G.J. N-Terminal 4-imidazolidinone prodrugs of Leu-enkephalin: Synthesis, chemical and enzymatic stability studies. Eur. J. Pharm. Sci. 1999, 7, 317–323. [Google Scholar] [CrossRef]
- Rasmussen, G.J.; Bundgaard, H. Prodrugs of peptides. 10. Protection of di-and tripeptides against aminopeptidase by formation of bioreversible 4-imidazolidinone derivatives. Int. J. Pharm. 1991, 71, 45–53. [Google Scholar] [CrossRef]
- Rasmussen, G.J.; Bundgaard, H. Prodrugs of peptides. 15. 4-Imidazolidinone prodrug derivatives of enkephalins to prevent aminopeptidase-catalyzed metabolism in plasma and absorptive mucosae. Int. J. Pharm. 1991, 76, 113–122. [Google Scholar] [CrossRef]
- Mathews, W.R.; Runge, T.A.; Haroldsen, P.E.; Gaskell, S.J. Characterization of impurities in a synthetic renin substrate peptide by fast-atom bombardment mass spectrometry and hybrid tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1989, 3, 314–319. [Google Scholar] [CrossRef]
- Fowles, L.F.; Beck, E.; Worrall, S.; Shanley, B.C.; de Jersey, J. The formation and stability of imidazolidinone adducts from acetaldehyde and model peptides A kinetic study with implications for protein modification in alcohol abuse. Biochem. Pharmacol. 1996, 51, 1259–1267. [Google Scholar] [CrossRef]
- Martín, J.; Crespo, G.; González-Menéndez, V.; Pérez-Moreno, G.; Sánchez-Carrasco, P.; Pérez-Victoria, I.; Ruiz-Pérez, L.M.; González-Pacanowska, D.; Vicente, F.; Genilloud, O.; et al. MDN-0104, an Antiplasmodial Betaine Lipid from Heterospora chenopodii. J. Nat. Prod. 2014, 77, 2118–2123. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.; Chater, K.; Butter, M.; Hopwood, D.; Bittner, M.; Buttner, M. Practical Streptomyces Genetics: A Laboratory Manual; John Innes Centre Foundation: Norwich, UK, 2000. [Google Scholar]
- Quail, M.A.; Kozarewa, I.; Smith, F.; Scally, A.; Stephens, P.J.; Durbin, R.; Swerdlow, H.; Turner, D.J. A Large genome center’s improvement to the Illumina sequencing system. Nat. Methods 2008, 5, 1005–1010. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Vaser, R.; Šikić, M. Time- and memory-efficient genome assembly with raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, 112963. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36 (Suppl. 2), W5–W9. [Google Scholar] [CrossRef]
- Oh, S.; Kim, S.; Kong, S.; Yang, G.; Lee, N.; Han, D.; Goo, J.; Siqueira-Neto, J.L.; Freitas-Junior, L.H.; Song, R. Synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-a]benzimidazole derivatives against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 2014, 12, 395–403. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Moon, S.; Jang, J.; Yang, G.; Lee, C.; Moon, H.K.; Chatelain, E.; Genovesio, A.; Cechetto, J.; Freitas-Junior, L.H. An image-based high-content screening assay for compounds targeting intracellular Leishmania donovani amastigotes in human macrophages. PLoS Negl. Trop. Dis. 2012, 6, e1671. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Position | δC, Type | δH, Mult. (J in Hz) |
|---|---|---|---|
| Salicylate | 1 | 160.1, C | - |
| 2 | 117.3, CH | 6.99, d (8.3) | |
| 3 | 136.0, CH | 7.54, ddd (8.8, 7.4, 1.7) | |
| 4 | 119.3, CH | 6.95, ddd (8.8, 7.3, 0.8) | |
| 5 | 131.0, CH | 7.94, dd (7.9, 1.6) | |
| 6 | 112.7, C | - | |
| 7 | 167.8, C | - | |
| Ser | 8 | NH | 8.53, br s |
| 9 | 51.4, CH | 4.37, br s | |
| 10 | 63.2, CH2 | 4.65, m | |
| 4.62, m | |||
| 11 | 166.0, C | - | |
| Gly | 12 | NH | 8.89, t (5.6) |
| 13 | 42.2, CH2 | 3.83, dd (16.5, 5.9) | |
| 3.74, dd (16.5, 5.4) | |||
| 14 | 167.9, C | - | |
| β-Ala | 15 | NH | 8.02, t (5.4) |
| 16 | 34.7, CH2 | 3.25, m | |
| 17 | 31.9, CH2 | 2.53, m | |
| 18 | 170.9, C | - | |
| Nα-Methyl Nδ-OH Orn | 19 | N | - |
| 20 | 47.1, CH2 | 3.50, m | |
| 21 | 22.0, CH2 | 1.83, m | |
| 1.57, m | |||
| 22 | 24.7, CH2 | 1.66, m | |
| 1.58, m | |||
| 23 | 61.7, CH | 3.58 * | |
| 24 | 167.8, C | - | |
| Nδ-OH Orn | 25 | N | - |
| 26 | 52.2, CH | 4.04, q (5.3) | |
| 27 | 162.9, C | - | |
| 28 | N | - | |
| 29 | 51.2, CH2 | 3.52, m | |
| 3.44, m | |||
| 30 | 20.8, CH2 | 1.90, m | |
| 31 | 25.3, CH2 | 2.37, m | |
| 1.76, m | |||
| 32 | 33.0, CH3 | 2.56, m | |
| 33 | 80.2, C | - | |
| 34 | 23.3, CH3 | 1.50, br s | |
| 35 | 20.3, CH3 | 1.32, br s |
| 33-epi-Madurastatin D1 (3) | Madurastatin D1 (4) | ||||
|---|---|---|---|---|---|
| Residue | Position | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) |
| Salicylate | 2 | 116.3, CH | 7.00, d (8.4) | 116.3, CH | 7.00, d (8.3) |
| 3 | 133.8, CH | 7.47, ddd (8.4, 7.4, 1.3) | 133.9, CH | 7.47, ddd (8.3, 7.4, 1.3) | |
| 4 | 118.8, CH | 6.95, dd (8.1, 7.3) | 118.8, CH | 6.95, dd (8.2, 7.5) | |
| 5 | 127.8, CH | 7.64, dd (7.7, 1.3) | 127.8, CH | 7.64, dd (7.8, 1.3) | |
| Ser | 9 | 67.2, CH | 5.01, dd (10.5, 7.8) | 67.1, CH | 5.01, dd (10.3, 7.8) |
| 10 | 69.2, CH2 | 4.65, dd (10.5, 8.5) | 69.2, CH2 | 4.65, dd (10.3, 8.5) | |
| 4.52, dd (8.8, 7.6) | 4.52, dd (8.5, 7.7) | ||||
| Gly | 13 | 41.7, CH2 | 3.75, dd (16.5, 5.7) | 41.9, CH2 | 375, dd (16.5, 5.9) |
| 3.66, dd (16.5, 5.4) | 3.66, dd (16.5, 5.7) | ||||
| β-Ala | 16 | 34.4, CH2 | 3.26, m | 34.3, CH2 | 3.25, m |
| 17 | 31.7, CH2 | 2.52, m | 31.6, CH2 | 2.53, m | |
| Nα-Methyl Nδ-OH Orn | 20 | 46.9, CH2 | 3.46, m | 47.1, CH2 | 3.46, m |
| 21 | 21.7, CH2 | 1.68, m | 21.3, CH2 | 1.68, m | |
| 1.46, m | 1.44, m | ||||
| 22 | 24.1, CH2 | 1.50, m | 26.3, CH2 | 1.46, m | |
| 23 | 62.1, CH | 3.14, m | 64.6, CH | 2.88, m | |
| Nδ-OH Orn | 26 | 51.2, CH | 4.13, q (5.9) | 51.5, CH | 4.27, m |
| 29 | 50.9, CH2 | 3.52, m | 50.8 CH2 | 3.54, m | |
| 3.43, m | 3.44, m | ||||
| 30 | 20.6, CH2 | 1.91, m | 20.5, CH2 | 1.87, m | |
| 31 | 25.2, CH2 | 2.17, m | 25.9, CH2 | 1.98, m | |
| 1.77, m | 1.84, m | ||||
| 32 | 33.4, CH3 | 2.27, s | 37.6, CH3 | 2.29, s | |
| 33 | 73.5, CH | 4.42, m | 74.4, CH | 4.01, q (5.5) | |
| 34 | 15.1, CH3 | 1.12, d (5.7) | 19.4, CH3 | 1.21, d (5.4) | |
| Area Target Compound × 106 | |||||||
|---|---|---|---|---|---|---|---|
| Acetonitrile Extract | n-Butanol Extract | Ethyl Acetate Extract | Methanol Extract | Acetone Extract | |||
| Targeted Compounds | [M + H]+ | RT (min) | Av. ± SD | Av. ± SD | Av. ± SD | Av. ± SD | Av. ± SD |
| H1 | 610.283 ± 0.005 | 0.8 ± 0.5 | 0.3 ± 0.1 | 0.2 ± 0.0 | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.2 ± 0.0 |
| C1 (1) | 592.273 ± 0.005 | 1.7 ± 0.5 | 11.0 ± 1.5 | 8.6 ± 1.5 | 8.9 ± 0.6 | 9.6 ± 0.3 | 8.3 ± 0.7 |
| H2 (2) | 650.314 ± 0.005 | 1.9 ± 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 |
| 33-epi D1 (3) | 618.288 ± 0.005 | 2.3 ± 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.9 ± 0.3 |
| D1 (4) | 618.288 ± 0.005 | 2.6 ± 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 1.5 ± 0.2 |
| D2 (5) | 632.304 ± 0.005 | 2.9 ± 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.1 ± 0.3 |
| Madurastatins | ||||||
|---|---|---|---|---|---|---|
| C1 (1) | H2 (2) | 33-epi D1 (3) | D1 (4) | D2 (5) | ||
| T. cruzi | Inhibition norm. inf. ratio IC50 (µM) | >50 | >50 | >50 | >50 | >50 |
| Inhibition norm. para. num. IC50 (µM) | 30.76 | >50 | 46.08 | 27.23 | 8.93 | |
| Cell ratio (%) CC50 (µM) | >50 | >50 | >50 | 46.77 | 24.74 | |
| L. donovani | Inhibition norm. inf. ratio IC50 (µM) | >50 | >50 | >50 | >50 | >50 |
| Inhibition norm. para. num. IC50 (µM) | >50 | >50 | >50 | >50 | >50 | |
| Cell ratio (%) CC50 (µM) | >50 | >50 | >50 | >50 | >50 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Bonilla, M.; Sánchez-Hidalgo, M.; González, I.; Oves-Costales, D.; Martín, J.; Murillo-Alba, J.; Tormo, J.R.; Cho, A.; Byun, S.-Y.; No, J.-H.; et al. Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents? Int. J. Mol. Sci. 2024, 25, 301. https://doi.org/10.3390/ijms25010301
Pérez-Bonilla M, Sánchez-Hidalgo M, González I, Oves-Costales D, Martín J, Murillo-Alba J, Tormo JR, Cho A, Byun S-Y, No J-H, et al. Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents? International Journal of Molecular Sciences. 2024; 25(1):301. https://doi.org/10.3390/ijms25010301
Chicago/Turabian StylePérez-Bonilla, Mercedes, Marina Sánchez-Hidalgo, Ignacio González, Daniel Oves-Costales, Jesús Martín, José Murillo-Alba, José R. Tormo, Ahreum Cho, Soo-Young Byun, Joo-Hwan No, and et al. 2024. "Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents?" International Journal of Molecular Sciences 25, no. 1: 301. https://doi.org/10.3390/ijms25010301
APA StylePérez-Bonilla, M., Sánchez-Hidalgo, M., González, I., Oves-Costales, D., Martín, J., Murillo-Alba, J., Tormo, J. R., Cho, A., Byun, S.-Y., No, J.-H., Shum, D., Ioset, J.-R., Genilloud, O., & Reyes, F. (2024). Madurastatins with Imidazolidinone Rings: Natural Products or Side-Reaction Products from Extraction Solvents? International Journal of Molecular Sciences, 25(1), 301. https://doi.org/10.3390/ijms25010301