
The Role of TAM Receptors in Bone
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of TAM Receptors in Bone Remodeling
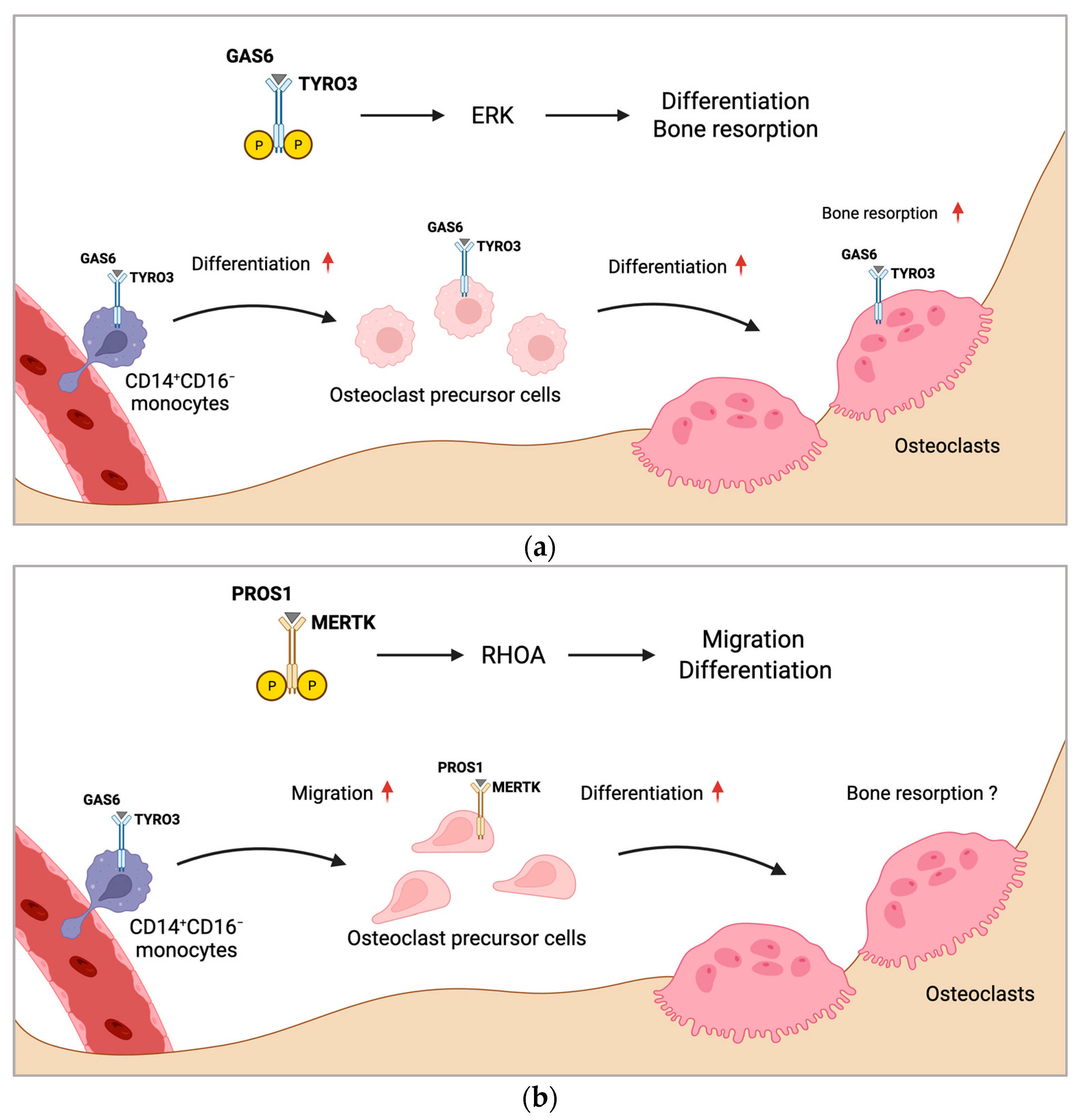
2.1. TAM Receptors in Osteoclasts
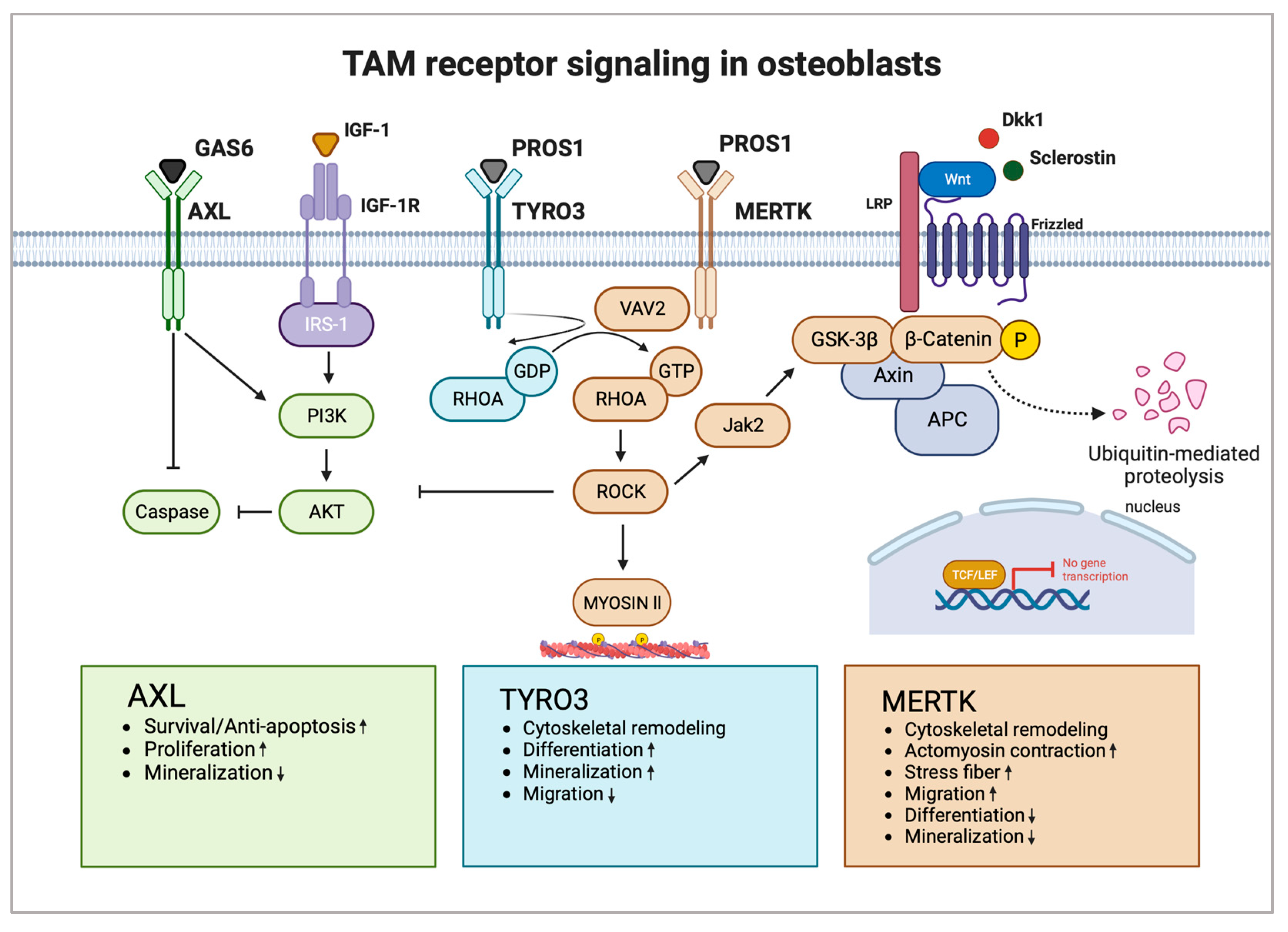
2.2. TAM Receptors in Osteoblasts
3. TAM Receptors in Bone Health and Disease
3.1. TAM Receptors in Postmenopausal Osteoporosis
3.2. TAM Receptors in Bone Healing
3.3. TAM Receptors in Inflammatory Bone Loss in Periodontal Disease
3.4. TAM Receptors in Rheumatoid Arthritis
4. TAM Receptors in Primary Malignant Bone Tumors and Cancer Bone Metastases
4.1. Osteosarcoma
4.2. Multiple Myeloma
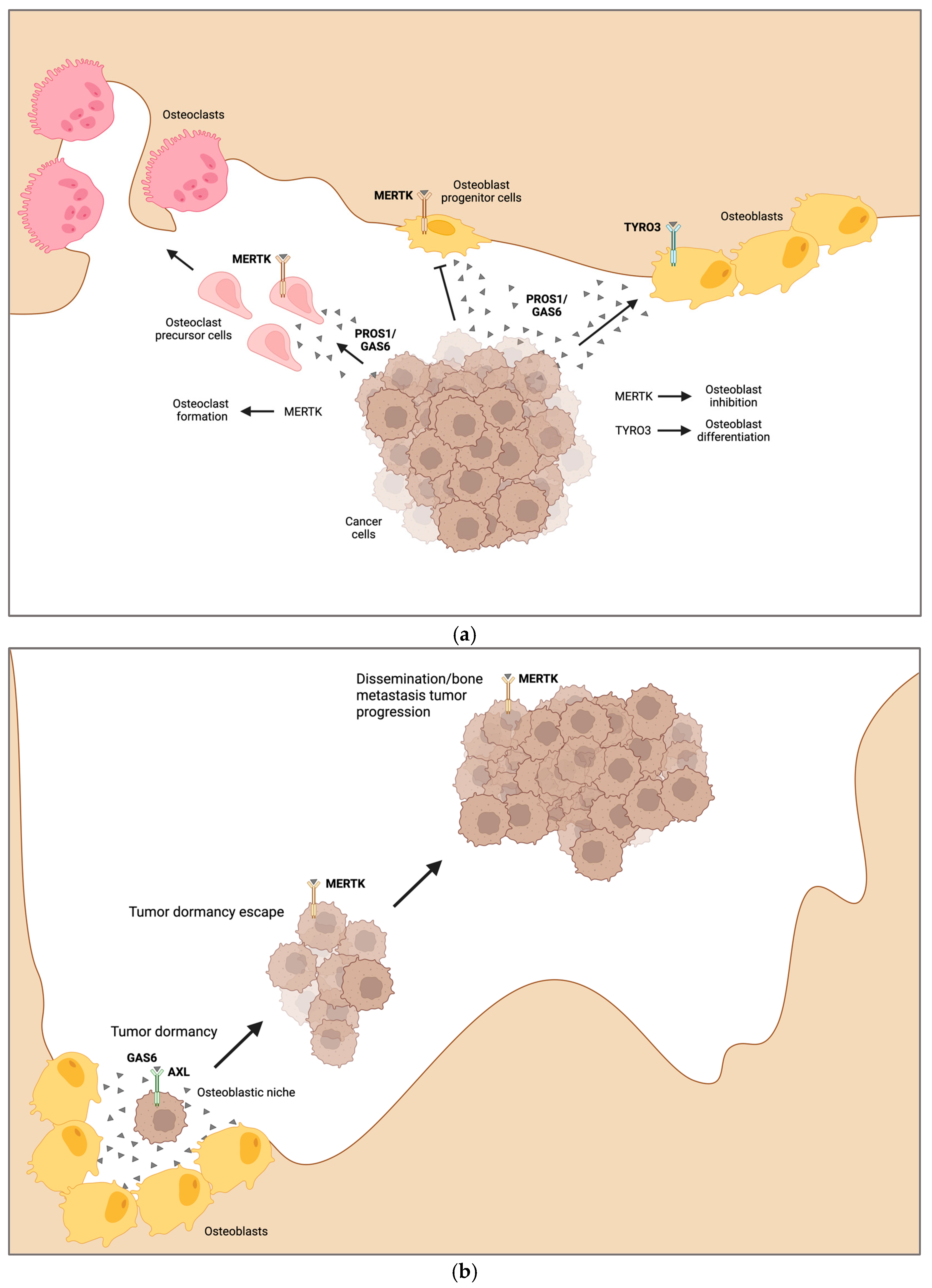
4.3. Bone Metastasis
4.4. Tumor Cell Dormancy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The Protein Encoded by a Growth Arrest-Specific Gene (Gas6) Is a New Member of the Vitamin-K-Dependent Proteins Related to Protein-S, a Negative Coregulator in the Blood-Coagulation Cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol.-Gastrointest. Liver 2016, 310, G899–G905. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Wium, M.; Paccez, J.D.; Zerbini, L.F. The Dual Role of TAM Receptors in Autoimmune Diseases and Cancer: An Overview. Cells 2018, 7, 166. [Google Scholar] [CrossRef]
- Bellan, M.; Pirisi, M.; Sainaghi, P.P. The Gas6/TAM System and Multiple Sclerosis. Int. J. Mol. Sci. 2016, 17, 1807. [Google Scholar] [CrossRef]
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002, 2, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Maimon, A. TAM receptors, Phosphatidylserine, inflammation, and Cancer. Cell Commun. Signal. 2019, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, C.E.J.; Beermann, S.; Broeren, M.G.A.; Bennink, M.B.; Koenders, M.I.; van Lent, P.L.E.M.; van den Berg, W.B.; van der Kraan, P.M.; van de Loo, F.A.J. Protective role of the Mer Tyrosine Kinase via Efferocytosis in Rheumatoid Arthritis Models. Front. Immunol. 2018, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ekman, C.; Jonsen, A.; Sturfelt, G.; Bengtsson, A.A.; Gottsater, A.; Lindblad, B.; Lindqvist, E.; Saxne, T.; Dahlback, B. Increased plasma levels of the soluble Mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res. Ther. 2011, 13, R62. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Guerrieri, J.; Dittman, L.M.; Merrill, J.T.; Cohen, P.L. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res. Ther. 2013, 15, R212. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Bosurgi, L.; Bernink, J.H.; Cuevas, V.D.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.A.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.G.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2012, 481, 190. [Google Scholar] [CrossRef]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, J.; Zarrer, J.; Gensch, V.; Riecken, K.; Berenbrok, N.; Luu, T.V.; Beitzen-Heineke, A.; Vargas-Delgado, M.E.; Pantel, K.; Bokemeyer, C.; et al. Regulation of bone homeostasis by MERTK and TYRO3. Nat. Commun. 2022, 13, 7689. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, J.; Zarrer, J.; Riecken, K.; Waizenegger, J.; Vargas-Delgado, M.E.; Meier, L.; Bokemeyer, C.; Pantel, K.; Alberto, E.; Ghosh, S.; et al. MERTK is a target in the microenviroenment of osteolytic bone metastasis. Am. Assoc. Cancer Res. Annu. Meet. 2023, 83, 1189. [Google Scholar]
- Decker, A.M.; Matsumoto, M.; Decker, J.T.; Roh, A.; Inohara, N.; Sugai, J.; Martin, K.; Taichman, R.; Kaigler, D.; Shea, L.D.; et al. Inhibition of Mertk Signaling Enhances Bone Healing after Tooth Extraction. J. Dent. Res. 2023, 102, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef]
- Taichman, R.S. Blood and bone: Two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood 2005, 105, 2631–2639. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.N.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part. B-Rev. 2017, 23, 268. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.D.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.T.; Lanyon, L.E. Osteoregulatory Nature of Mechanical Stimuli—Function as a Determinant for Adaptive Remodeling in Bone. J. Orthop. Res. 1987, 5, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Dykes, S.S.; Siemann, D.W. Inhibition of the Axl pathway impairs breast and prostate cancer metastasis to the bones and bone remodeling. Clin. Exp. Metastasis 2021, 38, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Nakamura, Y.S.; Hakeda, Y.; Takakura, N.; Kameda, T.; Hamaguchi, I.; Miyamoto, T.; Kakudo, S.; Nakano, T.; Kumegawa, M.; Suda, T. Tyro 3 receptor tyrosine kinase and its ligand, Gas6, stimulate the function of osteoclasts. Stem Cells 1998, 16, 229–238. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Katagiri, M.; Chikazu, D. Osteoclastic bone resorption through receptor tyrosine kinase and extracellular signal-regulated kinase signaling in mature osteoclasts. Mod. Rheumatol. 2004, 14, 1–5. [Google Scholar] [CrossRef]
- He, Y.; Staser, K.; Rhodes, S.D.; Liu, Y.; Wu, X.; Park, S.J.; Yuan, J.; Yang, X.; Li, X.; Jiang, L.; et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS ONE 2011, 6, e24780. [Google Scholar] [CrossRef]
- Choi, E.B.; Agidigbi, T.S.; Kang, I.S.; Kim, C. ERK Inhibition Increases RANKL-Induced Osteoclast Differentiation in RAW 264.7 Cells by Stimulating AMPK Activation and RANK Expression and Inhibiting Anti-Osteoclastogenic Factor Expression. Int. J. Mol. Sci. 2022, 23, 13512. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Heiland, G.; Zhao, Y.; Derer, A.; Braun, T.; Engelke, K.; Neumann, E.; Mueller-Ladner, U.; Liu, Y.; Zwerina, J.; Schett, G. Deletion of the receptor tyrosine kinase Tyro3 inhibits synovial hyperplasia and bone damage in arthritis. Ann. Rheum. Dis. 2014, 73, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, F.; Zhu, H.; Liu, X.; Shi, L.; Li, Y.; Zhong, H.; Su, Y. Soluble TAM receptor tyrosine kinases in rheumatoid arthritis: Correlation with disease activity and bone destruction. Clin. Exp. Immunol. 2018, 192, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Xu, L.; Zhu, H.; Bai, M.; Li, X.; Zhao, Z.; Zhong, H.; Cheng, G.; Li, X.; Hu, F.; et al. CD14(+)CD16(−) monocytes are the main precursors of osteoclasts in rheumatoid arthritis via expressing Tyro3TK. Arthritis Res. Ther. 2020, 22, 221. [Google Scholar] [CrossRef] [PubMed]
- Yahara, Y.; Barrientos, T.; Tang, Y.J.; Puviindran, V.; Nadesan, P.; Zhang, H.; Gibson, J.R.; Gregory, S.G.; Diao, Y.; Xiang, Y.; et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat. Cell Biol. 2020, 22, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Yahara, Y.; Nguyen, T.; Ishikawa, K.; Kamei, K.; Alman, B.A. The origins and roles of osteoclasts in bone development, homeostasis and repair. Development 2022, 149, dev199908. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, J.; Kawamura, S.; Okiji, F.; Shirazaki, M.; Sakai, S.; Saito, H.; Ishii, M. Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc. Natl. Acad. Sci. USA 2013, 110, 7009–7013. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Saitoh, Y.; Minami, T.; Takeno, N.; Tsuneyama, K.; Miyahara, T.; Nakayama, T.; Sakurai, H.; Takano, Y.; Nishimura, M.; et al. Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J. Immunol. 2009, 183, 7825–7831. [Google Scholar] [CrossRef]
- Lee, J.; Park, C.; Kim, H.J.; Lee, Y.D.; Lee, Z.H.; Song, Y.W.; Kim, H.H. Stimulation of osteoclast migration and bone resorption by C-C chemokine ligands 19 and 21. Exp. Mol. Med. 2017, 49, e358. [Google Scholar] [CrossRef]
- Yu, X.; Huang, Y.; Collin-Osdoby, P.; Osdoby, P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J. Bone Miner. Res. 2003, 18, 1404–1418. [Google Scholar] [CrossRef]
- Ishii, M.; Kikuta, J.; Shimazu, Y.; Meier-Schellersheim, M.; Germain, R.N. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J. Exp. Med. 2010, 207, 2793–2798. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Egen, J.G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R.L.; Germain, R.N. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, C.; Zhang, J.; Bao, Y.; Tang, Y.; Lv, X.; Ma, B.; Wu, X.; Mao, G. RhoA promotes osteoclastogenesis and regulates bone remodeling through mTOR-NFATc1 signaling. Mol. Med. 2023, 29, 49. [Google Scholar] [CrossRef] [PubMed]
- Destaing, O.; Saltel, F.; Gilquin, B.; Chabadel, A.; Khochbin, S.; Ory, S.; Jurdic, P. A novel Rho-mDia2-HDAC6 pathway controls podosome patterning through microtubule acetylation in osteoclasts. J. Cell Sci. 2005, 118 Pt 13, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Chellaiah, M.A.; Soga, N.; Swanson, S.; McAllister, S.; Alvarez, U.; Wang, D.; Dowdy, S.F.; Hruska, K.A. Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J. Biol. Chem. 2000, 275, 11993–12002. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Macrophage-Osteoclast Associations: Origin, Polarization, and Subgroups. Front. Immunol. 2021, 12, 778078. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Huang, R.; Wang, X.; Zhou, Y.; Xiao, Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Res. 2017, 5, 17019. [Google Scholar] [CrossRef]
- Dou, C.; Ding, N.; Zhao, C.; Hou, T.; Kang, F.; Cao, Z.; Liu, C.; Bai, Y.; Dai, Q.; Ma, Q.; et al. Estrogen Deficiency-Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J. Bone Miner. Res. 2018, 33, 899–908. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Movila, A.; Kataoka, S.; Wisitrasameewong, W.; Ruiz Torruella, M.; Murakoshi, M.; Murakami, S.; Kawai, T. Proinflammatory M1 Macrophages Inhibit RANKL-Induced Osteoclastogenesis. Infect. Immun. 2016, 84, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Caskey, L.; Holtzhausen, A.; Hunter, D.; Story, C.; Earp, H.S. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J. Clin. Investig. 2018, 128, 2356–2369. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Glinton, K.; Subramanian, M.; Wilsbacher, L.D.; Rothlin, C.V.; Tabas, I.; Thorp, E.B. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Investig. 2021, 131, e139576. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; MacDonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, T.S.; Vellozo, N.S.; Guimaraes-Pinto, K.; Cabral-Piccin, M.; Fabiano-Coelho, L.; Matos-Silva, T.C.; Filardy, A.A.; Takiya, C.M.; Lopes, M.F. Axl receptor induces efferocytosis, dampens M1 macrophage responses and promotes heart pathology in Trypanosoma cruzi infection. Commun. Biol. 2022, 5, 1421. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of tumor-associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral. Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef]
- Lin, J.; Xu, A.; Jin, J.; Zhang, M.; Lou, J.; Qian, C.; Zhu, J.; Wang, Y.; Yang, Z.; Li, X.; et al. MerTK-mediated efferocytosis promotes immune tolerance and tumor progression in osteosarcoma through enhancing M2 polarization and PD-L1 expression. Oncoimmunology 2022, 11, 2024941. [Google Scholar] [CrossRef]
- Li, Z.H.; Chen, J.F.; Zhang, J.; Lei, Z.Y.; Wu, L.L.; Meng, S.B.; Wang, J.L.; Xiong, J.; Lin, D.N.; Wang, J.Y.; et al. Mesenchymal stem cells promote polarization of M2 macrophages in mice with acute-on-chronic liver failure via Mertk/JAK1/STAT6 signaling. Stem Cells 2023, 41, 1171–1184. [Google Scholar] [CrossRef]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Zizzo, G.; Cohen, P.L. Antibody Cross-Linking of CD14 Activates MerTK and Promotes Human Macrophage Clearance of Apoptotic Neutrophils: The Dual Role of CD14 at the Crossroads Between M1 and M2c Polarization. Inflammation 2018, 41, 2206–2221. [Google Scholar] [CrossRef]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [PubMed]
- Collett, G.; Wood, A.; Alexander, M.Y.; Varnum, B.C.; Boot-Handford, R.P.; Ohanian, V.; Ohanian, J.; Fridell, Y.W.; Canfield, A.E. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ. Res. 2003, 92, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Collett, G.D.; Sage, A.P.; Kirton, J.P.; Alexander, M.Y.; Gilmore, A.P.; Canfield, A.E. Axl/phosphatidylinositol 3-kinase signaling inhibits mineral deposition by vascular smooth muscle cells. Circ. Res. 2007, 100, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Kugimiya, F.; Oshima, Y.; Ohba, S.; Ikeda, T.; Saito, T.; Shinoda, Y.; Kawasaki, Y.; Ogata, N.; Hoshi, K.; et al. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2007, 2, e1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bruxvoort, K.J.; Zylstra, C.R.; Liu, J.; Cichowski, R.; Faugere, M.C.; Bouxsein, M.L.; Wan, C.; Williams, B.O.; Clemens, T.L. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc. Natl. Acad. Sci. USA 2007, 104, 2259–2264. [Google Scholar] [CrossRef]
- Karner, C.M.; Long, F. Wnt signaling and cellular metabolism in osteoblasts. Cell Mol. Life Sci. 2017, 74, 1649–1657. [Google Scholar] [CrossRef]
- Shi, W.; Xu, C.; Gong, Y.; Wang, J.; Ren, Q.; Yan, Z.; Mei, L.; Tang, C.; Ji, X.; Hu, X.; et al. RhoA/Rock activation represents a new mechanism for inactivating Wnt/beta-catenin signaling in the aging-associated bone loss. Cell Regen. 2021, 10, 8. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Russow, G.; Jahn, D.; Appelt, J.; Mardian, S.; Tsitsilonis, S.; Keller, J. Anabolic Therapies in Osteoporosis and Bone Regeneration. Int. J. Mol. Sci. 2018, 20, 83. [Google Scholar] [CrossRef]
- Eastell, R.; O’Neill, T.W.; Hofbauer, L.C.; Langdahl, B.; Reid, I.R.; Gold, D.T.; Cummings, S.R. Postmenopausal osteoporosis. Nat. Rev. Dis. Primers 2016, 2, 16069. [Google Scholar] [CrossRef] [PubMed]
- Foger-Samwald, U.; Dovjak, P.; Azizi-Semrad, U.; Kerschan-Schindl, K.; Pietschmann, P. Osteoporosis: Pathophysiology and therapeutic options. EXCLI J. 2020, 19, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Sanda, N.; Miyawaki, Y.; Fujimori, Y.; Yamada, T.; Takagi, A.; Murate, T.; Saito, H.; Kojima, T. Down-regulation of PROS1 gene expression by 17beta-estradiol via estrogen receptor alpha (ERalpha)-Sp1 interaction recruiting receptor-interacting protein 140 and the corepressor-HDAC3 complex. J. Biol. Chem. 2010, 285, 13444–13453. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.W.; Romeo, G.; Hughes, Q.W.; Baker, R.I. Micro-ribonucleic Acid 494 regulation of protein S expression. J. Thromb. Haemost. 2013, 11, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Comp, P.C.; Thurnau, G.R.; Welsh, J.; Esmon, C.T. Functional and immunologic protein S levels are decreased during pregnancy. Blood 1986, 68, 881–885. [Google Scholar] [CrossRef]
- Malm, J.; Laurell, M.; Dahlback, B. Changes in the plasma levels of vitamin K-dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br. J. Haematol. 1988, 68, 437–443. [Google Scholar] [CrossRef]
- Tans, G.; Curvers, J.; Middeldorp, S.; Thomassen, M.C.; Meijers, J.C.; Prins, M.H.; Bouma, B.N.; Buller, H.R.; Rosing, J. A randomized cross-over study on the effects of levonorgestrel- and desogestrel-containing oral contraceptives on the anticoagulant pathways. Thromb. Haemost. 2000, 84, 15–21. [Google Scholar] [CrossRef]
- Mo, R.; Tony Zhu, Y.; Zhang, Z.; Rao, S.M.; Zhu, Y.J. GAS6 is an estrogen-inducible gene in mammary epithelial cells. Biochem. Biophys. Res. Commun. 2007, 353, 189–194. [Google Scholar] [CrossRef]
- Son, B.K.; Akishita, M.; Iijima, K.; Ogawa, S.; Maemura, K.; Yu, J.; Takeyama, K.; Kato, S.; Eto, M.; Ouchi, Y. Androgen receptor-dependent transactivation of growth arrest-specific gene 6 mediates inhibitory effects of testosterone on vascular calcification. J. Biol. Chem. 2010, 285, 7537–7544. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Li, Y.; Dai, W. Serum sex hormone and growth arrest-specific protein 6 levels in male patients with coronary heart disease. Asian J. Androl. 2016, 18, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.J.; Lee, C.H.; Shieh, Y.S.; Hsiao, F.C.; Lin, F.H.; Hsieh, C.H. Gender differences in plasma growth arrest-specific protein 6 levels in adult subjects. Clin. Chim. Acta 2015, 441, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Xiao, C.; Wu, X.; Zhu, J.; Qin, C.; He, L.; Cui, H.; Zhang, L.; Zhang, W.; Yang, C.; et al. Genetic Correlation, Shared Loci, and Causal Association Between Sex Hormone-Binding Globulin and Bone Mineral Density: Insights From a Large-Scale Genomewide Cross-Trait Analysis. J. Bone Miner. Res. 2023, 38, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, T.A.; Gerstenfeld, L.C. Fracture healing: Mechanisms and interventions. Nat. Rev. Rheumatol. 2015, 11, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wendler, S.; Schlundt, C.; Bucher, C.H.; Birkigt, J.; Schipp, C.J.; Volk, H.D.; Duda, G.N.; Schmidt-Bleek, K. Immune Modulation to Enhance Bone Healing -A New Concept to Induce Bone Using Prostacyclin to Locally Modulate Immunity. Front. Immunol. 2019, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Cai, B.S.; Kasikara, C. TAM receptors and their ligand-mediated activation: Role in atherosclerosis. Int. Rev. Cel. Mol. Bio 2020, 357, 21–33. [Google Scholar] [CrossRef]
- Vago, J.P.; Amaral, F.A.; van de Loo, F.A.J. Resolving inflammation by TAM receptor activation. Pharmacol. Ther. 2021, 227, 107893. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, X.Q.; Gao, M.J.; Lee, W.; Zhou, J.; Zhao, Y.F.; Wang, G.D. The Pros1/Tyro3 axis protects against periodontitis by modulating STAT/SOCS signalling. J. Cell. Mol. Med. 2019, 23, 2769–2781. [Google Scholar] [CrossRef]
- Nassar, M.; Tabib, Y.; Capucha, T.; Mizraji, G.; Nir, T.; Pevsner-Fischer, M.; Zilberman-Schapira, G.; Heyman, O.; Nussbaum, G.; Bercovier, H.; et al. GAS6 is a key homeostatic immunological regulator of host-commensal interactions in the oral mucosa. Proc. Natl. Acad. Sci. USA 2017, 114, E337–E346. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Y.X.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J.K. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. Ther. 2002, 4, S265–S272. [Google Scholar] [CrossRef] [PubMed]
- Matteson, E.L. Current treatment strategies for rheumatoid arthritis. Mayo Clin. Proc. 2000, 75, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Qindeel, M.; Ullah, M.H.; Fakhar-ud-Din; Ahmed, N.; Rehman, A.U. Recent trends, challenges and future outlook of transdermal drug delivery systems for rheumatoid arthritis therapy. J. Control. Release 2020, 327, 595–615. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Cells of the synovium in rheumatoid arthritis—Osteoclasts. Arthritis Res. Ther. 2007, 9, 203. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am. J. Pathol. 1998, 152, 943–951. [Google Scholar]
- Gravallese, E.M.; Manning, C.; Tsay, A.; Naito, A.; Pan, C.; Amento, E.; Goldring, S.R. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000, 43, 250–258. [Google Scholar] [CrossRef]
- Yokota, K. Osteoclast differentiation in rheumatoid arthritis. Immunol. Med. 2023, 1–6. [Google Scholar] [CrossRef]
- Brandstrom, H.; Bjorkman, T.; Ljunggren, O. Regulation of osteoprotegerin secretion from primary cultures of human bone marrow stromal cells. Biochem. Biophys. Res. Commun. 2001, 280, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappa B ligand: Modulation of the expression by osteotropic factors and cytokines. Biochem. Biophys. Res. Commun. 2000, 275, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Danks, L.; Sabokbar, A.; Gundle, R.; Athanasou, N.A. Synovial macrophage-osteoclast differentiation in inflammatory arthritis. Ann. Rheum. Dis. 2002, 61, 916–921. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Harkes, I.C.; Dougherty, L.; Wicks, I.P. Expression of receptor tyrosine kinase Axl and its ligand Gas6 in rheumatoid arthritis—Evidence for a novel endothelial cell survival pathway. Am. J. Pathol. 1999, 154, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Vago, J.P.; Valdrighi, N.; Blaney-Davidson, E.N.; Hornikx, D.; Neefjes, M.; Barba-Sarasua, M.E.; Thielen, N.G.M.; van den Bosch, M.H.J.; van der Kraan, P.M.; Koenders, M.I.; et al. Gas6/Axl Axis Activation Dampens the Inflammatory Response in Osteoarthritic Fibroblast-like Synoviocytes and Synovial Explants. Pharmaceuticals 2023, 16, 703. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; He, C.; Yang, A.; Zhou, H.; Lu, Q.; Birge, R.B.; Wu, Y. Receptor tyrosine kinases Tyro3, Axl, and Mertk differentially contribute to antibody-induced arthritis. Cell Commun. Signal. 2023, 21, 195. [Google Scholar] [CrossRef]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Y.J.; Man, Y.; Pan, F.; Li, Z.H.; Jia, L.S. Knockdown of AXL receptor tyrosine kinase in osteosarcoma cells leads to decreased proliferation and increased apoptosis. Int. J. Immunopathol. Pharmacol. 2013, 26, 179–188. [Google Scholar] [CrossRef]
- Han, J.; Tian, R.; Yong, B.; Luo, C.; Tan, P.; Shen, J.; Peng, T. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem. Biophys. Res. Commun. 2013, 435, 493–500. [Google Scholar] [CrossRef]
- Truong, D.D.; Lamhamedi-Cherradi, S.E.; Ludwig, J.A. Targeting the IGF/PI3K/mTOR pathway and AXL/YAP1/TAZ pathways in primary bone cancer. J. Bone Oncol. 2022, 33, 100419. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.E.; Mohiuddin, S.; Mishra, D.K.; Krishnan, S.; Velasco, A.R.; Vetter, A.M.; Pence, K.; McCall, D.; Truong, D.D.; Cuglievan, B.; et al. Transcriptional activators YAP/TAZ and AXL orchestrate dedifferentiation, cell fate, and metastasis in human osteosarcoma. Cancer Gene Ther. 2021, 28, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, J.S.; Ben-Batalla, I.; Weinhold, N.; Meissner, T.; Wroblewski, M.; Janning, M.; Riecken, K.; Binder, M.; Atanackovic, D.; Taipaleenmaeki, H.; et al. Role of Growth arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia 2015, 29, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Ohkawara, H.; Ogawa, K.; Ikeda, K.; Ueda, K.; Shichishima-Nakamura, A.; Ito, E.; Imai, J.I.; Yanagisawa, Y.; Honma, R.; et al. Autocrine and Paracrine Interactions between Multiple Myeloma Cells and Bone Marrow Stromal Cells by Growth Arrest-specific Gene 6 Cross-talk with Interleukin-6. J. Biol. Chem. 2017, 292, 4280–4292. [Google Scholar] [CrossRef] [PubMed]
- Kosta, A.; Mekhloufi, A.; Lucantonio, L.; Zingoni, A.; Soriani, A.; Cippitelli, M.; Gismondi, A.; Fazio, F.; Petrucci, M.T.; Santoni, A.; et al. GAS6/TAM signaling pathway controls MICA expression in multiple myeloma cells. Front. Immunol. 2022, 13, 942640. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Voevoda-Dimenshtein, V.; Zoabi, A.; Rosenberg, E.; Magen, H.; Ostrovsky, O.; Shimoni, A.; Weiss, L.; Abraham, M.; Peled, A.; et al. CXCL13 chemokine is a novel player in multiple myeloma osteolytic microenvironment, M2 macrophage polarization, and tumor progression. J. Hematol. Oncol. 2022, 15, 144. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clezardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83. [Google Scholar] [CrossRef]
- Yin, J.J.; Pollock, C.B.; Kelly, K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005, 15, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E172–E181. [Google Scholar] [CrossRef] [PubMed]
- Granata, V.; Crisafulli, L.; Nastasi, C.; Ficara, F.; Sobacchi, C. Bone Marrow Niches and Tumour Cells: Lights and Shadows of a Mutual Relationship. Front. Immunol. 2022, 13, 884024. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Han, Y.; Kang, Y. Bone marrow niches in the regulation of bone metastasis. Br. J. Cancer 2021, 124, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xia, F.; Wei, Y.; Wei, X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Wikman, H.; Vessella, R.; Pantel, K. Cancer micrometastasis and tumour dormancy. APMIS 2008, 116, 754–770. [Google Scholar] [CrossRef]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Widner, D.B.; Park, S.H.; Eber, M.R.; Shiozawa, Y. Interactions Between Disseminated Tumor Cells and Bone Marrow Stromal Cells Regulate Tumor Dormancy. Curr. Osteoporos. Rep. 2018, 16, 596–602. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Jung, Y.; Shiozawa, Y.; Wang, J.; McGregor, N.; Dai, J.; Park, S.I.; Berry, J.E.; Havens, A.M.; Joseph, J.; Kim, J.K.; et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia 2012, 14, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef] [PubMed]
- Dormady, S.P.; Zhang, X.M.; Basch, R.S. Hematopoietic progenitor cells grow on 3T3 fibroblast monolayers that overexpress growth arrest-specific gene-6 (GAS6). Proc. Natl. Acad. Sci. USA 2000, 97, 12260–12265. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engelmann, J.; Ragipoglu, D.; Ben-Batalla, I.; Loges, S. The Role of TAM Receptors in Bone. Int. J. Mol. Sci. 2024, 25, 233. https://doi.org/10.3390/ijms25010233
Engelmann J, Ragipoglu D, Ben-Batalla I, Loges S. The Role of TAM Receptors in Bone. International Journal of Molecular Sciences. 2024; 25(1):233. https://doi.org/10.3390/ijms25010233
Chicago/Turabian StyleEngelmann, Janik, Deniz Ragipoglu, Isabel Ben-Batalla, and Sonja Loges. 2024. "The Role of TAM Receptors in Bone" International Journal of Molecular Sciences 25, no. 1: 233. https://doi.org/10.3390/ijms25010233
APA StyleEngelmann, J., Ragipoglu, D., Ben-Batalla, I., & Loges, S. (2024). The Role of TAM Receptors in Bone. International Journal of Molecular Sciences, 25(1), 233. https://doi.org/10.3390/ijms25010233