
RNA-Seq Analysis of Trans-Differentiated ARPE-19 Cells Transduced by AAV9-AIPL1 Vectors

 , , and
, , and
Abstract
:1. Introduction
2. Results
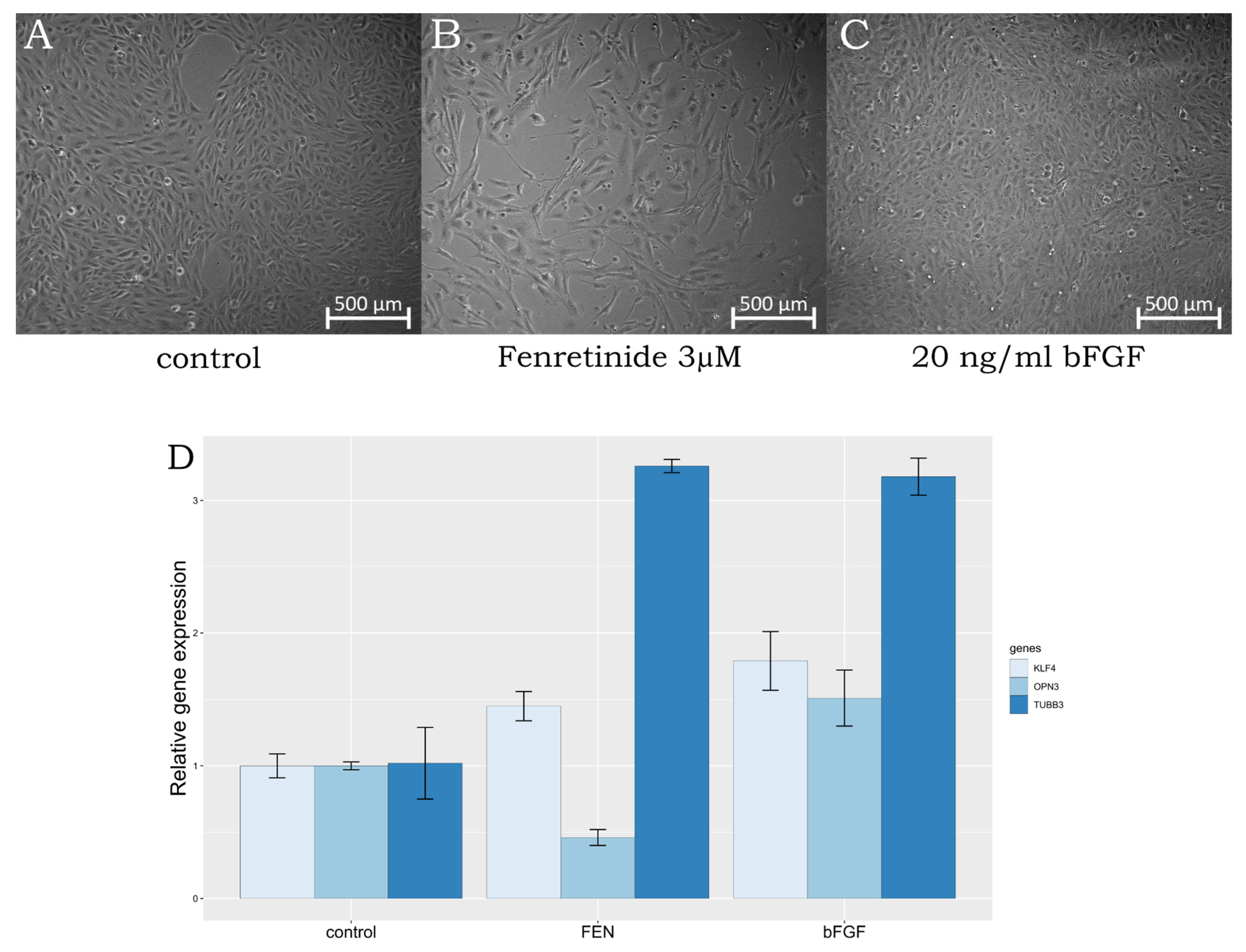
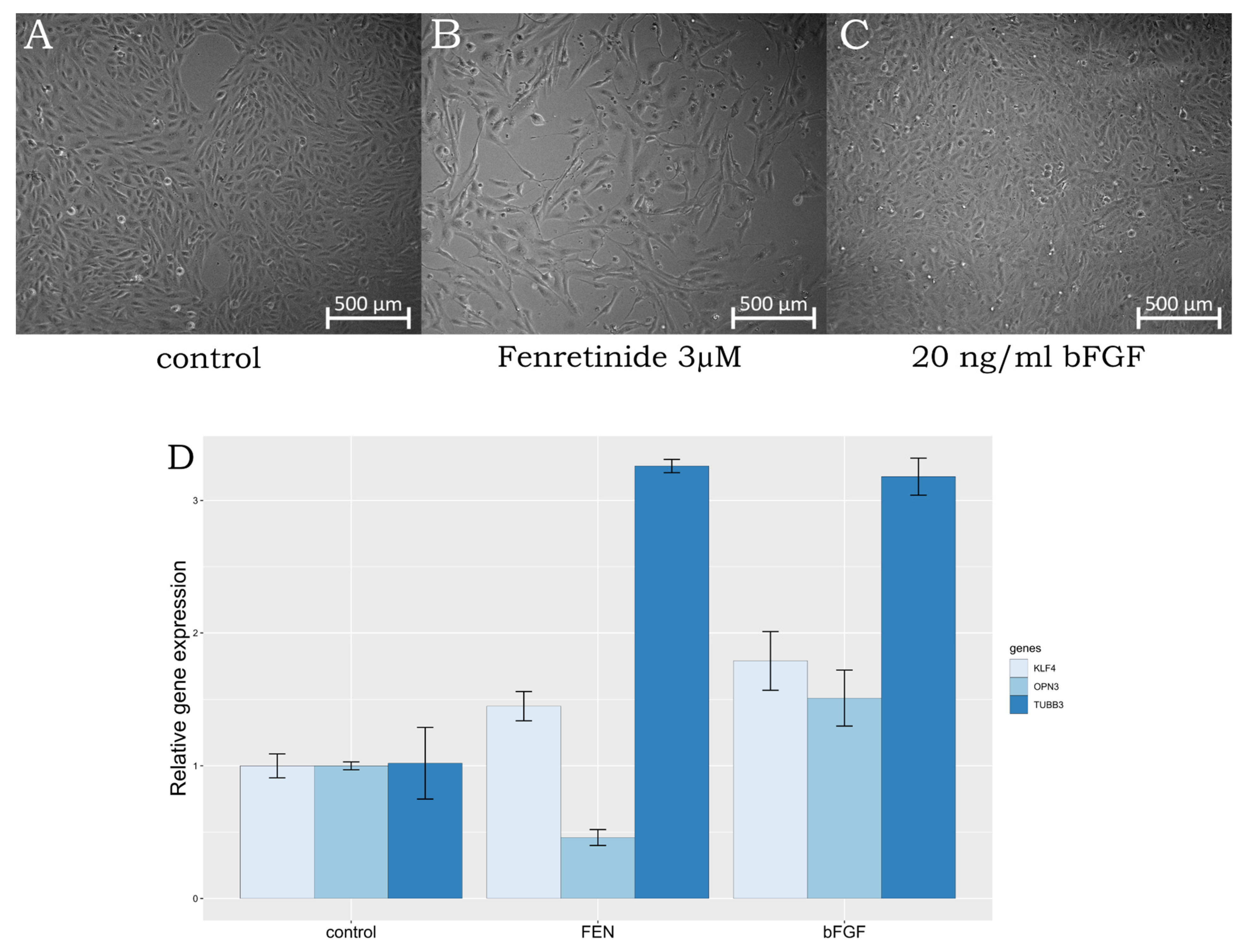
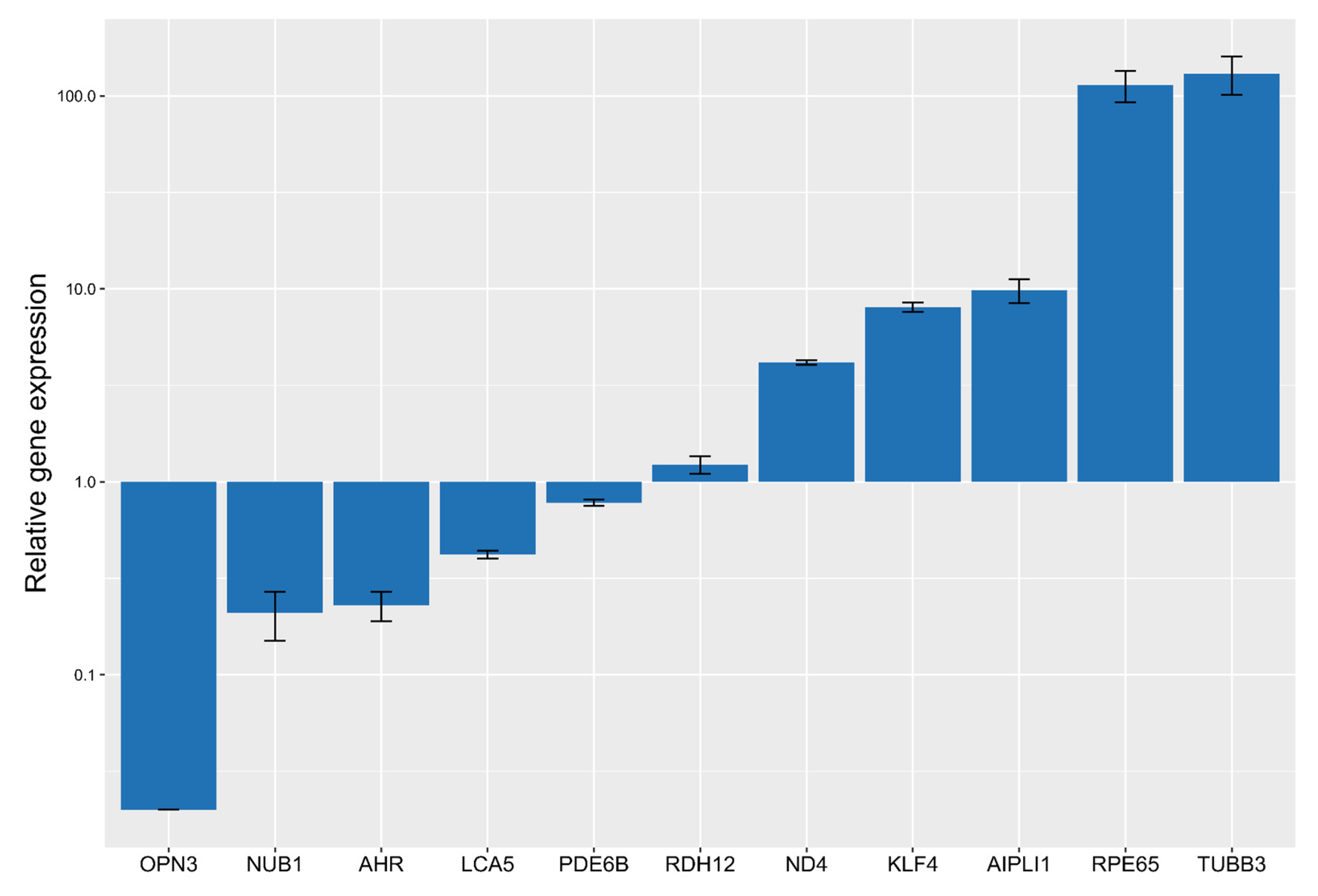
2.1. ARPE-19 Cells Trans-Differentiation
2.2. AIPL1co and AIPLwt Expressed in HEK-293 Cells
2.3. qPCR Screening
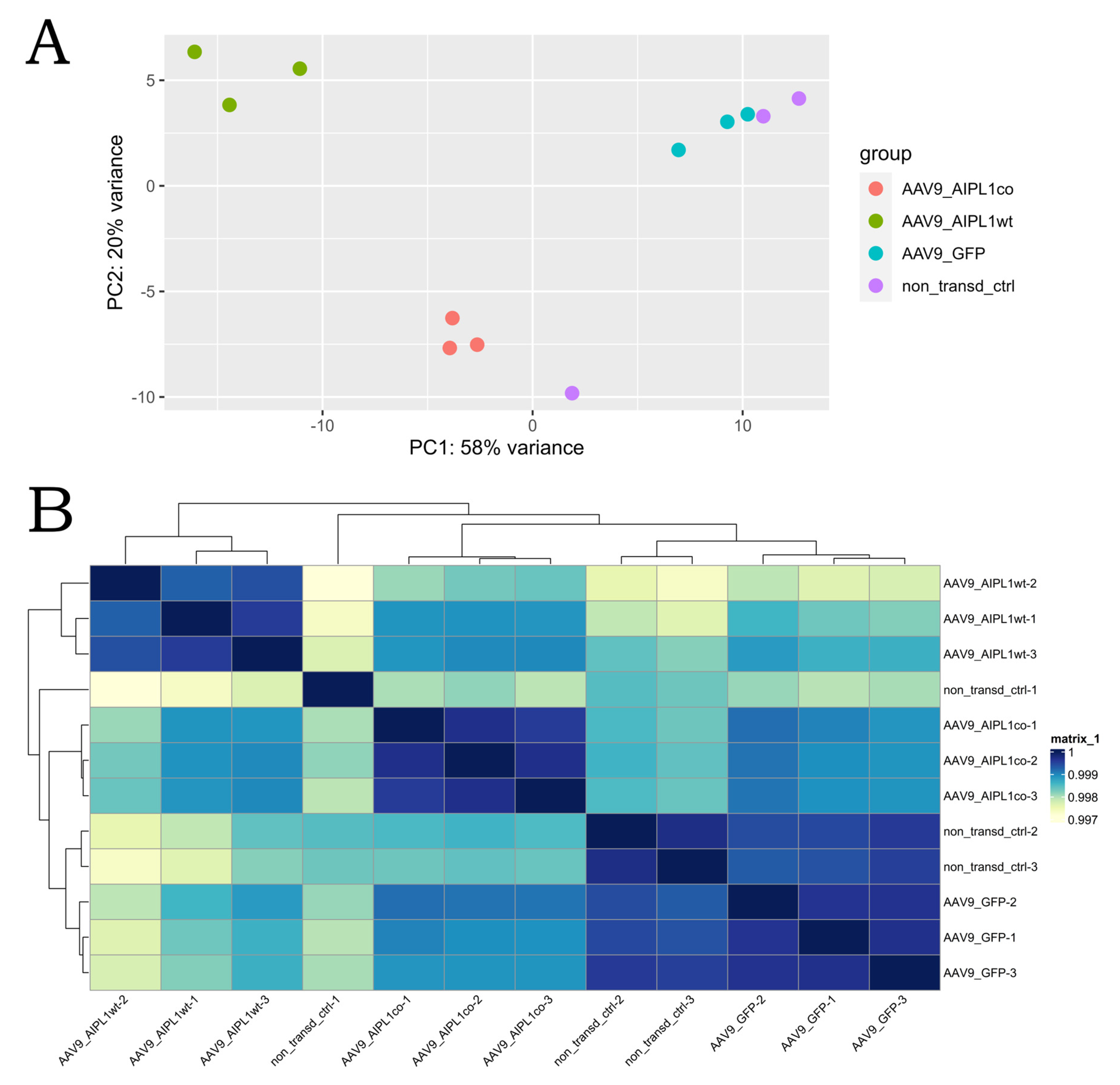
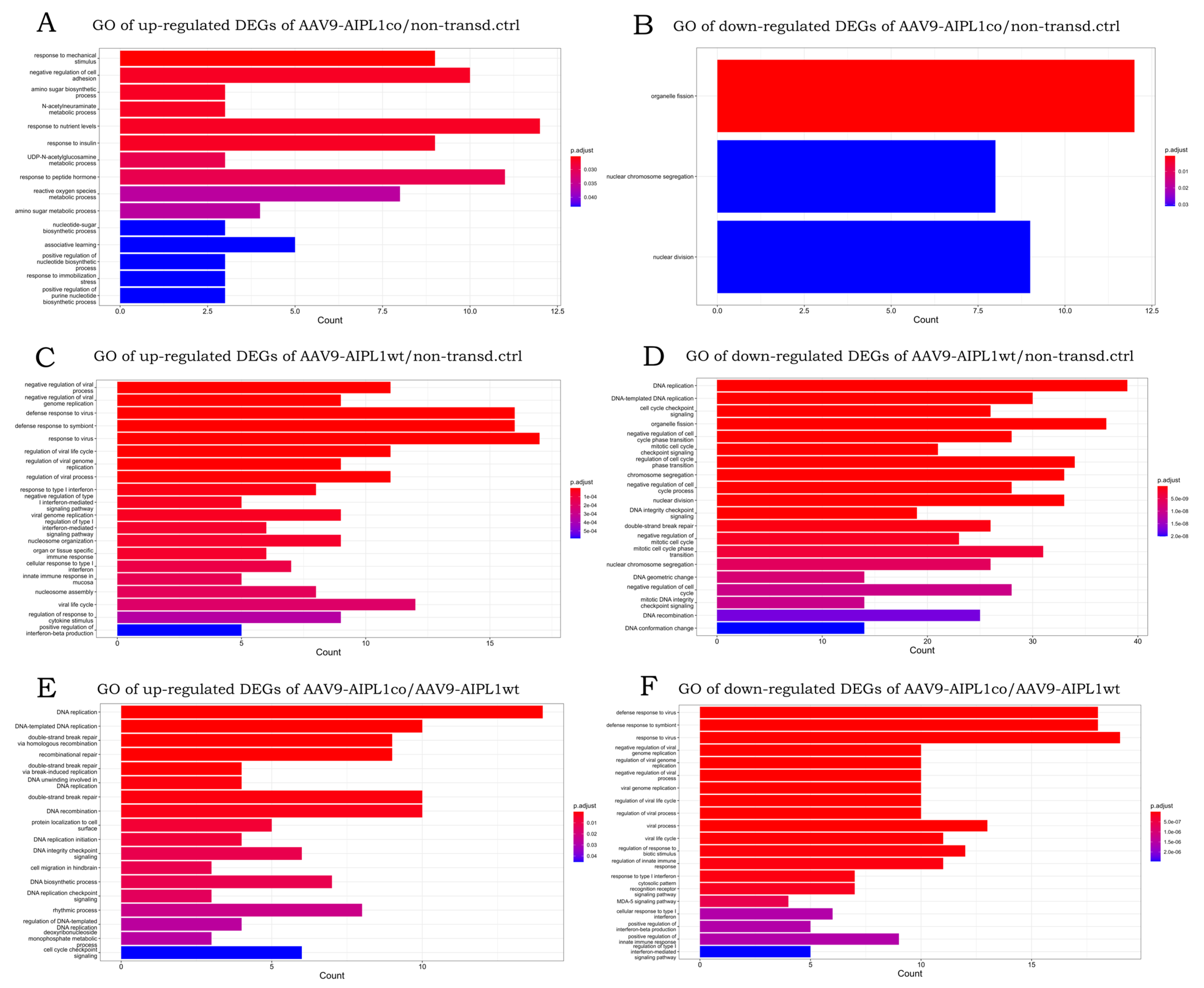
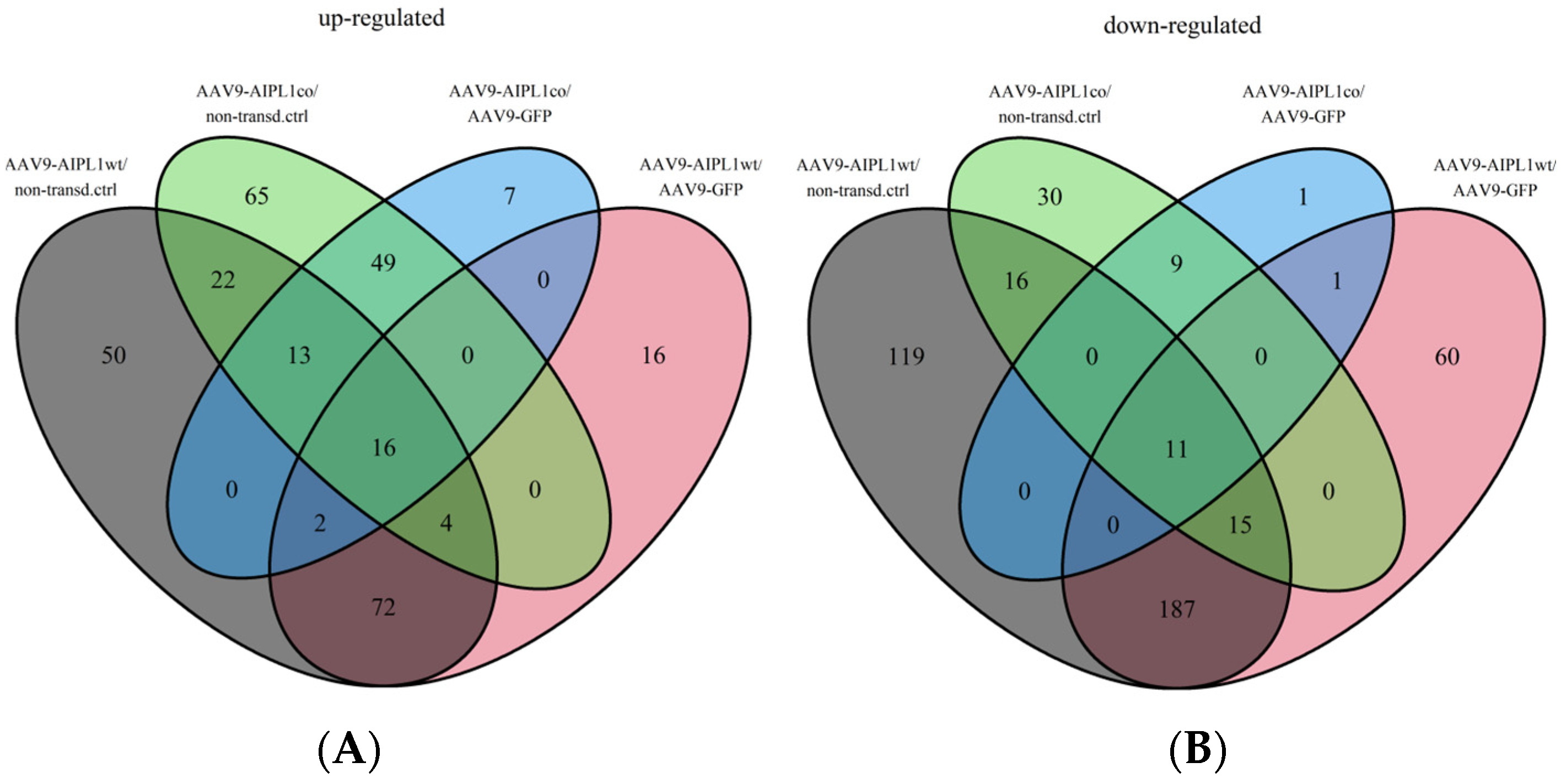
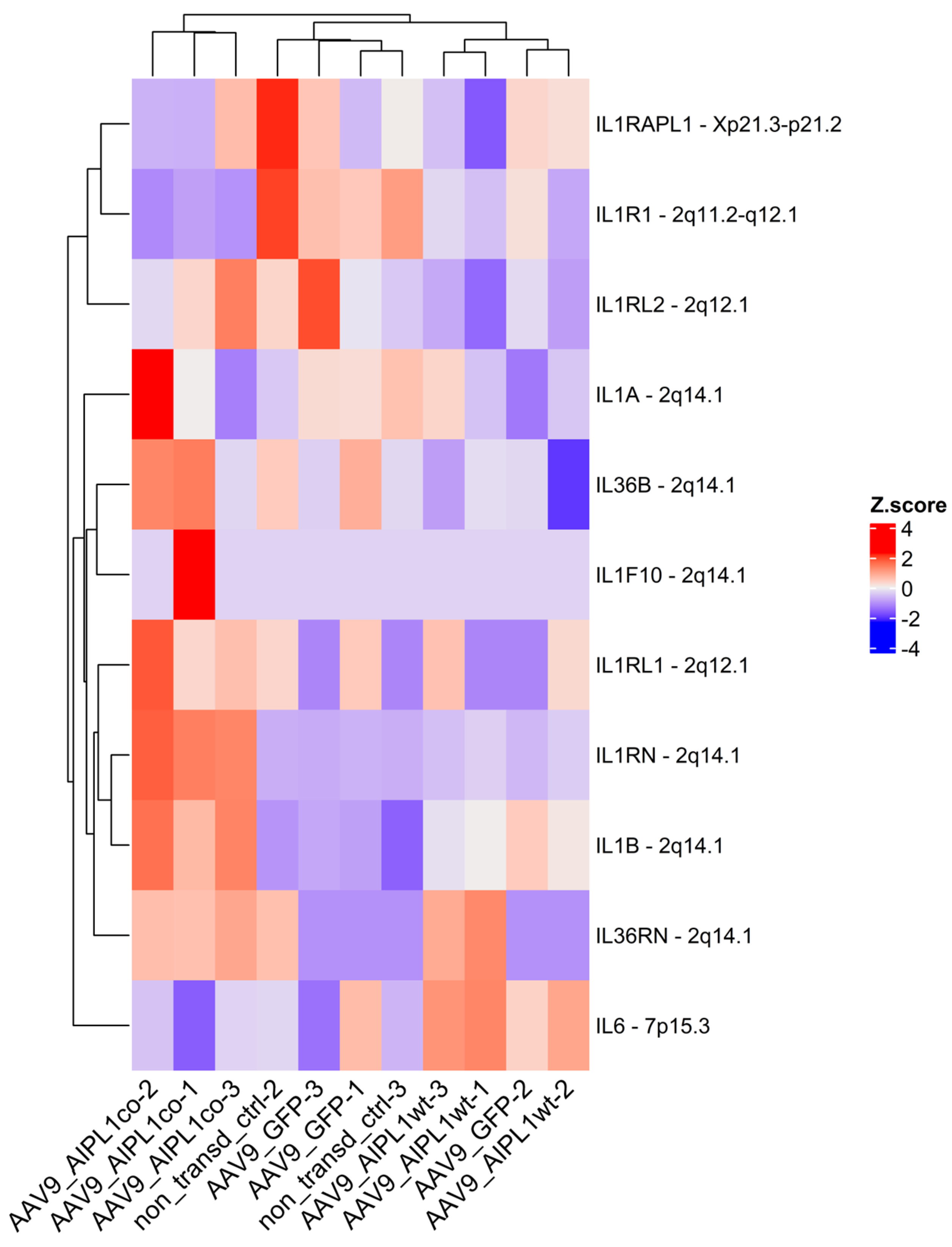
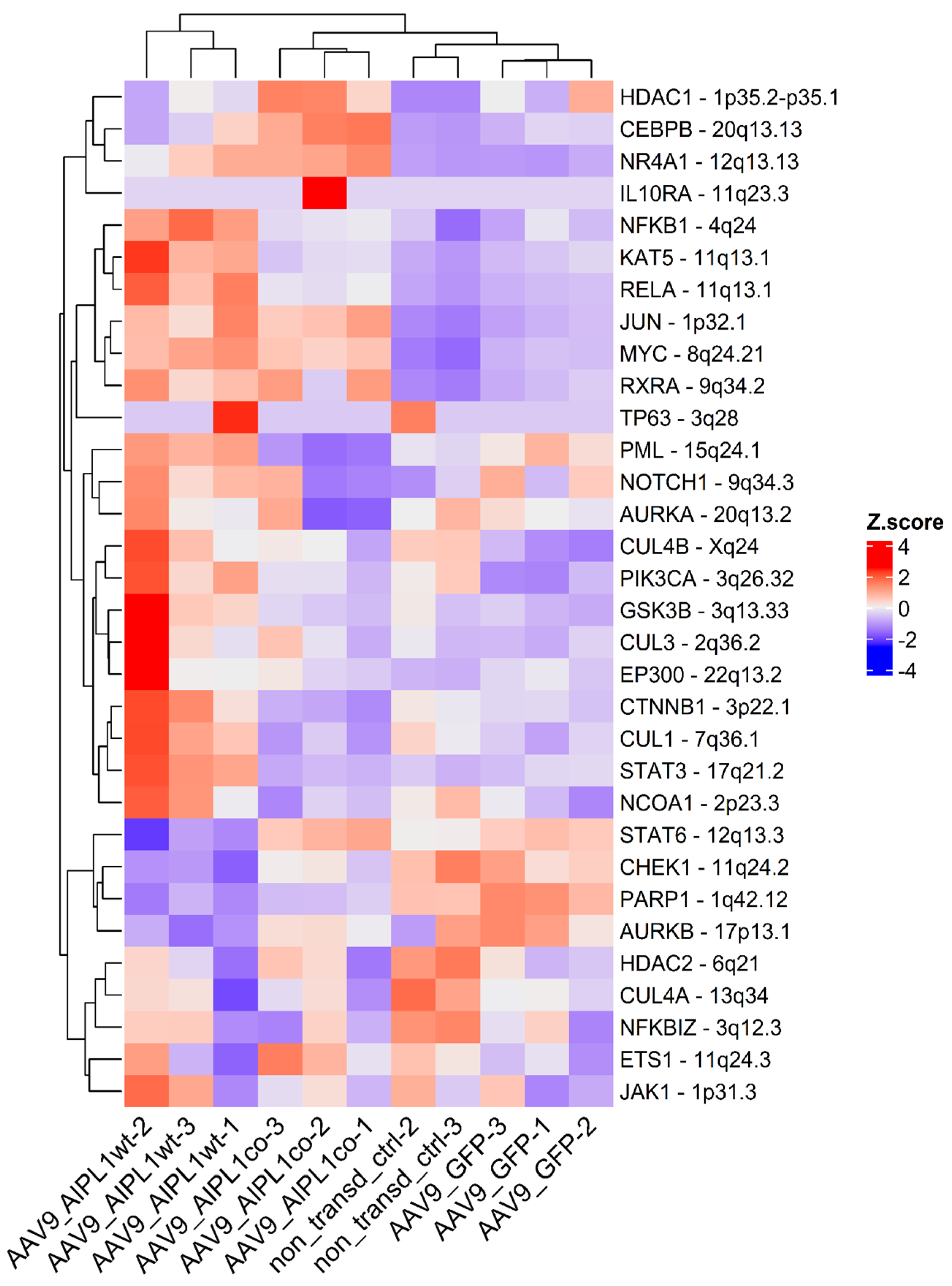
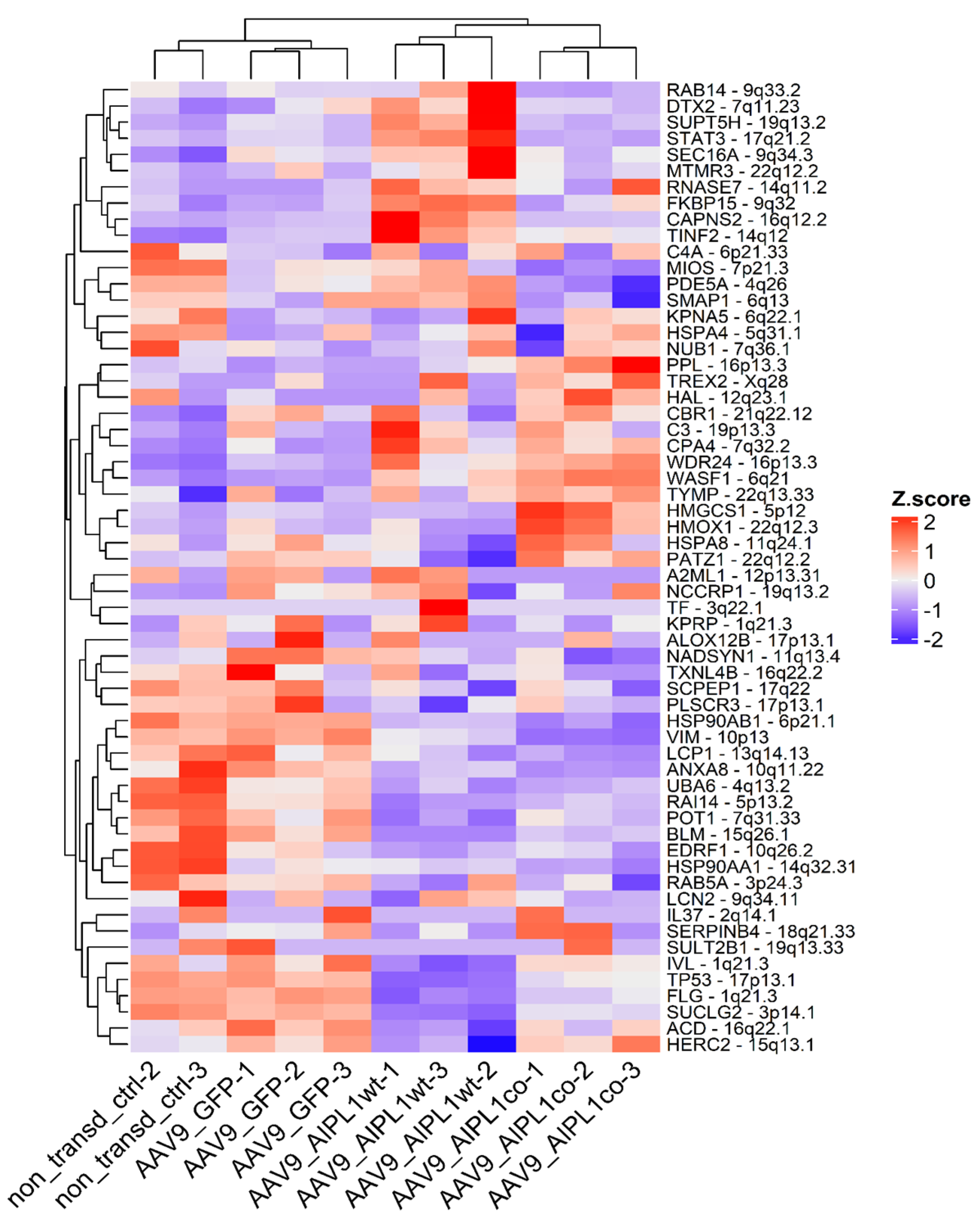
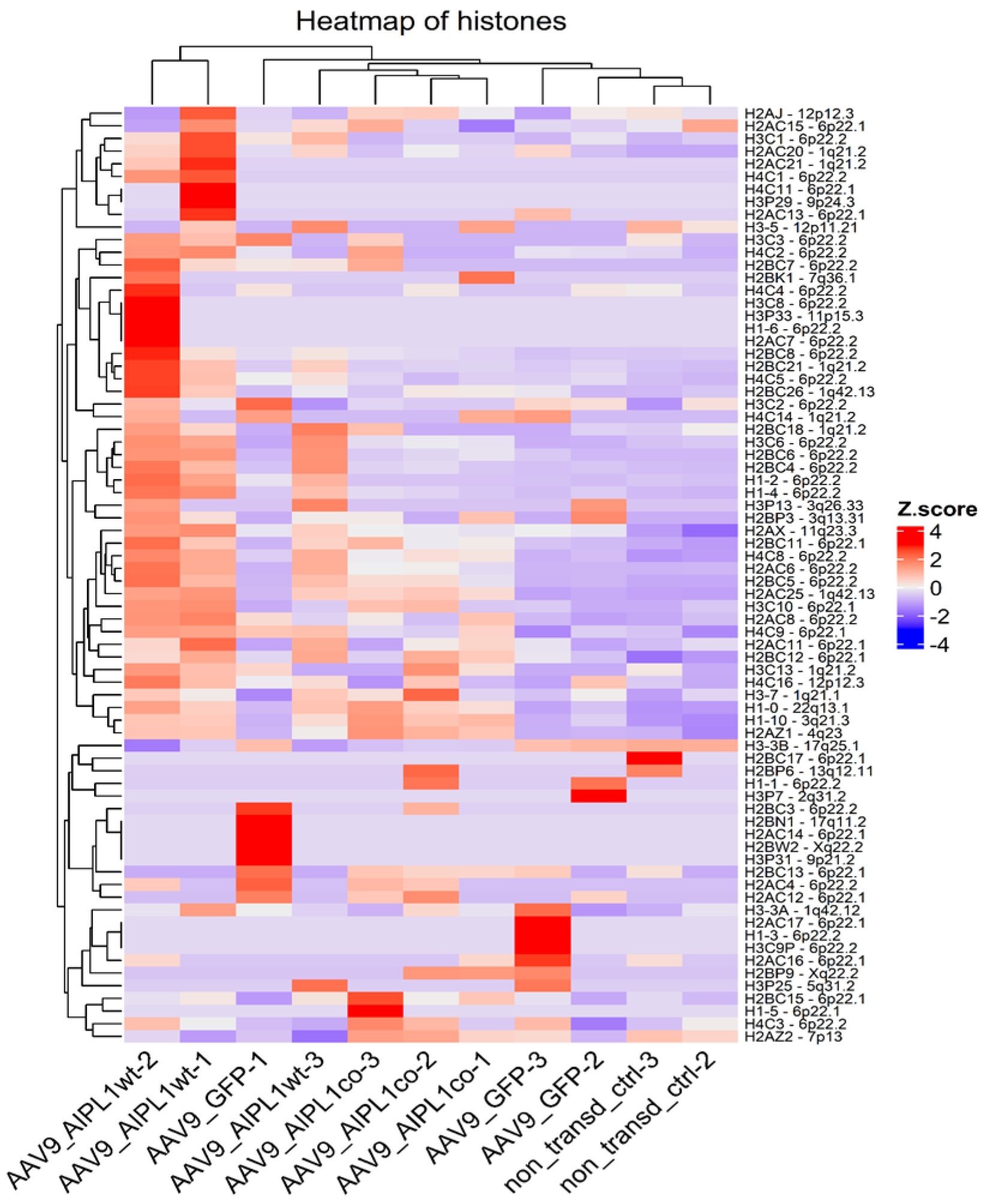
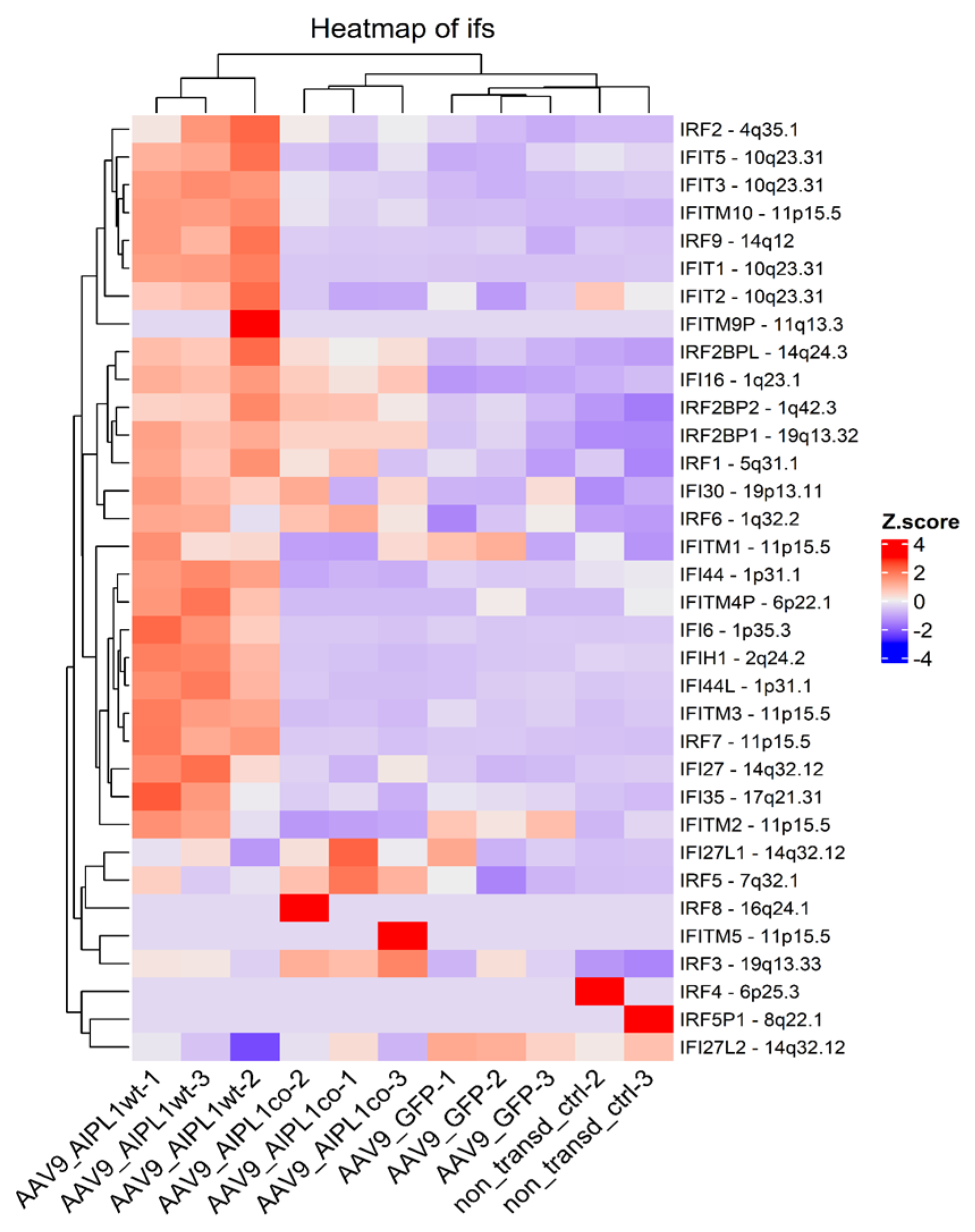
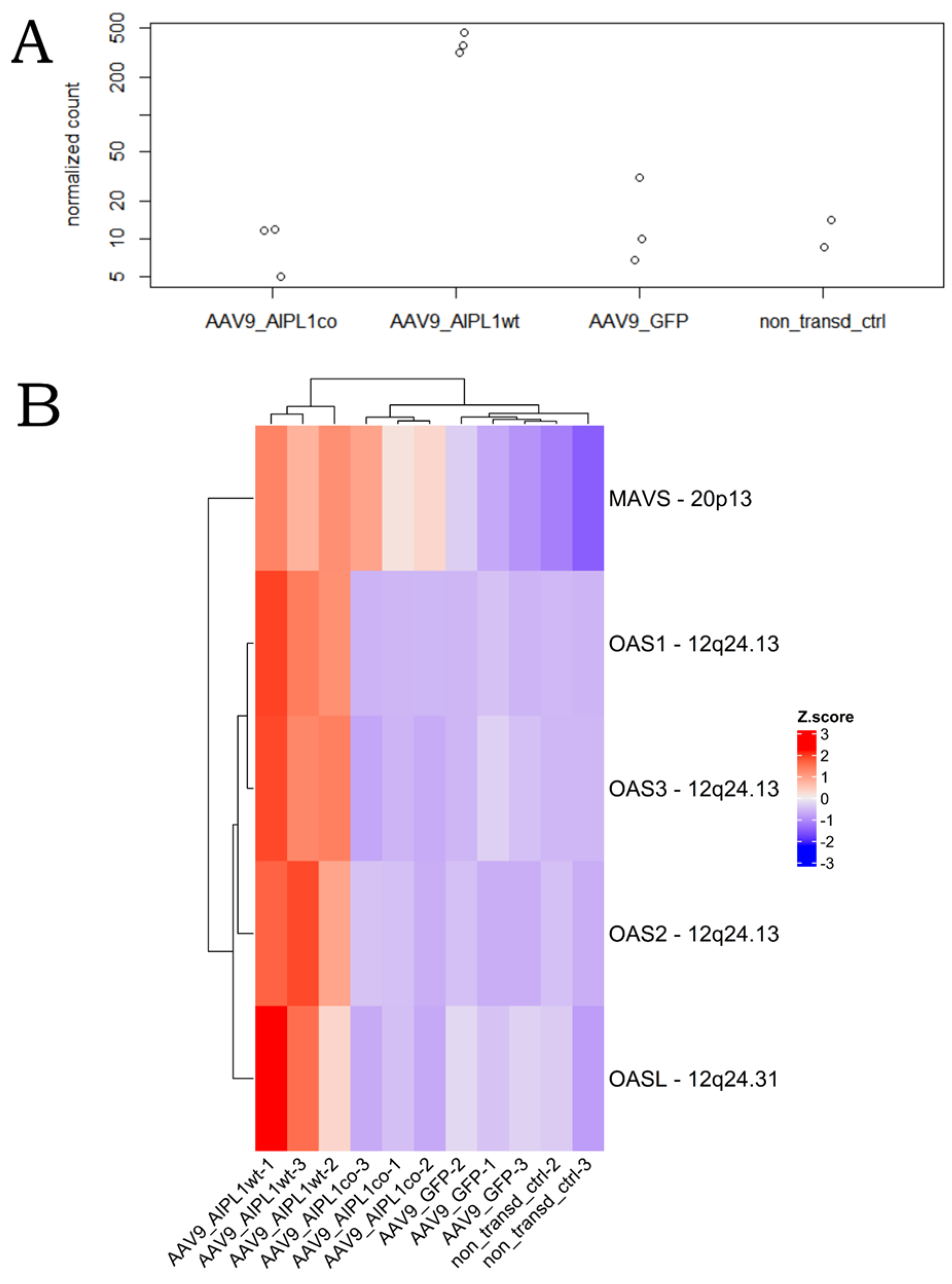
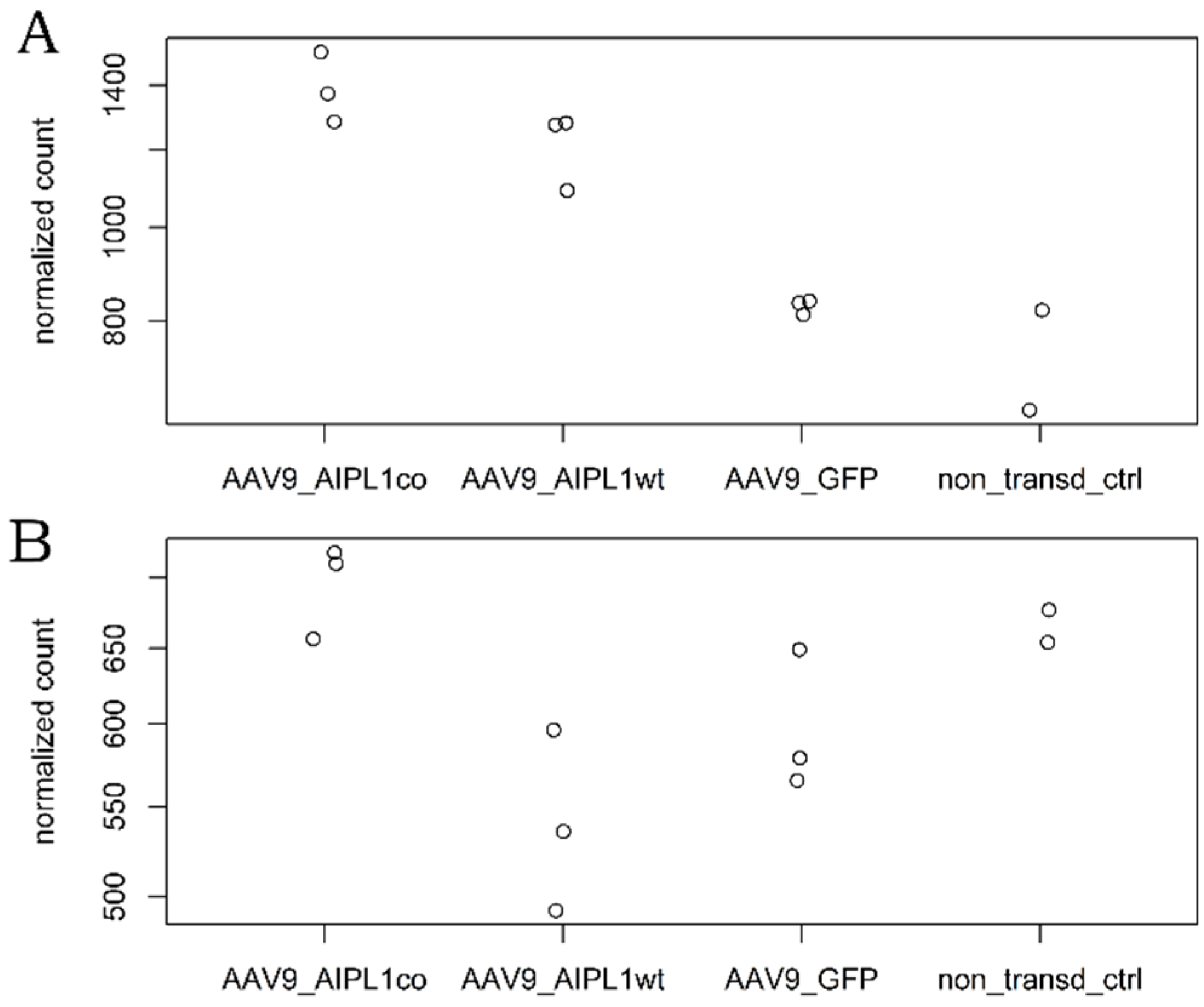
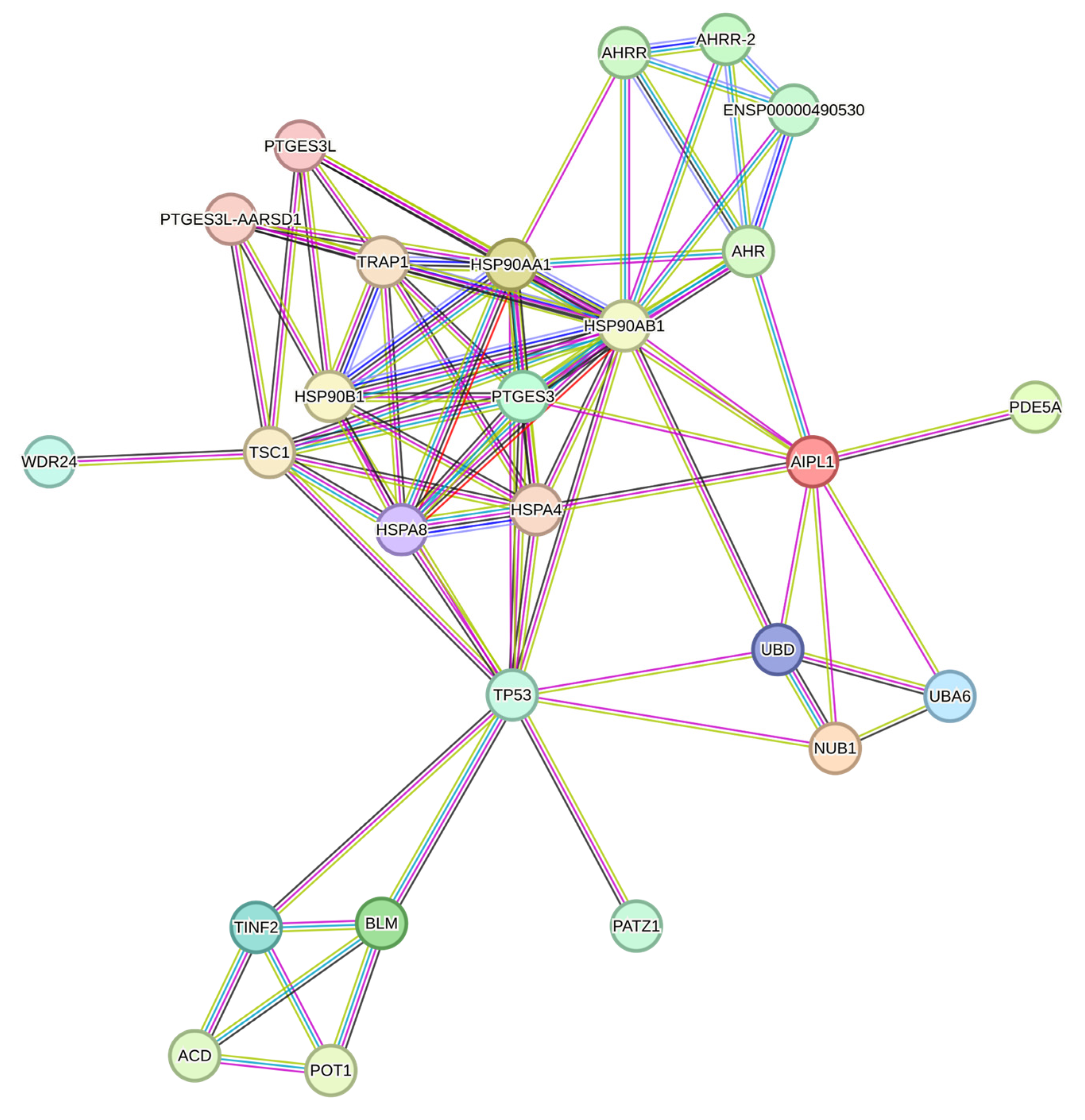
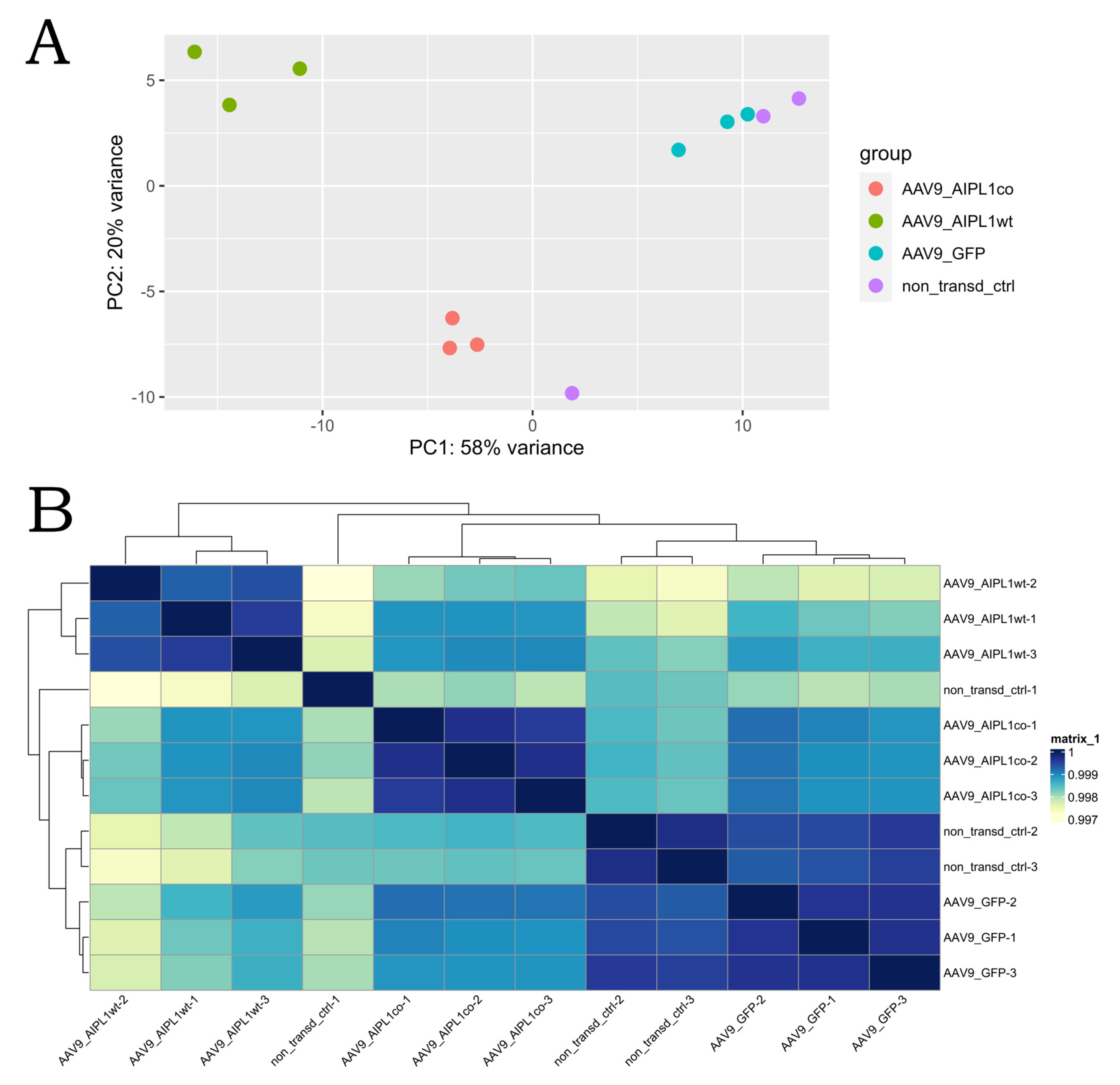
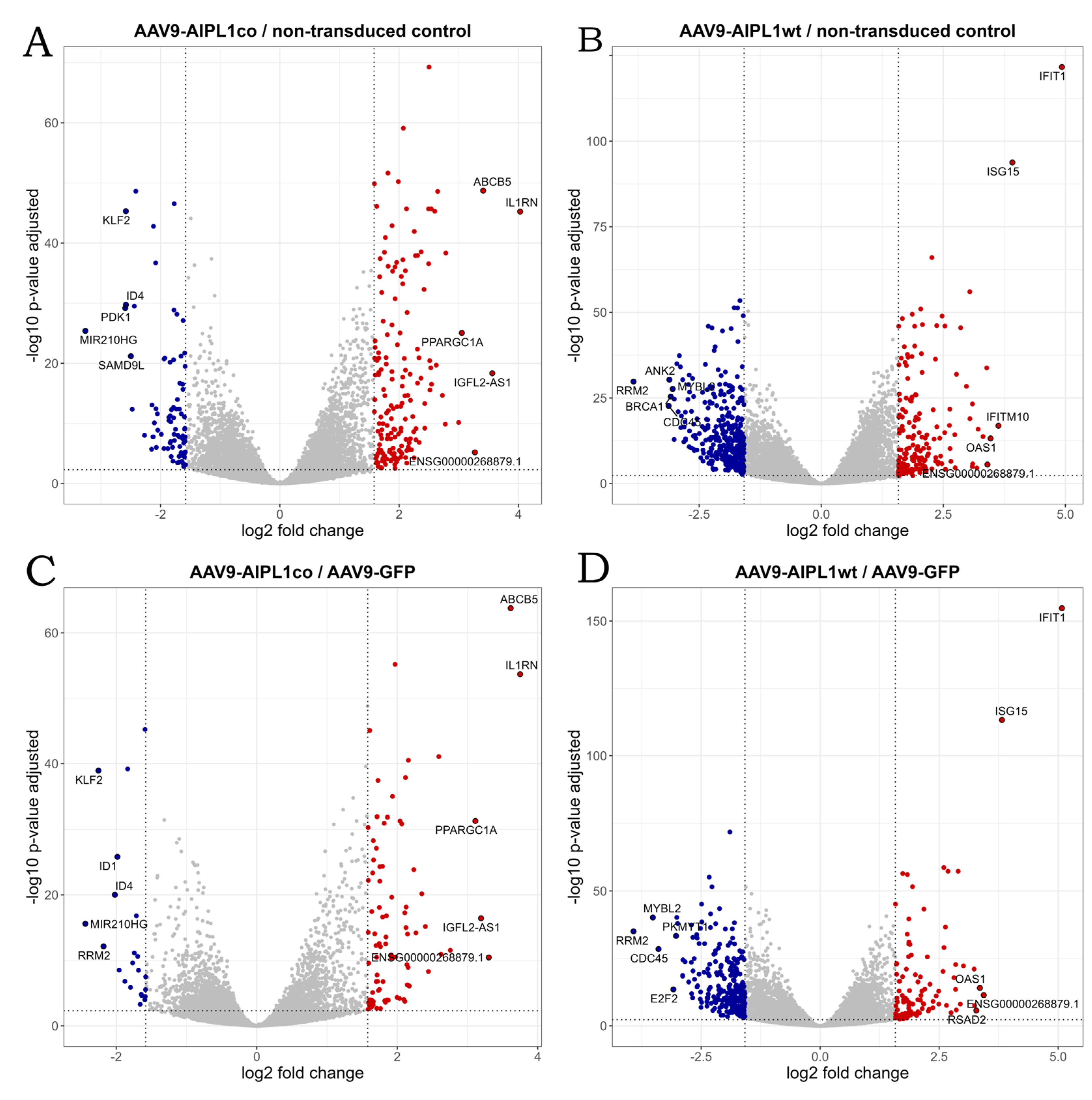
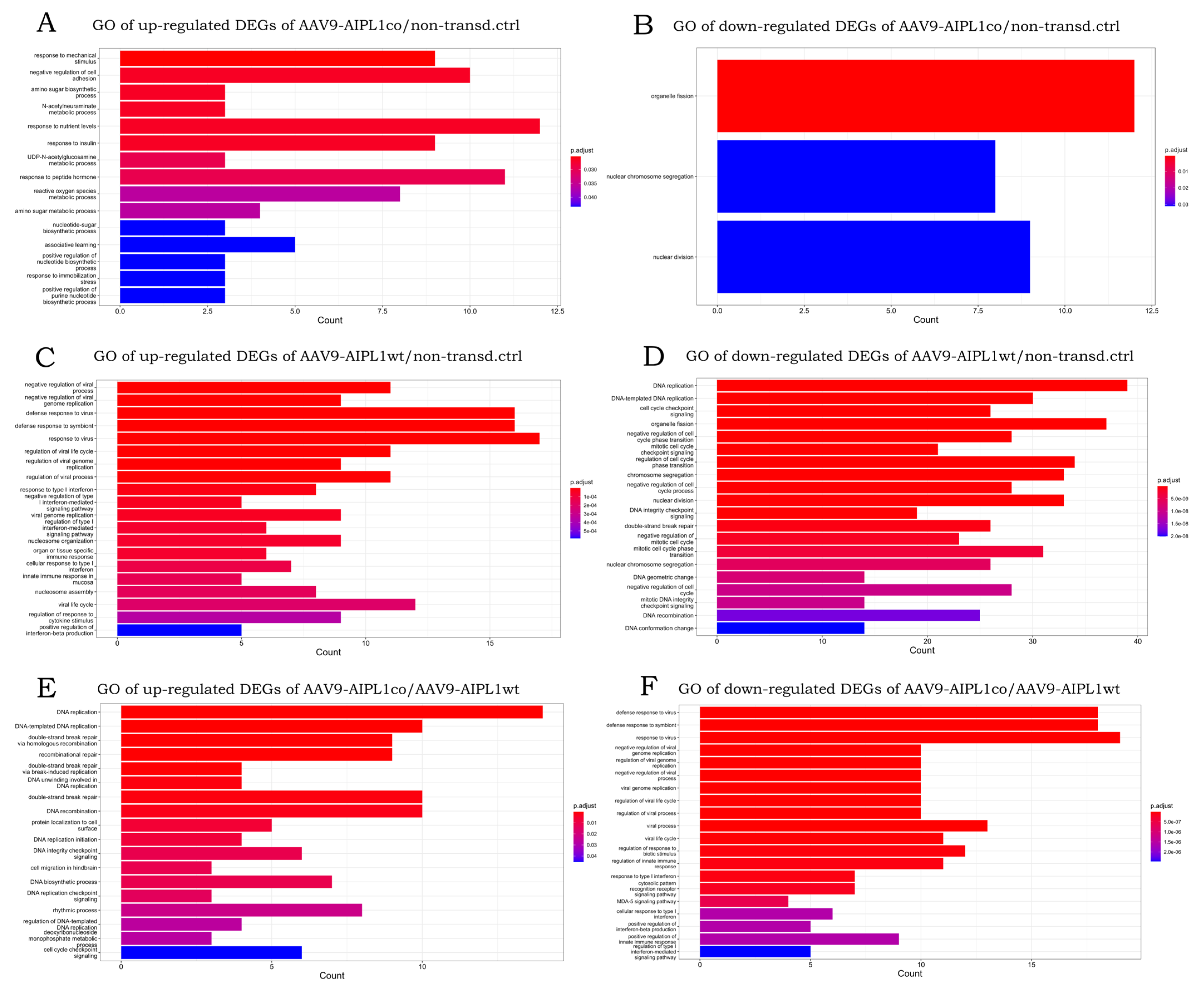
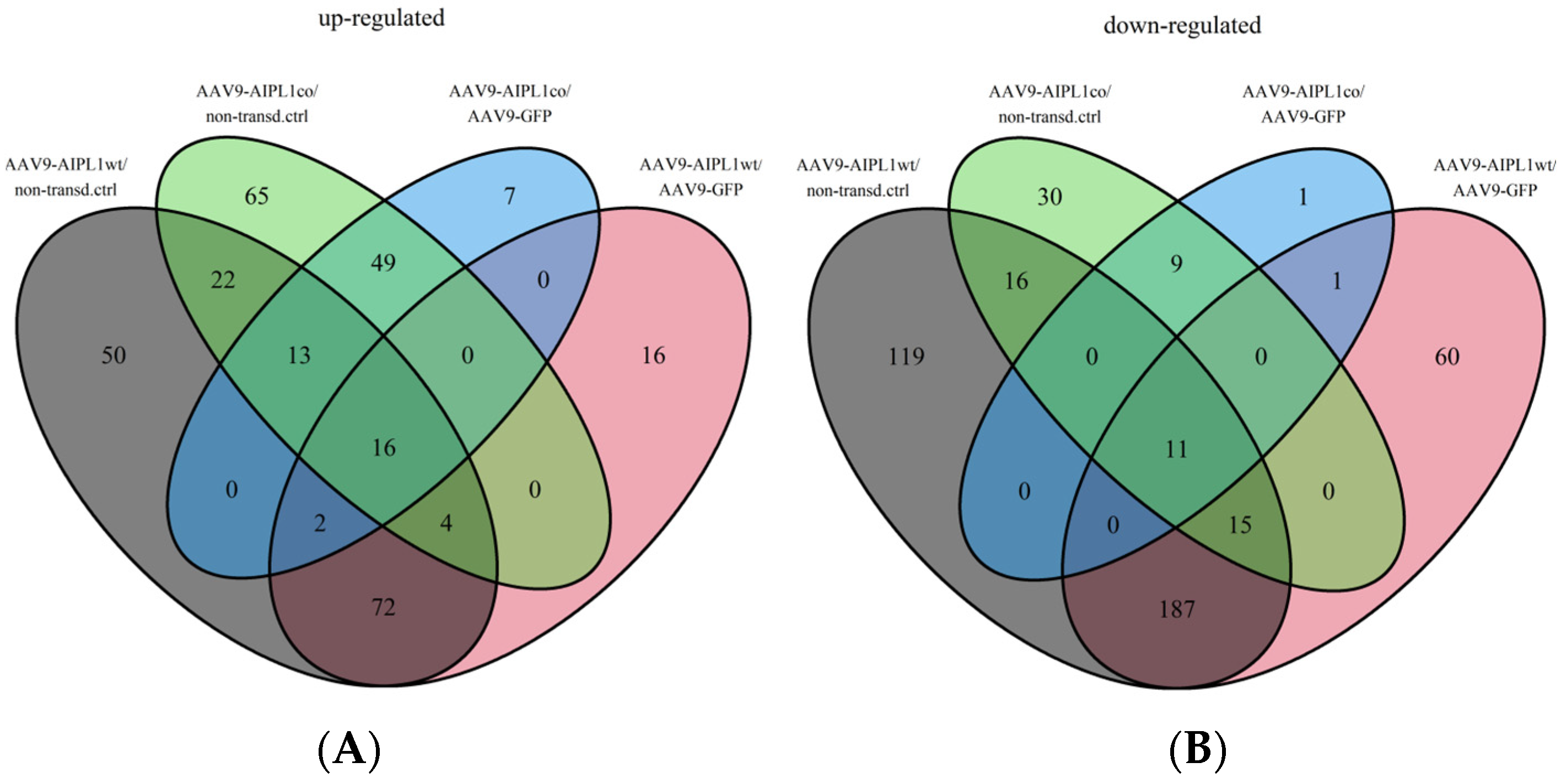
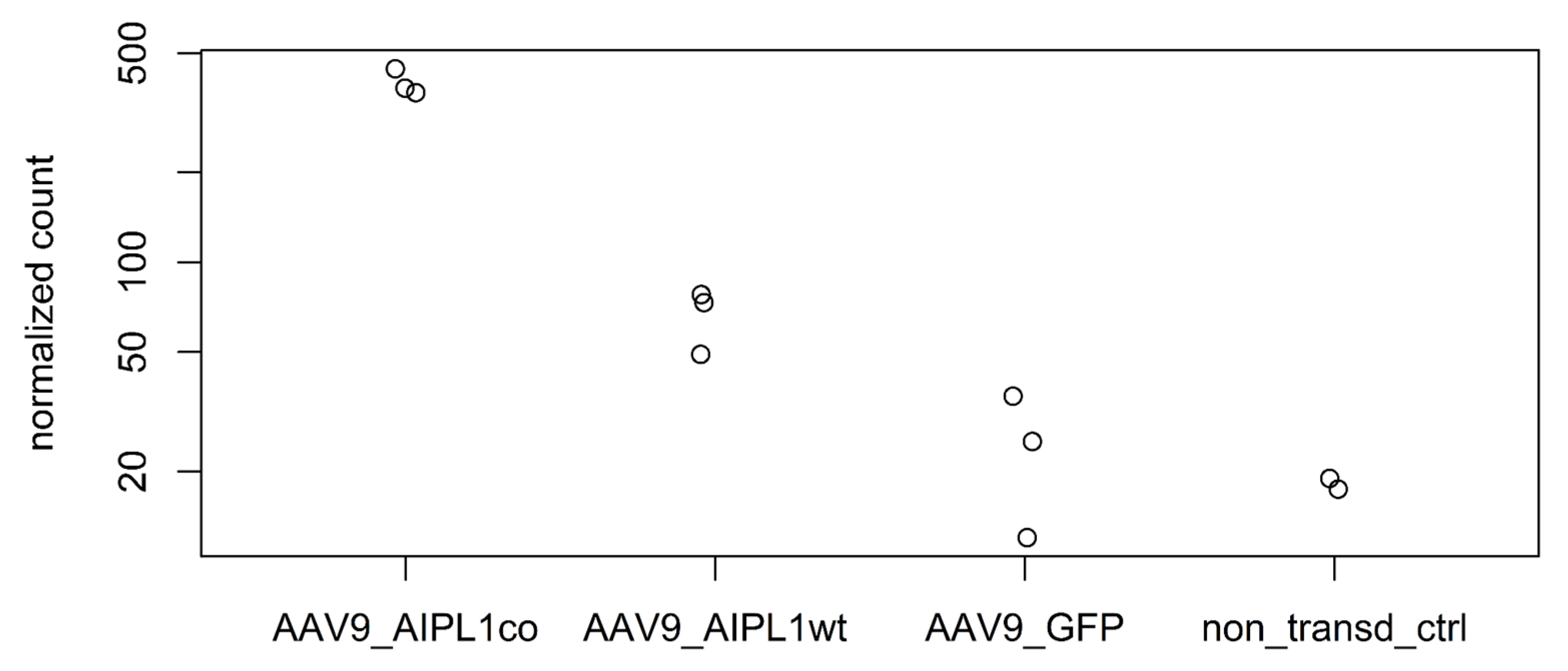
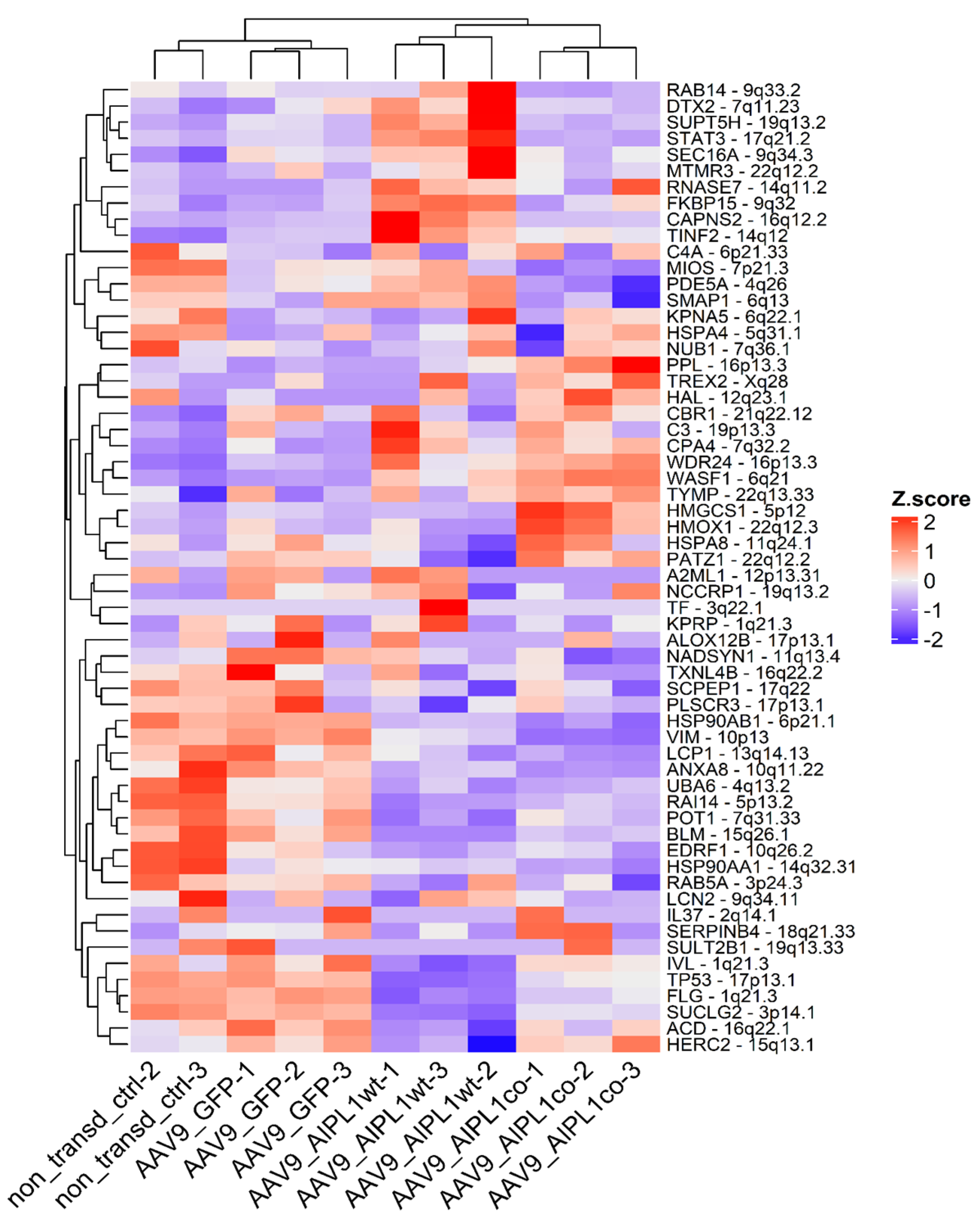
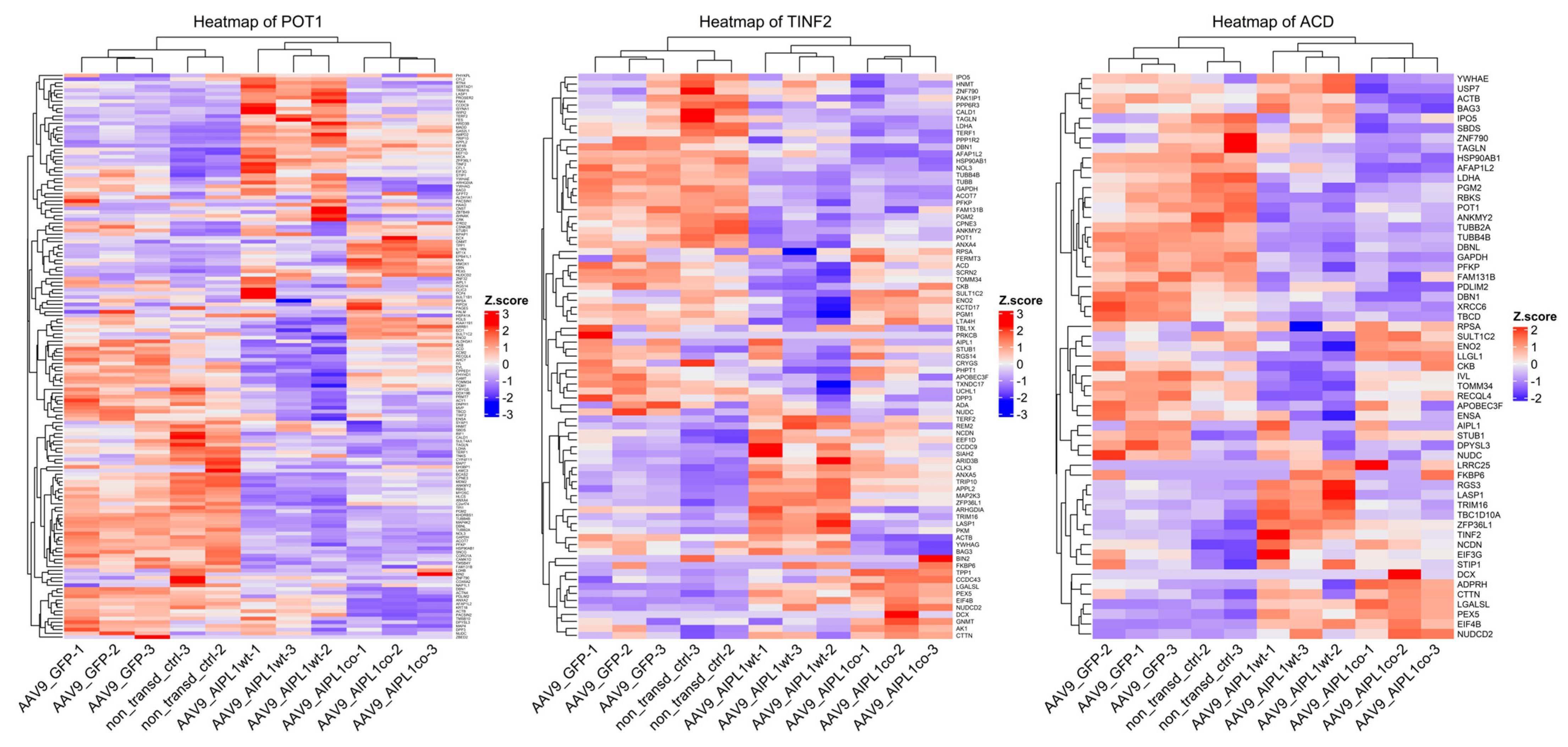
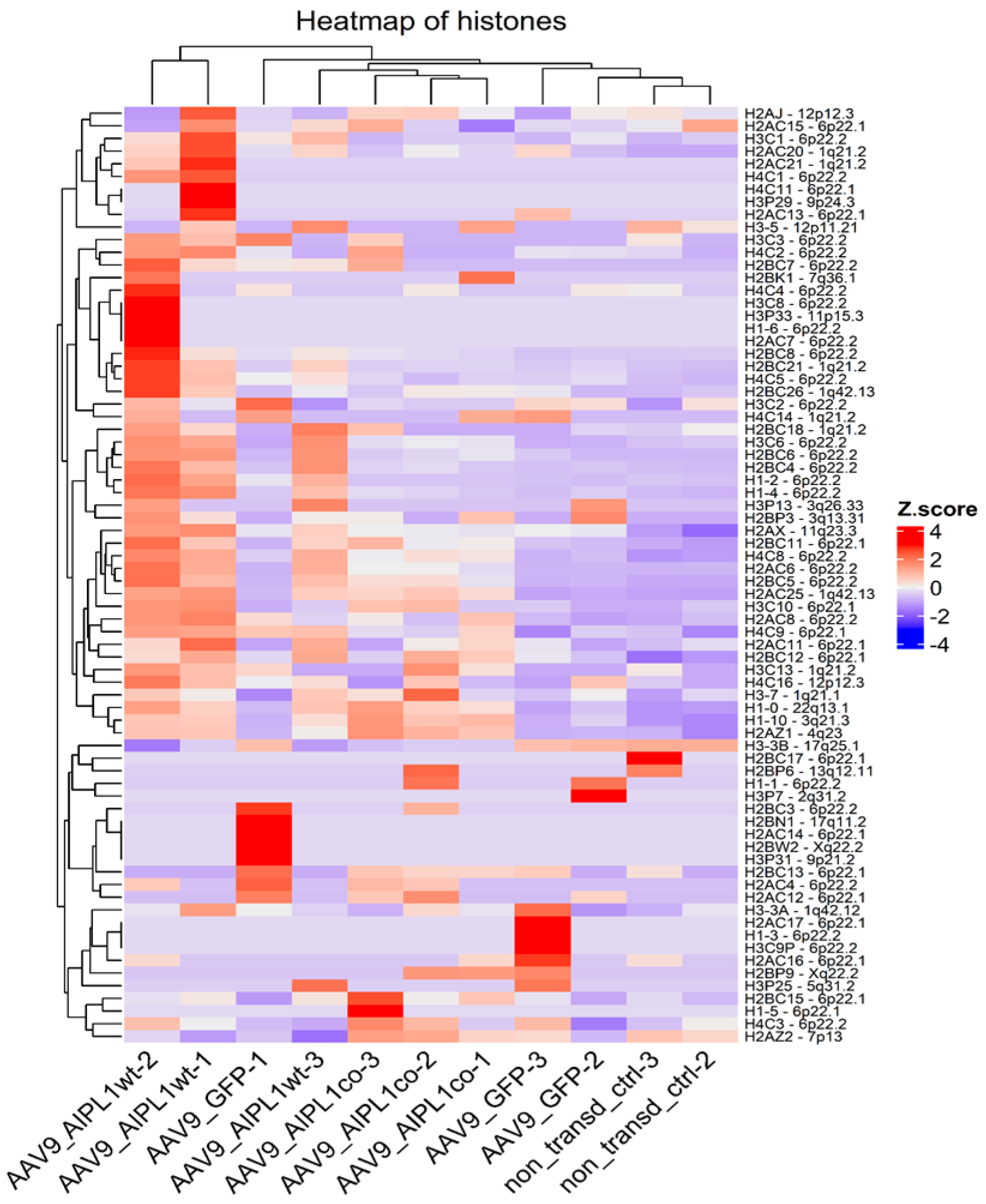
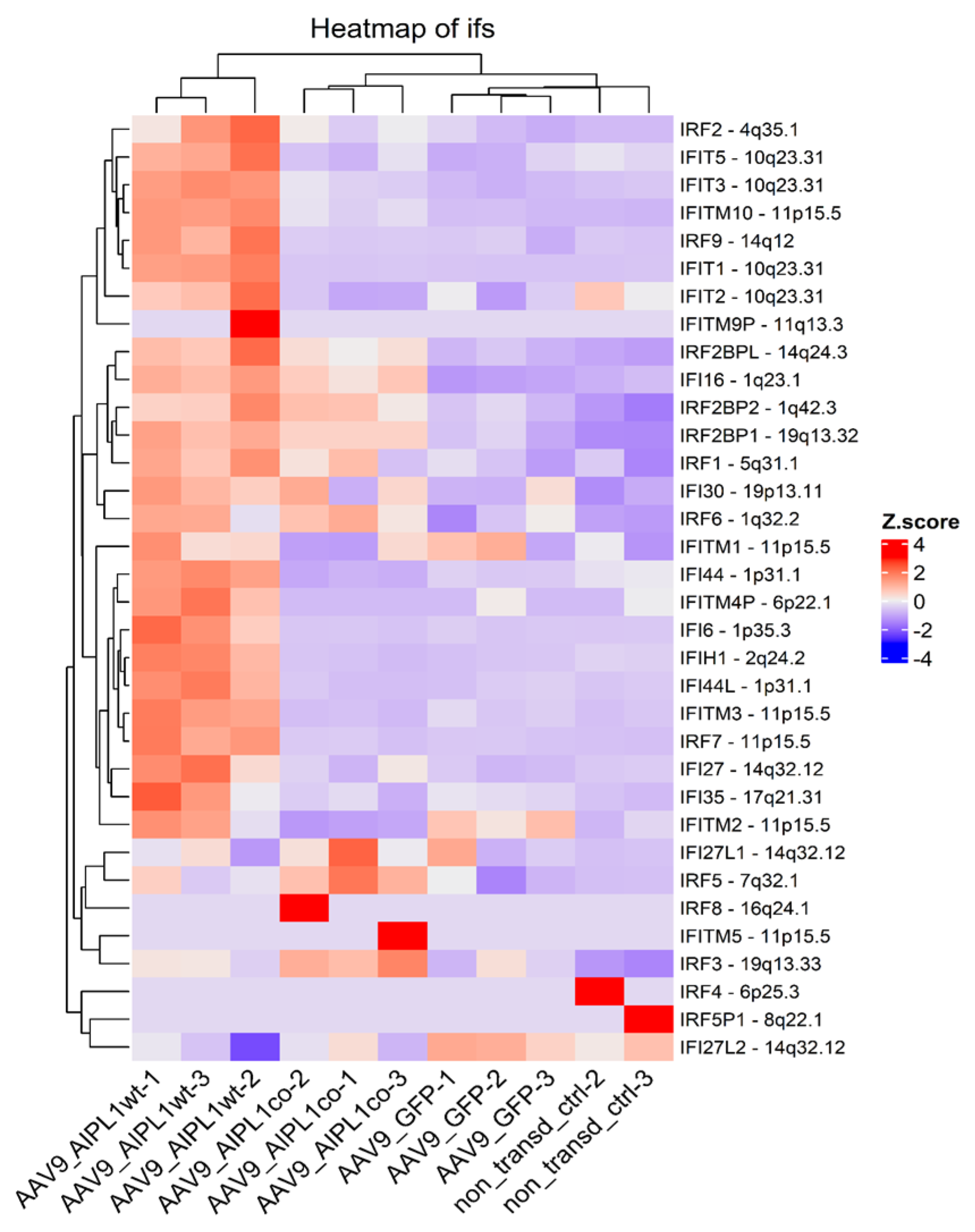
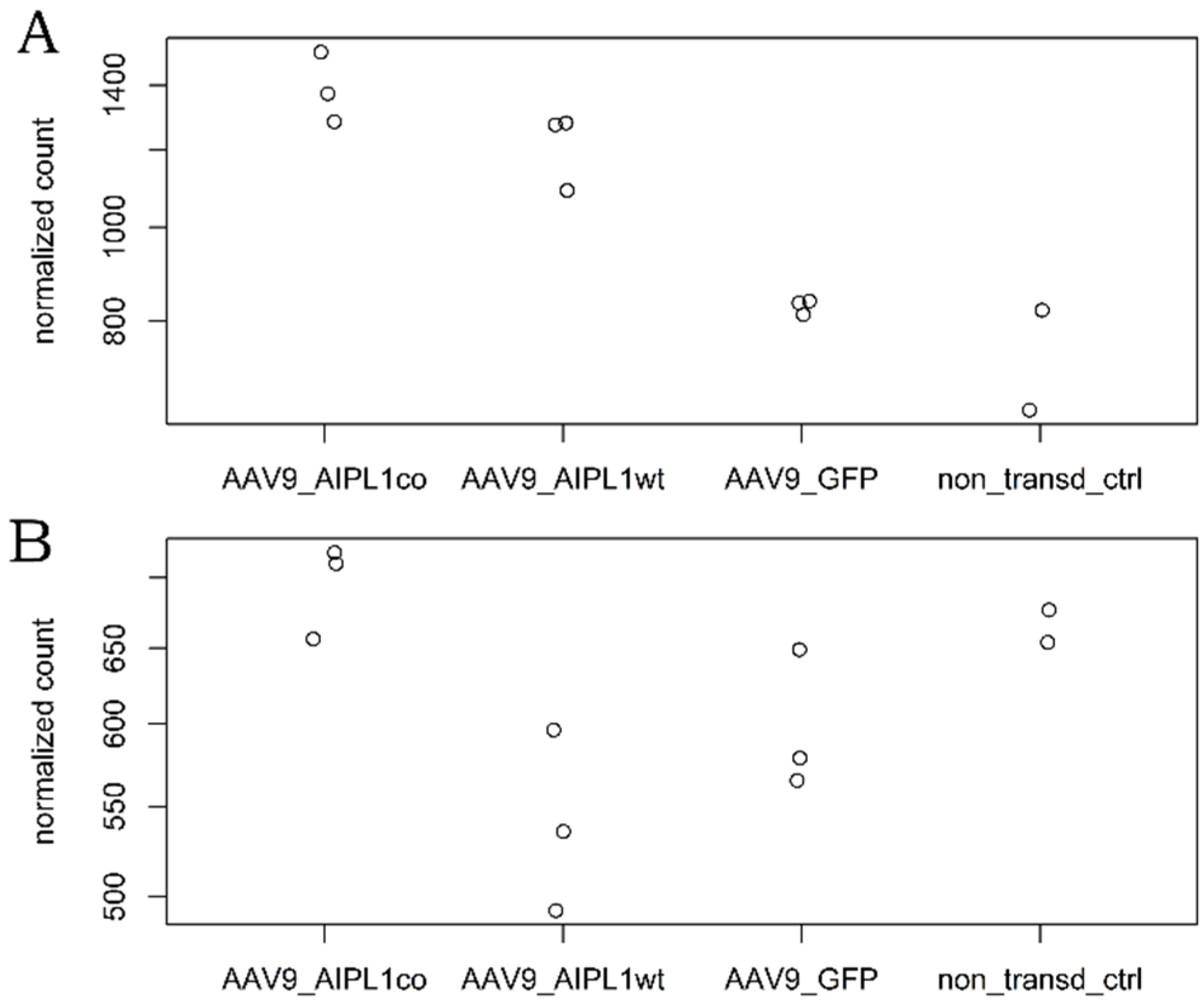
2.4. RNA-Seq Data Analysis
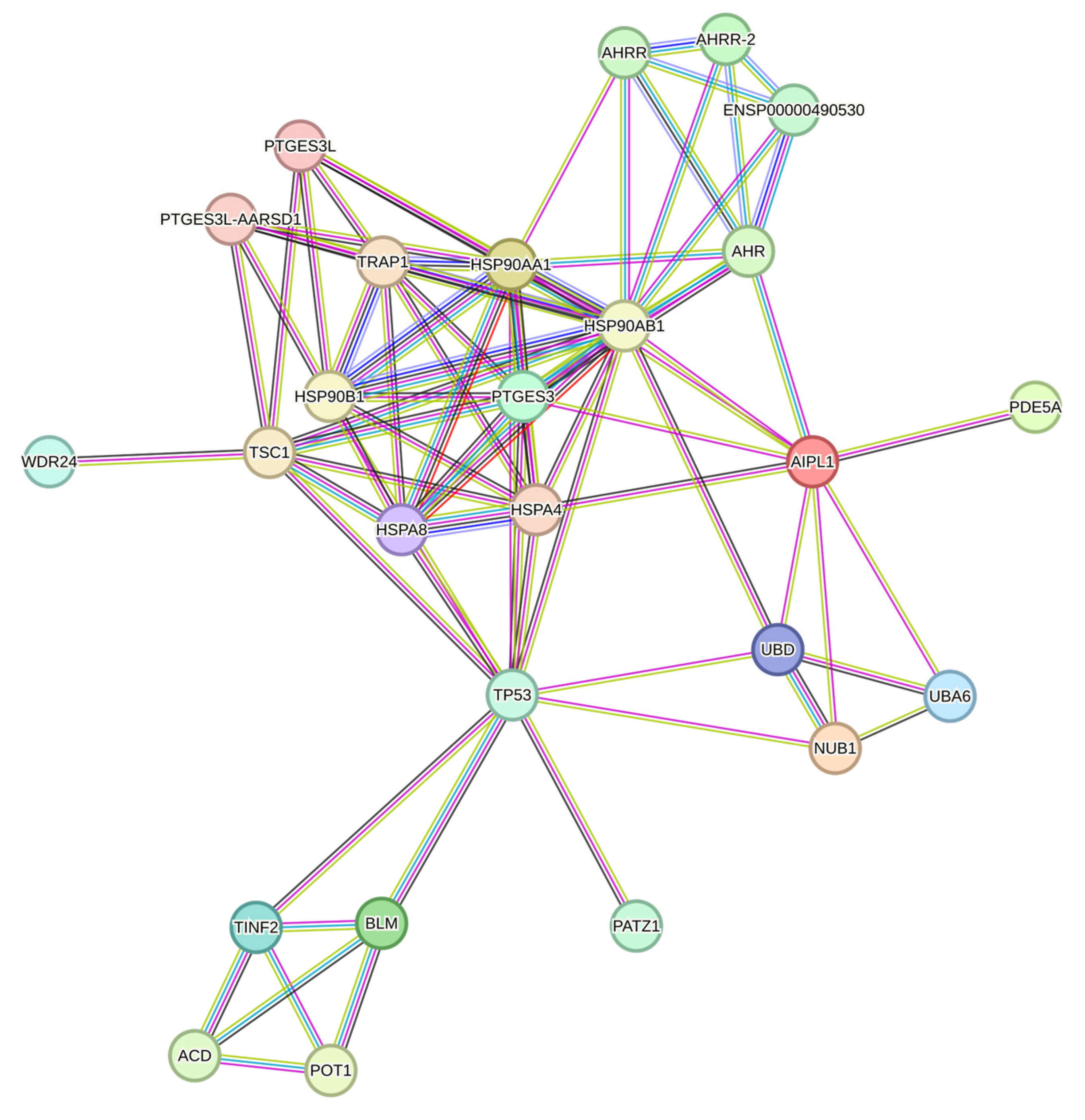
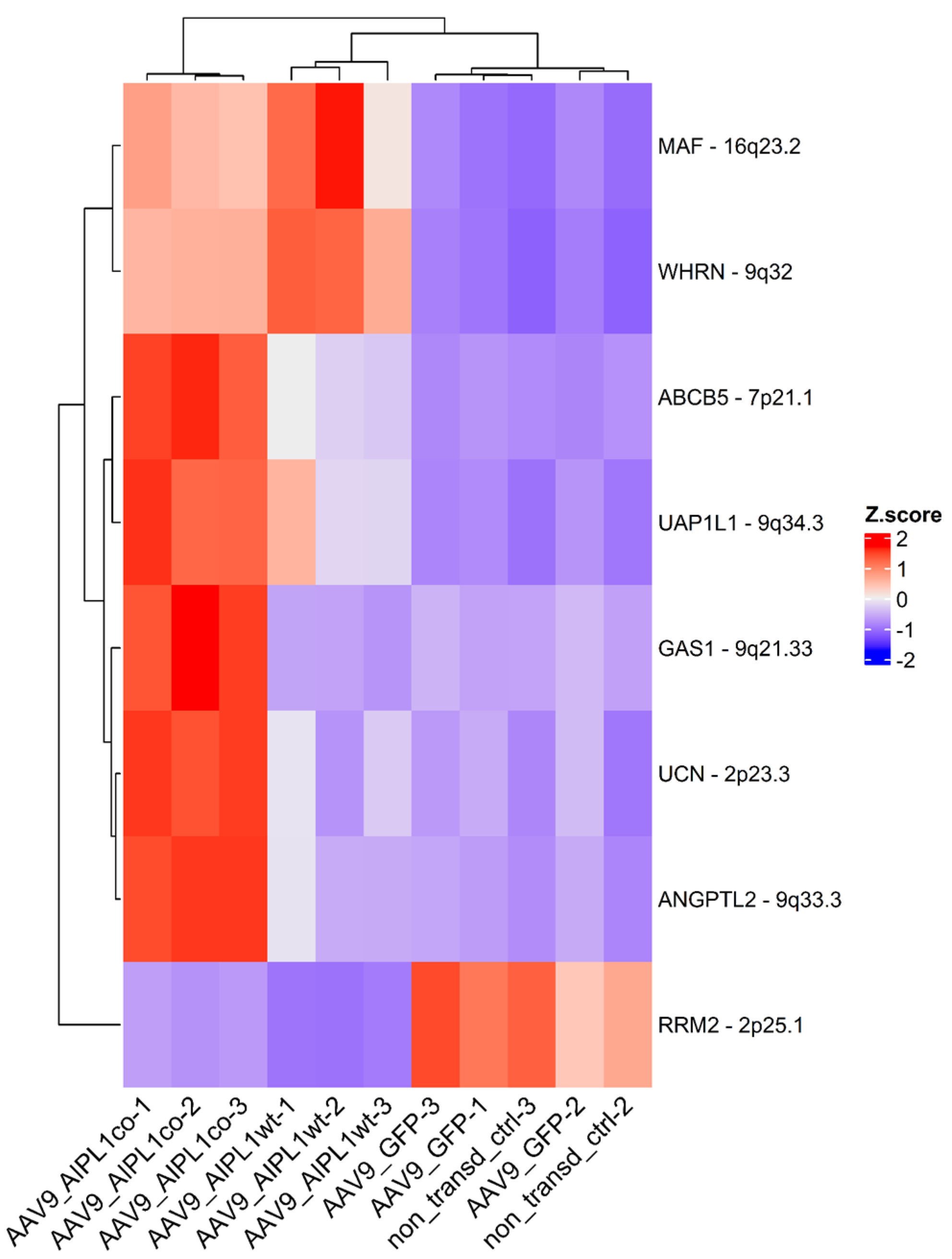
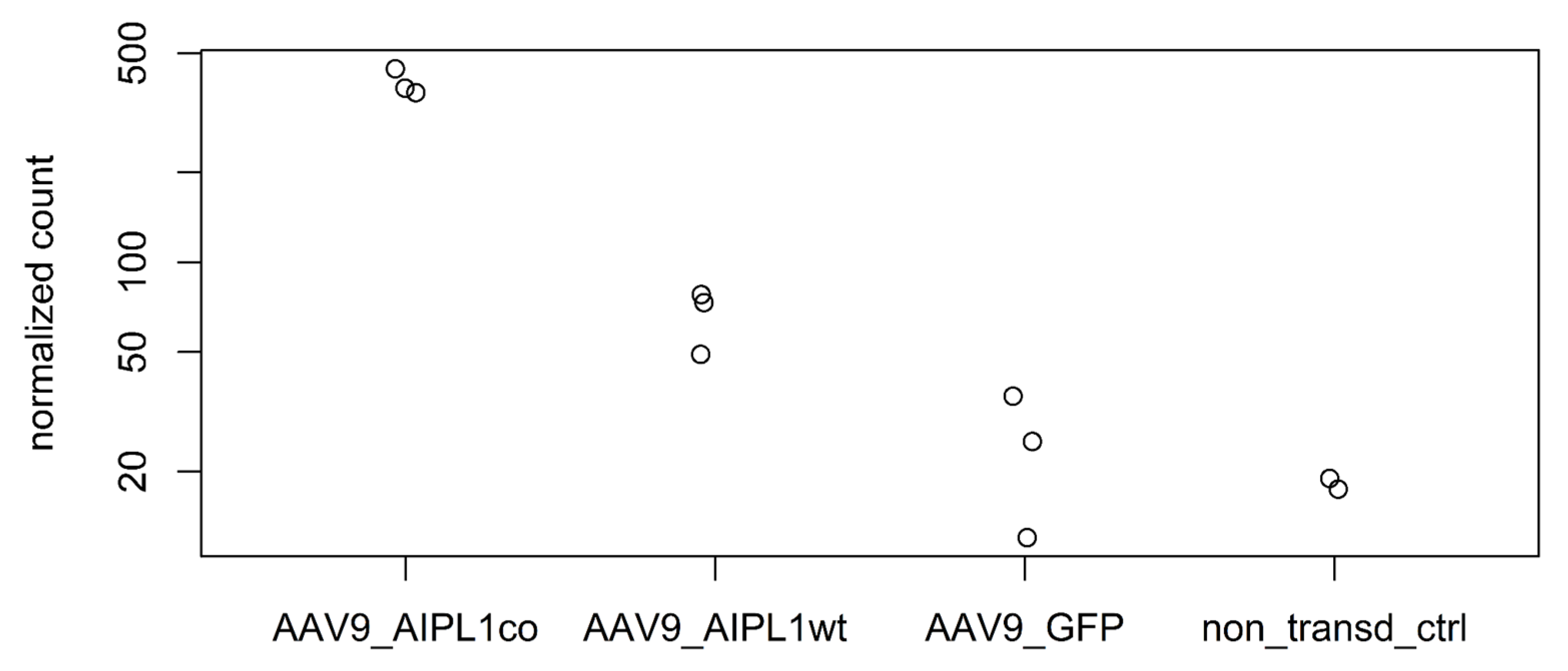
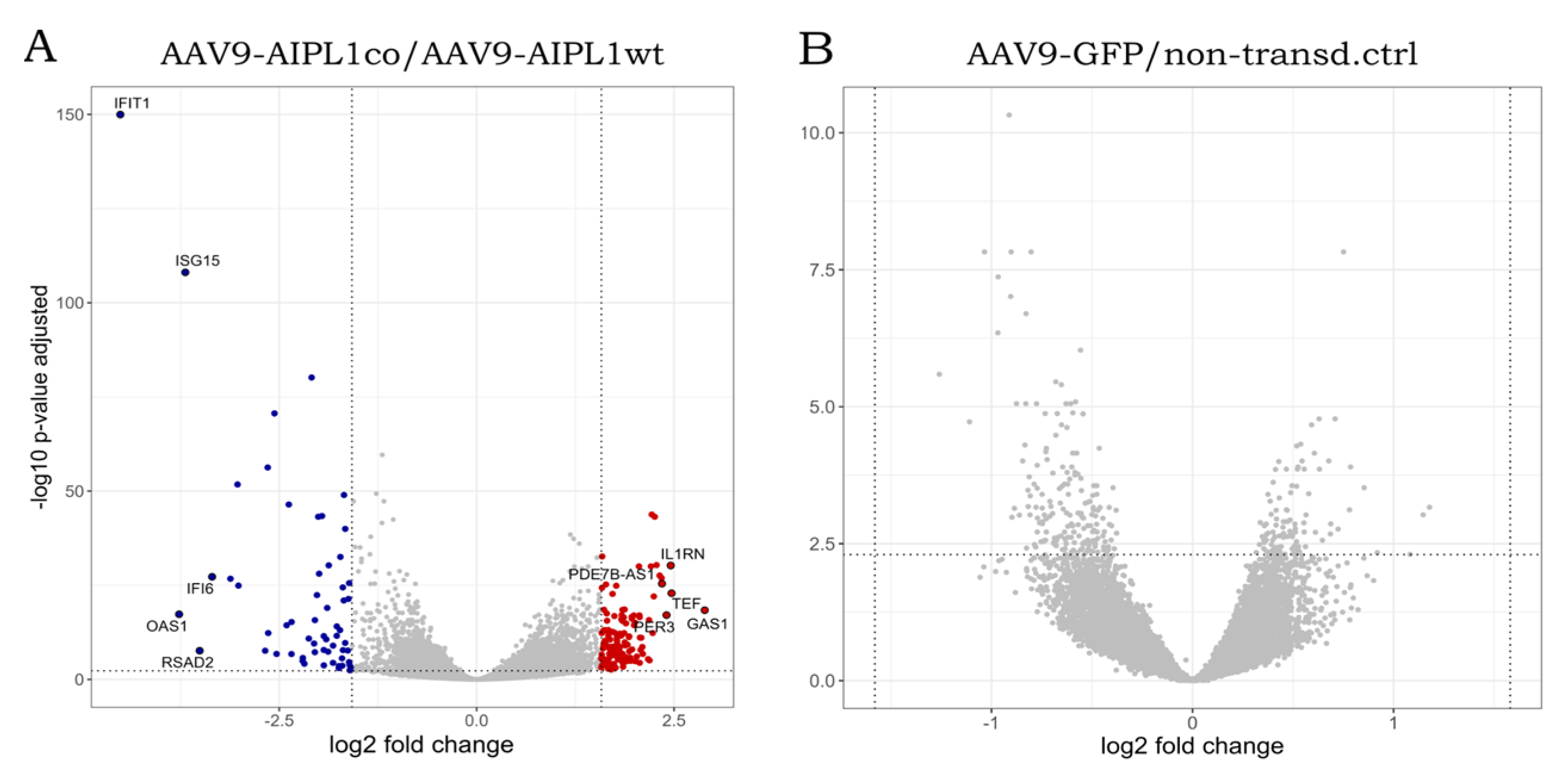
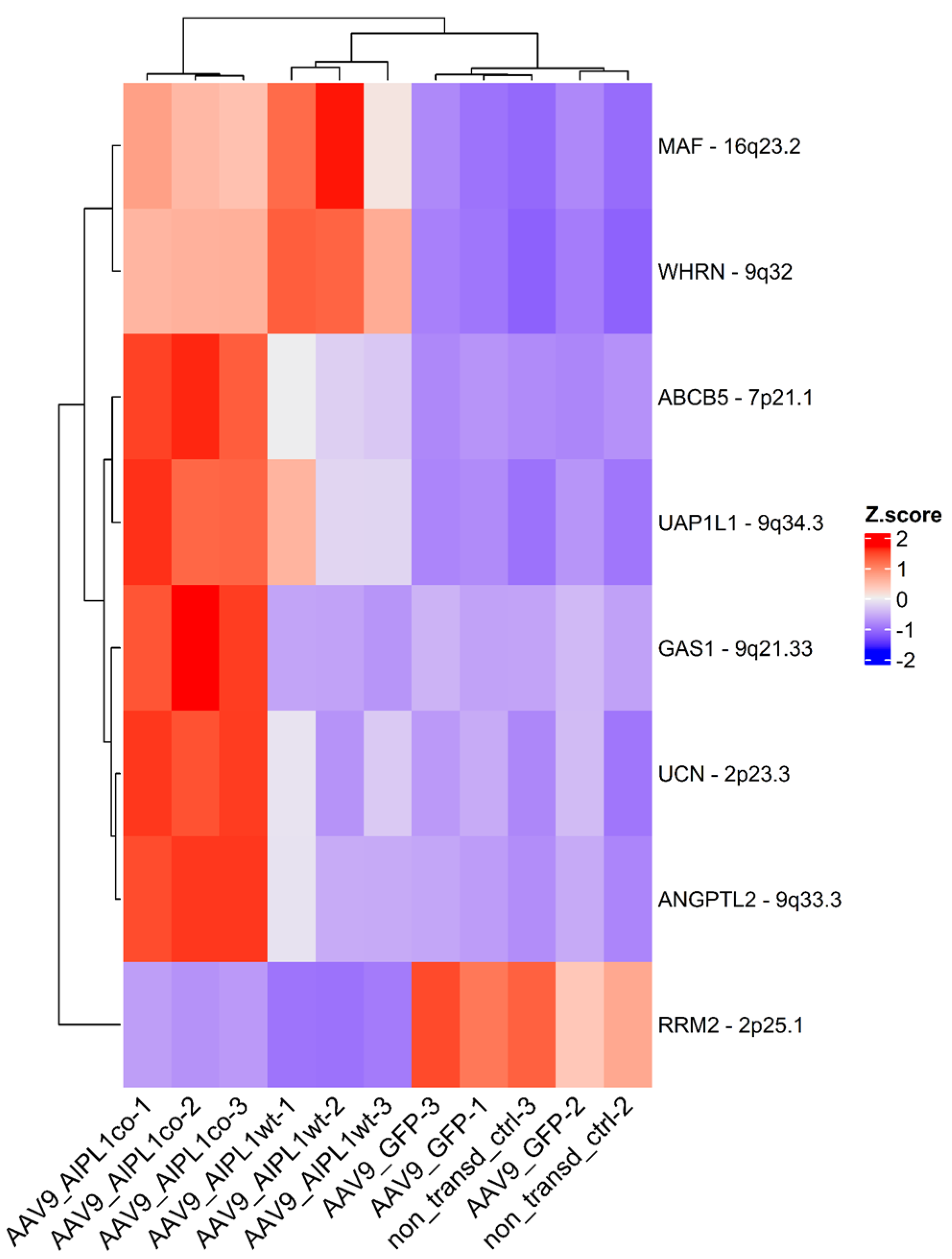
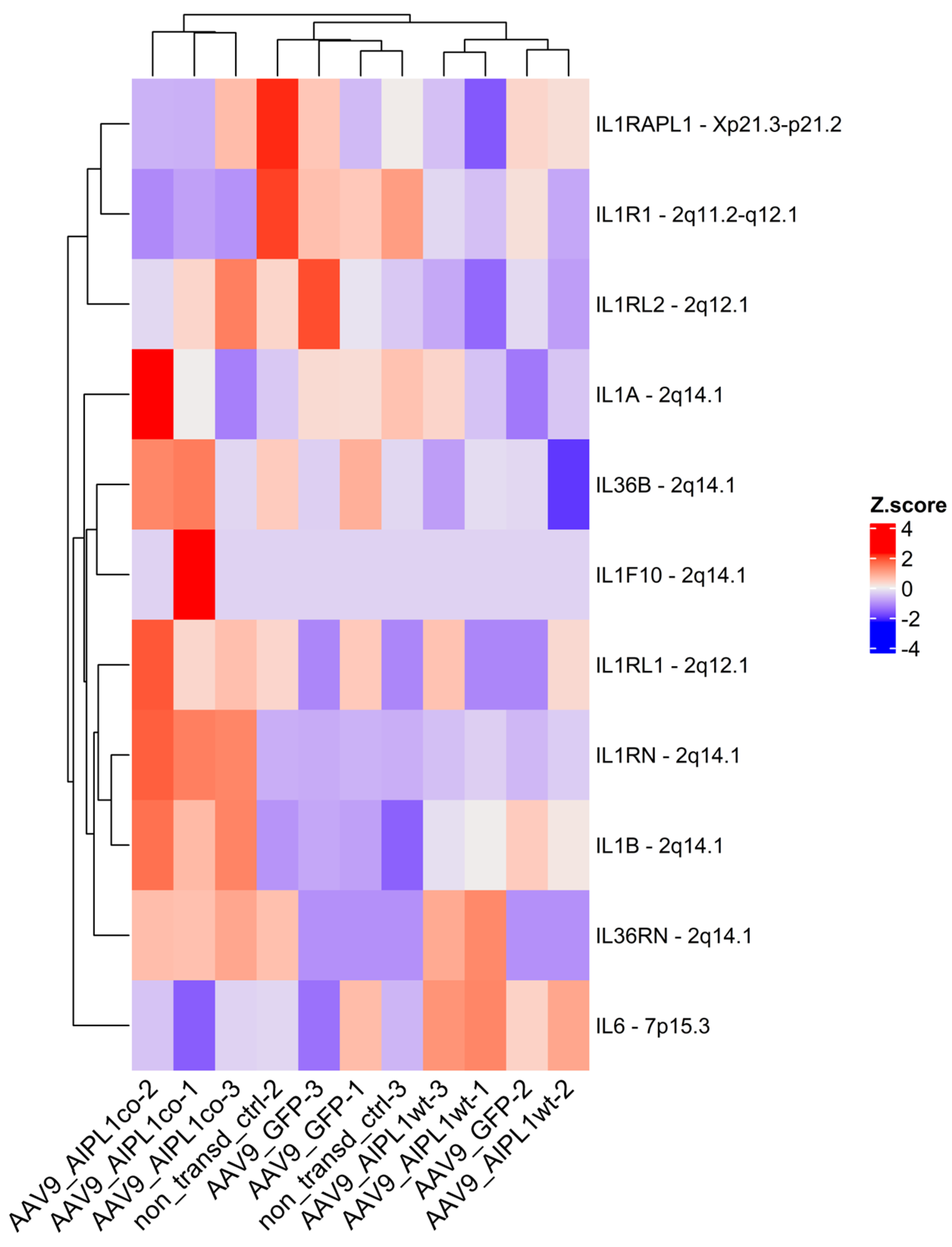
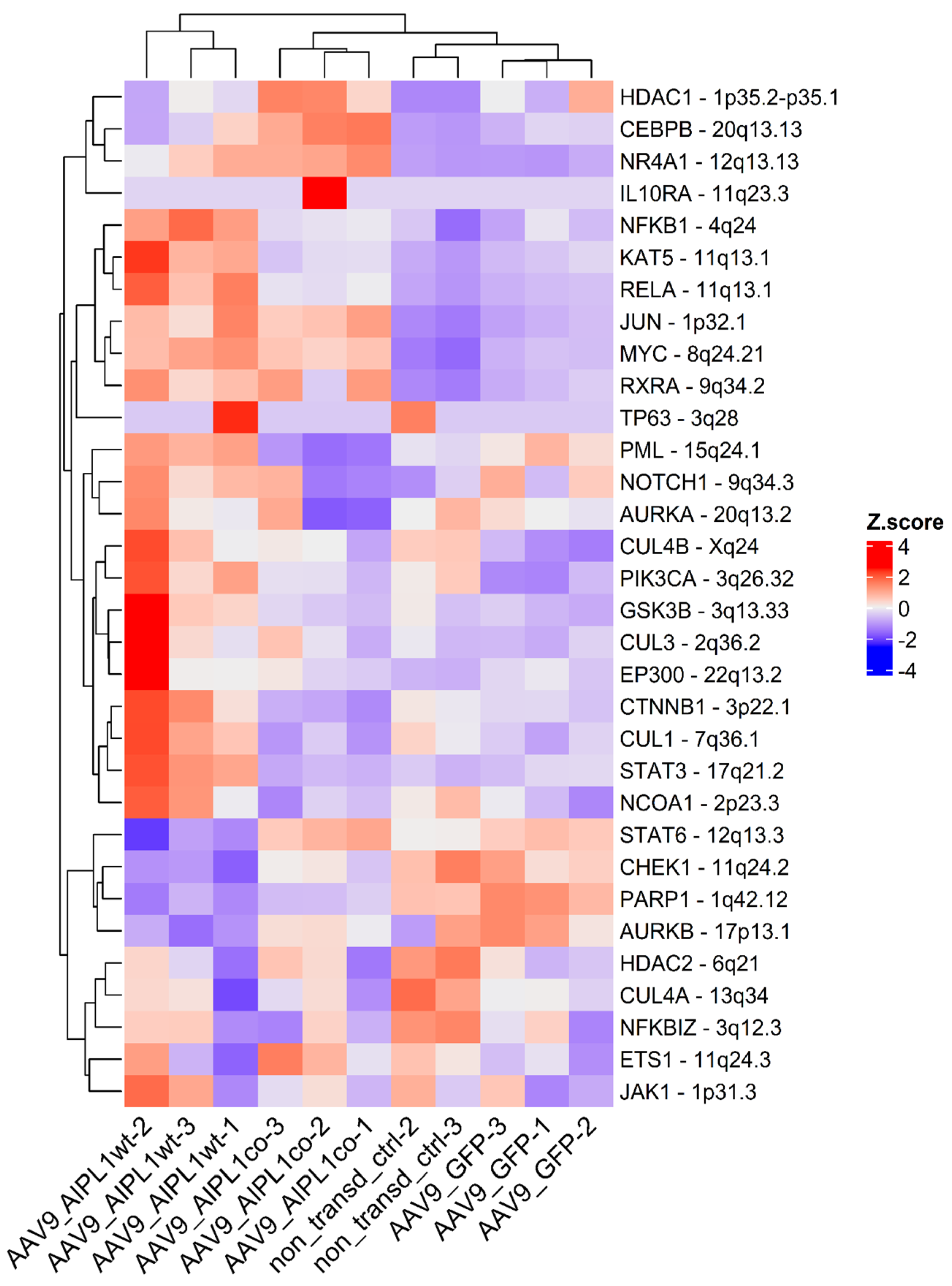
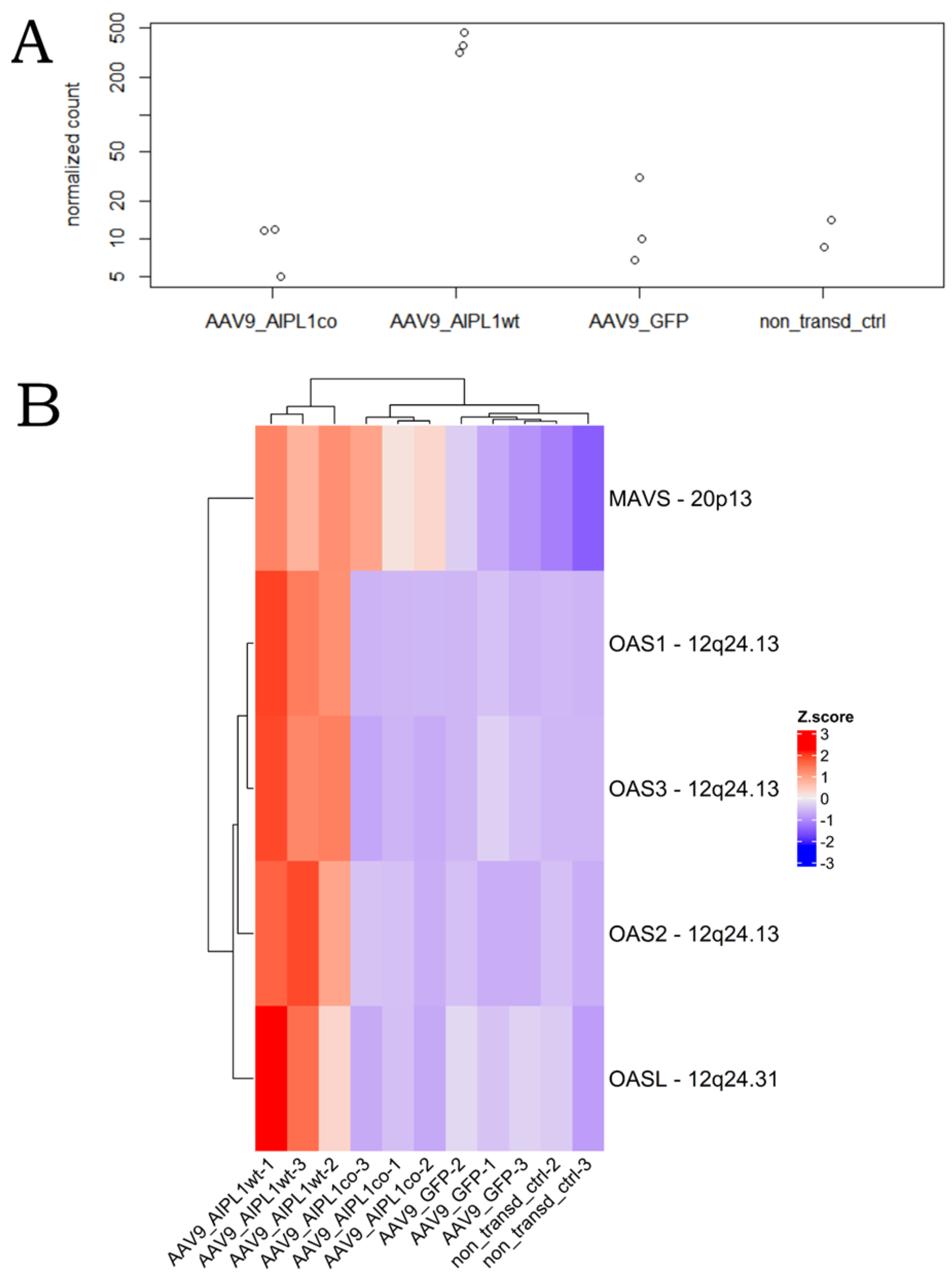
2.5. Genes Relevant for Both AAV9-AIPL1wt and AAV9-AIPL1co Transduced Cells
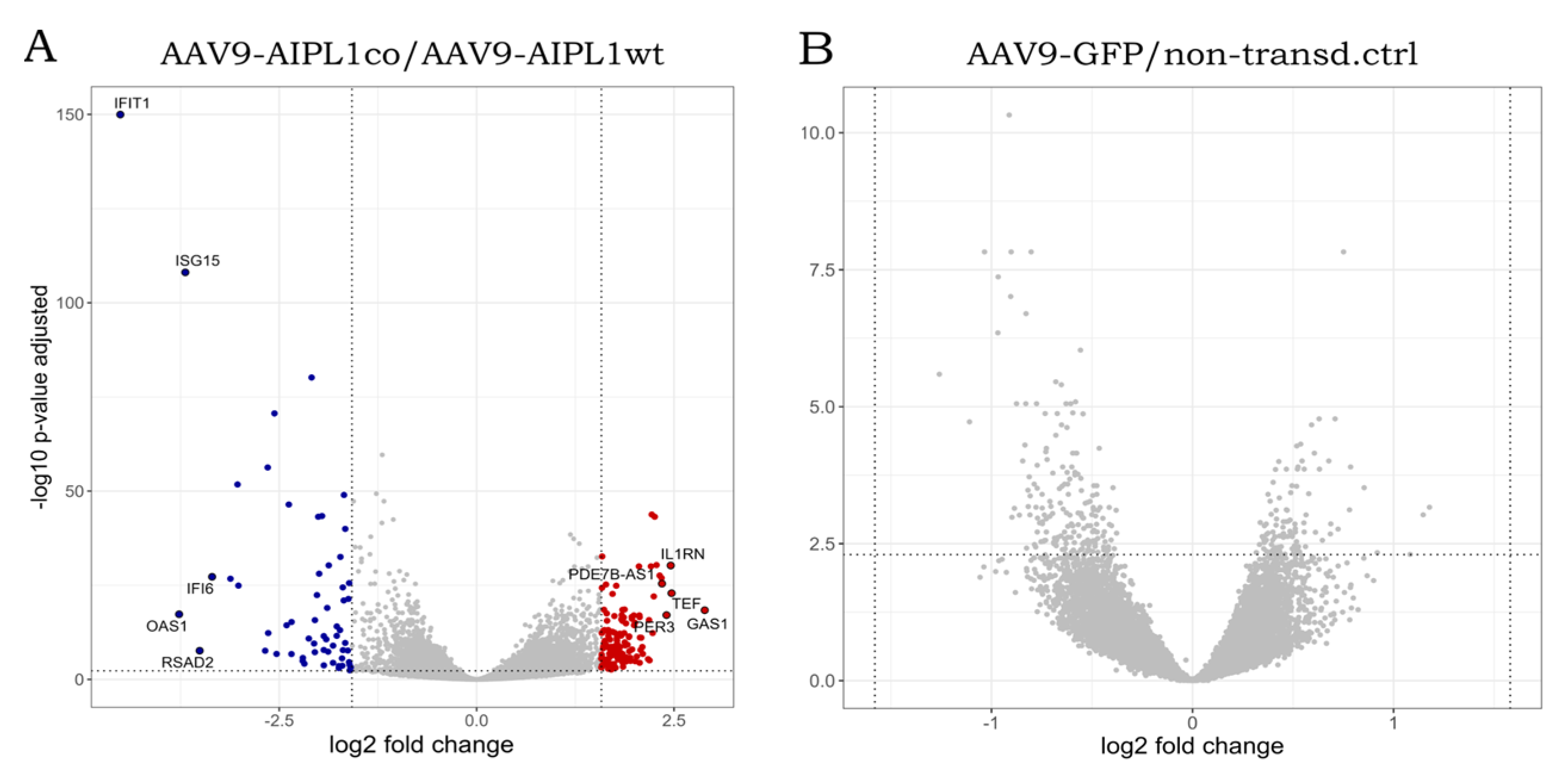
2.6. Genes Overexpressed in ARPE-19 Cells Transduced with AAV9-AIPL1co
2.7. Genes Overexpressed in ARPE-19 Cells Transduced with AAV9-AIPL1wt
3. Discussion
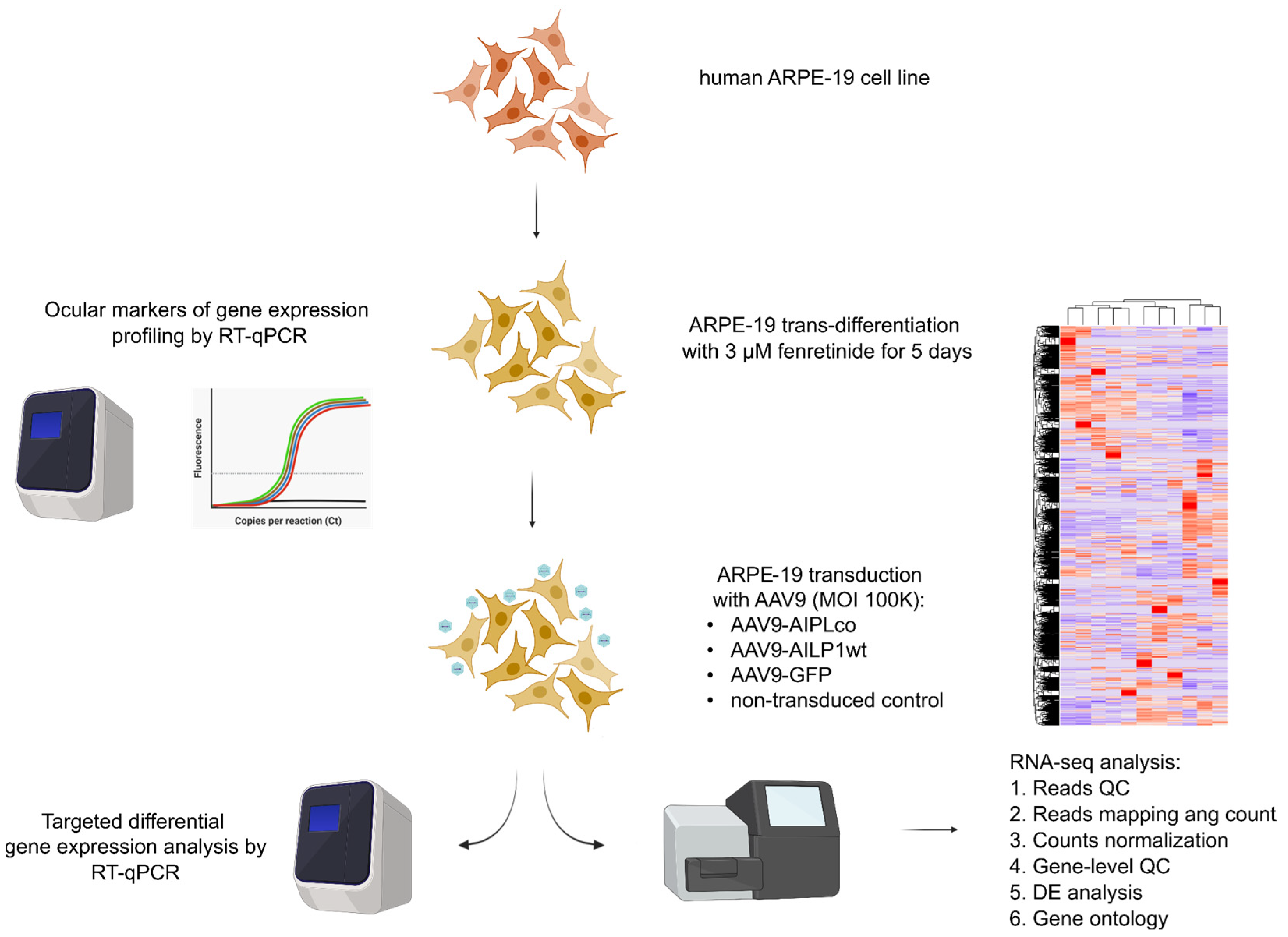
4. Materials and Methods
4.1. Genes and Plasmids
4.2. AAV Production and Quality Control
4.3. RT-qPCR and Gene Expression Analysis
4.4. Western Blot
4.5. ARPE-19 Cells
4.6. ARPE-19 Transduction and RNA Preparation
4.7. Transcriptome Sequencing
4.8. Processing of Sequencing Results
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raimondi, R.; Zollet, P.; De Rosa, F.P.; Tsoutsanis, P.; Stravalaci, M.; Paulis, M.; Inforzato, A.; Romano, M.R. Where Are We with RPE Replacement Therapy? A Translational Review from the Ophthalmologist Perspective. Int. J. Mol. Sci. 2022, 23, 682. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber Congenital Amaurosis: Genes, Proteins and Disease Mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef] [PubMed]
- RetNet—Retinal Information Network. Available online: https://web.sph.uth.edu/RetNet/ (accessed on 17 November 2023).
- Auricchio, F.; Scavone, C.; Cimmaruta, D.; Di Mauro, G.; Capuano, A.; Sportiello, L.; Rafaniello, C. Drugs Approved for the Treatment of Multiple Sclerosis: Review of Their Safety Profile. Expert Opin. Drug Saf. 2017, 16, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Artemyev, N.O. AIPL1: A Specialized Chaperone for the Phototransduction Effector. Cell. Signal. 2017, 40, 183–189. [Google Scholar] [CrossRef]
- Ocugen, Inc. Ocugen Receives Fourth FDA Orphan Drug Designation for the Same Product, OCU400 (AAV-NR2E3) Gene Therapy, for the Treatment of Another Key Inherited Retinal Disease (IRD), PDE6B Gene Mutation-Associated Retinal Diseases. Available online: https://ir.ocugen.com/news-releases/news-release-details/ocugen-receives-fourth-fda-orphan-drug-designation-same-product (accessed on 17 November 2023).
- Chen, S.; Samuel, W.; Fariss, R.N.; Duncan, T.; Kutty, R.K.; Wiggert, B. Differentiation of Human Retinal Pigment Epithelial Cells into Neuronal Phenotype by N-(4-Hydroxyphenyl)Retinamide. J. Neurochem. 2003, 84, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.J.M.; Kuhlmann, E.D.; van Vugt, A.H.M.; Winkens, H.J.; Janssen, B.P.M.; Deutman, A.F.; Driessen, C.A.G.G. Retinoic Acid Delays Transcription of Human Retinal Pigment Neuroepithelium Marker Genes in ARPE-19 Cells. Neuroreport 2000, 11, 1571–1579. [Google Scholar] [CrossRef]
- Carr, A.-J.; Vugler, A.A.; Yu, L.; Semo, M.; Coffey, P.; Moss, S.E.; Greenwood, J. The Expression of Retinal Cell Markers in Human Retinal Pigment Epithelial Cells and Their Augmentation by the Synthetic Retinoid Fenretinide. Mol. Vis. 2011, 17, 1701–1715. [Google Scholar]
- Amemiya, K.; Haruta, M.; Takahashi, M.; Kosaka, M.; Eguchi, G. Adult Human Retinal Pigment Epithelial Cells Capable of Differentiating into Neurons. Biochem. Biophys. Res. Commun. 2004, 316, 1–5. [Google Scholar] [CrossRef]
- Shafei, E.V.; Kurinov, A.M.; Kuznetsova, A.V.; Aleksandrova, M.A. Reprogramming of Human Retinal Pigment Epithelial Cells under the Effect of BFGF In Vitro. Bull. Exp. Biol. Med. 2017, 163, 574–582. [Google Scholar] [CrossRef]
- Yan, R.T.; Ma, W.X.; Wang, S.Z. Neurogenin2 Elicits the Genesis of Retinal Neurons from Cultures of Nonneural Cells. Proc. Natl. Acad. Sci. USA 2001, 98, 15014. [Google Scholar] [CrossRef]
- Rios, M.N.; Marchese, N.A.; Guido, M.E. Expression of Non-Visual Opsins Opn3 and Opn5 in the Developing Inner Retinal Cells of Birds. Light-Responses in Müller Glial Cells. Front. Cell. Neurosci. 2019, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Martins, M.; de Toledo, B.C.; Santos-França, P.L.; Oliveira-Valença, V.M.; Vieira-Vieira, C.H.; Matos-Rodrigues, G.E.; Linden, R.; Norden, C.; Martins, R.A.P.; Silveira, M.S. De Novo Genesis of Retinal Ganglion Cells by Targeted Expression of Klf4 in Vivo. Development 2019, 146, dev176586. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.I.L.; Sghari, S.; Upton, B.A.; Nord, C.; Hahn, M.; Ahlgren, U.; Lang, R.A.; Gunhaga, L. Distinct Opsin 3 (Opn3) Expression in the Developing Nervous System during Mammalian Embryogenesis. eNeuro 2021, 8, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Samuel, W.; Jaworski, C.; Postnikova, O.A.; Kutty, R.K.; Duncan, T.; Tan, L.X.; Poliakov, E.; Lakkaraju, A.; Redmond, T.M. Appropriately Differentiated ARPE-19 Cells Regain Phenotype and Gene Expression Profiles Similar to Those of Native RPE Cells. Mol. Vis. 2017, 23, 60. [Google Scholar] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Phred-Scaled Quality Scores—GATK. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360035531872-Phred-scaled-quality-scores (accessed on 17 November 2023).
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the Length, Weight and GC Content of the Human Genome. BMC Res. Notes 2019, 12, 106. [Google Scholar] [CrossRef]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-Content Normalization for RNA-Seq Data. BMC Bioinformatics 2011, 12, 480. [Google Scholar] [CrossRef]
- Benjamini, Y.; Speed, T.P. Summarizing and Correcting the GC Content Bias in High-Throughput Sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef]
- Versteeg, R.; van Schaik, B.D.C.; van Batenburg, M.F.; Roos, M.; Monajemi, R.; Caron, H.; Bussemaker, H.J.; van Kampen, A.H.C. The Human Transcriptome Map Reveals Extremes in Gene Density, Intron Length, GC Content, and Repeat Pattern for Domains of Highly and Weakly Expressed Genes. Genome Res. 2003, 13, 1998. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, J.; White, K.P. RNA-Seq Differential Expression Studies: More Sequence or More Replication? Bioinformatics 2014, 30, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Package “Pheatmap.”. 2022. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 17 November 2023).
- Koch, C.M.; Chiu, S.F.; Akbarpour, M.; Bharat, A.; Ridge, K.M.; Bartom, E.T.; Winter, D.R. A Beginner’s Guide to Analysis of RNA Sequencing Data. Am. J. Respir. Cell Mol. Biol. 2018, 59, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.M.; Skoumal, R.; Komka, Z.; Tóth, M.; Kerkelä, R.; Georgiadi, A.; Kersten, S.; Hakkola, J. Energy-Sensing Factors Coactivator Peroxisome Proliferator-Activated Receptor γ Coactivator 1-α (PGC-1α) and AMP-Activated Protein Kinase Control Expression of Inflammatory Mediators in Liver: Induction of Interleukin 1 Receptor Antagonist. J. Biol. Chem. 2012, 287, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, N.; Nicklin, M.J.; Di Giovine, F.S. Long-Range DNA Interactions at the IL-1/IL-36/IL-37 Gene Cluster (2q13) Are Induced by Activation of Monocytes. Cytokine 2014, 68, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, N.; Horvath, C.M. Histone H2A.Z Suppression of Interferon-Stimulated Transcription and Antiviral Immunity Is Modulated by GCN5 and BRD2. iScience 2018, 6, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.C.; Aotaki-Keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, a Human Retinal Pigment Epithelial Cell Line with Differentiated Properties. Exp. Eye Res. 1996, 62, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Behar-Cohen, F.; Gelizé, E.; Jonet, L.; Patricia, L. Anatomie de La Rétine. médecine/sciences 2020, 36, 594–599. [Google Scholar] [CrossRef]
- Jamieson, R.V.; Munier, F.; Balmer, A.; Farrar, N.; Perveen, R. Pulverulent Cataract with Variably Associated Microcornea and Iris Coloboma in a MAF Mutation Family. Br. J. Ophthalmol. 2003, 87, 411–412. [Google Scholar] [CrossRef]
- Jamieson, R.V.; Perveen, R.; Kerr, B.; Carette, M.; Yardley, J.; Heon, E.; Wirth, M.G.; Van Heyningen, V.; Donnai, D.; Munier, F.; et al. Domain Disruption and Mutation of the BZIP Transcription Factor, MAF, Associated with Cataract, Ocular Anterior Segment Dysgenesis and Coloboma. Hum. Mol. Genet. 2002, 11, 33–42. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, T.; Zeng, S.; Zhang, X.; Zhou, F.; Gillies, M.C.; Longoni, B.; Zhang, J.; Zhang, T.; Zeng, S.; et al. The Role of Nrf2/SMAF Signalling in Retina Ageing and Retinal Diseases. Biomedicines 2023, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Ksander, B.R.; Kolovou, P.E.; Wilson, B.J.; Saab, K.R.; Guo, Q.; Ma, J.; McGuire, S.P.; Gregory, M.S.; Vincent, W.J.B.; Perez, V.L.; et al. ABCB5 Is a Limbal Stem Cell Gene Required for Corneal Development and Repair. Nature 2014, 511, 353. [Google Scholar] [CrossRef] [PubMed]
- Norrick, A.; Esterlechner, J.; Niebergall-Roth, E.; Dehio, U.; Sadeghi, S.; Schröder, H.M.; Ballikaya, S.; Stemler, N.; Ganss, C.; Dieter, K.; et al. Process Development and Safety Evaluation of ABCB5+ Limbal Stem Cells as Advanced-Therapy Medicinal Product to Treat Limbal Stem Cell Deficiency. Stem Cell Res. Ther. 2021, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; May, N.R.; Fan, C.M. Growth Arrest Specific Gene 1 Is a Positive Growth Regulator for the Cerebellum. Dev. Biol. 2001, 236, 30–45. [Google Scholar] [CrossRef]
- Coutelle, O.; Blagden, C.S.; Hampson, R.; Halai, C.; Rigby, P.W.J.; Hughes, S.M. Transdifferentiation of the Ventral Retinal Pigmented Epithelium to Neural Retina in the Growth Arrest Specific Gene 1 Mutant. Dev. Biol. 2001, 236, 17–29. [Google Scholar] [CrossRef]
- Shinde, V.; Hu, N.; Mahale, A.; Maiti, G.; Daoud, Y.; Eberhart, C.G.; Maktabi, A.; Jun, A.S.; Al-Swailem, S.A.; Chakravarti, S. RNA Sequencing of Corneas from Two Keratoconus Patient Groups Identifies Potential Biomarkers and Decreased NRF2-Antioxidant Responses. Sci. Rep. 2020, 10, 9907. [Google Scholar] [CrossRef]
- Chang, H.Y.P.; Chodosh, J. The Genetics of Keratoconus. Semin. Ophthalmol. 2013, 28, 275–280. [Google Scholar] [CrossRef]
- Mathur, P.D.; Yang, J. Usher Syndrome and Non-Syndromic Deafness: Functions of Different Whirlin Isoforms in the Cochlea, Vestibular Organs, and Retina. Hear. Res. 2019, 375, 14–24. [Google Scholar] [CrossRef]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e12. [Google Scholar] [CrossRef]
- Tsakiri, N.; Kimber, I.; Rothwell, N.J.; Pinteaux, E. Interleukin-1-Induced Interleukin-6 Synthesis Is Mediated by the Neutral Sphingomyelinase/Src Kinase Pathway in Neurones. Br. J. Pharmacol. 2008, 153, 775–783. [Google Scholar] [CrossRef]
- Dripps, D.J.; Brandhuber, B.J.; Thompson, R.C.; Eisenberg, S.P. Interleukin-1 (IL-1) Receptor Antagonist Binds to the 80-KDa IL-1 Receptor but Does Not Initiate IL-1 Signal Transduction. J. Biol. Chem. 1991, 266, 10331–10336. [Google Scholar] [CrossRef]
- Tahtinen, S.; Tong, A.J.; Himmels, P.; Oh, J.; Paler-Martinez, A.; Kim, L.; Wichner, S.; Oei, Y.; McCarron, M.J.; Freund, E.C.; et al. IL-1 and IL-1ra Are Key Regulators of the Inflammatory Response to RNA Vaccines. Nat. Immunol. 2022, 23, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Barber, K.D.; Yan, B. Clash of the Cytokine Titans: Counter-Regulation of Interleukin-1 and Type I Interferon-Mediated Inflammatory Responses. Cell. Mol. Immunol. 2016, 14, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Gander-Bui, H.T.T.; Schläfli, J.; Baumgartner, J.; Walthert, S.; Genitsch, V.; van Geest, G.; Galván, J.A.; Cardozo, C.; Graham Martinez, C.; Grans, M.; et al. Targeted Removal of Macrophage-Secreted Interleukin-1 Receptor Antagonist Protects against Lethal Candida Albicans Sepsis. Immunity 2023, 56, 1743–1760.e9. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.H.; Kim, H.; He, Q.; Baek, H.J.; Yang, D.; Chen, L.Y.; Liang, J.; Chae, H.K.; Safari, A.; Liu, D.; et al. Genome-Wide YFP Fluorescence Complementation Screen Identifies New Regulators for Telomere Signaling in Human Cells. Mol. Cell. Proteom. 2011, 10, 1–11. [Google Scholar] [CrossRef]
- Prado, F.; Jimeno-González, S.; Reyes, J.C. Histone Availability as a Strategy to Control Gene Expression. RNA Biol. 2017, 14, 281–286. [Google Scholar] [CrossRef]
- Kassem, S.; Ferrari, P.; Hughes, A.L.; Soudet, J.; Rando, O.J.; Strubin, M. Histone Exchange Is Associated with Activator Function at Transcribed Promoters and with Repression at Histone Loci. Sci. Adv. 2020, 6, eabb0333. [Google Scholar] [CrossRef]
- Zhao, H.; Boije, H.; Granberg, F.; Pettersson, U.; Svensson, C. Activation of the Interferon-Induced STAT Pathway during an Adenovirus Type 12 Infection. Virology 2009, 392, 186–195. [Google Scholar] [CrossRef]
- Xie, R.; Van Wijnen, A.J.; Van Der Meijden, C.; Luong, M.X.; Stein, J.L.; Stein, G.S. The Cell Cycle Control Element of Histone H4 Gene Transcription Is Maximally Responsive to Interferon Regulatory Factor Pairs IRF-1/IRF-3 and IRF-1/IRF-7. J. Biol. Chem. 2001, 276, 18624–18632. [Google Scholar] [CrossRef]
- Izquierdo-Bouldstridge, A.; Bustillos, A.; Bonet-Costa, C.; Aribau-Miralbés, P.; García-Gomis, D.; Dabad, M.; Esteve-Codina, A.; Pascual-Reguant, L.; Peiró, S.; Esteller, M.; et al. Histone H1 Depletion Triggers an Interferon Response in Cancer Cells via Activation of Heterochromatic Repeats. Nucleic Acids Res. 2017, 45, 11622–11642. [Google Scholar] [CrossRef]
- Kari, V.; Karpiuk, O.; Tieg, B.; Kriegs, M.; Dikomey, E.; Krebber, H.; Begus-Nahrmann, Y.; Johnsen, S.A. A Subset of Histone H2B Genes Produces Polyadenylated MRNAs under a Variety of Cellular Conditions. PLoS ONE 2013, 8, e63745. [Google Scholar] [CrossRef]
- Vaughan, P.S.; Aziz, F.; van Wijnen, A.J.; Wu, S.; Harada, H.; Taniguchi, T.; Soprano, K.J.; Stein, J.L.; Stein, G.S. Activation of a Cell-Cycle-Regulated Histone Gene by the Oncogenic Transcription Factor IRF-2. Nature 1995, 377, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.L.; Park, E.N.; Vachon, V.K.; Danzy, S.; Lowen, A.C.; Conn, G.L. Human OAS1 Activation Is Highly Dependent on Both RNA Sequence and Context of Activating RNA Motifs. Nucleic Acids Res. 2020, 48, 7520–7531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jha, B.K.; Ogden, K.M.; Dong, B.; Zhao, L.; Elliott, R.; Patton, J.T.; Silverman, R.H.; Weiss, S.R. Homologous 2′,5′-Phosphodiesterases from Disparate RNA Viruses Antagonize Antiviral Innate Immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 13114–13119. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Earley, L.F.; Chai, Z.; Chen, X.; Sun, J.; He, T.; Deng, M.; Hirsch, M.L.; Ting, J.; Saailpmulski, R.J.; et al. Double-Stranded RNA Innate Immune Response Activation from Long-Term Adeno-Associated Virus Vector Transduction. JCI Insight 2018, 3, e120474. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and Activity of Histone in Diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, Y.; Peng, K.; Zeng, Z.; Chen, L.; Zeng, Y. Histones: The Critical Players in Innate Immunity. Front. Immunol. 2022, 13, 1030610. [Google Scholar] [CrossRef]
- Zhao, H.; Raines, L.N.; Huang, S.C.C. Molecular Chaperones: Molecular Assembly Line Brings Metabolism and Immunity in Shape. Metabolites 2020, 10, 394. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Beltran, W.A.; Sumaroka, A.; Swider, M.; Iwabe, S.; Roman, A.J.; Olivares, M.B.; Schwartz, S.B.; Komáromy, A.M.; et al. Human Retinal Gene Therapy for Leber Congenital Amaurosis Shows Advancing Retinal Degeneration despite Enduring Visual Improvement. Proc. Natl. Acad. Sci. USA 2013, 110, E517–E525. [Google Scholar] [CrossRef]
- Gcua—Graphical Codon Usage Analyser. Available online: https://gcua.schoedl.de/ (accessed on 17 November 2023).
- Lemma, S.; Avnet, S.; Meade, M.J.; Chano, T.; Baldini, N. Validation of Suitable Housekeeping Genes for the Normalization of MRNA Expression for Studying Tumor Acidosis. Int. J. Mol. Sci. 2018, 19, 2930. [Google Scholar] [CrossRef] [PubMed]
- PureLink TM RNA Micro Kit For Purification of Total RNA from Variety of Samples in Low Elution Volume Version B 19 December 2008 100003660 Invitrogen Corporation. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/purelink_rna_micro_kit_man.pdf (accessed on 17 November 2023).
- Agilent. RNA Electrophoresis, RNA Integrity, Bioanalyzer RIN. Available online: https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-rna-kits-reagents/bioanalyzer-rna-analysis-228256 (accessed on 17 November 2023).
- Instruction Manual NEBNext® Ultra TM II RNA Library Prep Kit for Illumina®. Version 3.1_5/20. Available online: https://www.neb.com/en/-/media/nebus/files/manuals/manuale7770.pdf (accessed on 17 November 2023).
- Agilent Technologies Agilent High Sensitivity DNA Kit Guide Agilent High Sensitivity DNA. Document Number SD-UF0000034 Rev. D 02/2020. Available online: https://www.agilent.com/cs/library/usermanuals/public/HighSensitivity_DNA_KG.pdf.pdf (accessed on 17 November 2023).
- Illumina HiSeq Systems Denature and Dilute Libraries Guide. Available online: www.illumina.com/company/legal.html (accessed on 17 November 2023).
- Anaconda.Org. Available online: https://anaconda.org/ (accessed on 17 November 2023).
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 November 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- RStudio Desktop—Posit. Available online: https://posit.co/download/rstudio-desktop/ (accessed on 17 November 2023).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 17 November 2023).
- ClusterProfiler: Statistical Analysis and Visualization of Functional Profiles for Genes and Gene Clusters Version 3.18.1 from Bioconductor. Available online: https://rdrr.io/bioc/clusterProfiler/ (accessed on 17 November 2023).
- SpringerLink. Ggplot2: Elegant Graphics for Data Analysis. Available online: https://link.springer.com/book/10.1007/978-3-319-24277-4 (accessed on 17 November 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Number of Upregulated DEGs | Number of Downregulated DEGs | Summary Number of DEGs |
|---|---|---|---|
| AAV9-AIPL1co vs. non-transduced control | 169 | 81 | 250 |
| AAV9-AIPL1wt vs. non-transduced control | 179 | 348 | 527 |
| AAV9-AIPL1co vs. AAV9-GFP | 87 | 22 | 109 |
| AAV9-AIPL1wt vs. AAV9-GFP | 110 | 274 | 384 |
| AAV9-AIPL1co vs. AAV9-AIPL1wt | 129 | 59 | 188 |
| AAV9-GFP vs. non-transduced control | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galieva, A.; Egorov, A.; Malogolovkin, A.; Brovin, A.; Karabelsky, A. RNA-Seq Analysis of Trans-Differentiated ARPE-19 Cells Transduced by AAV9-AIPL1 Vectors. Int. J. Mol. Sci. 2024, 25, 197. https://doi.org/10.3390/ijms25010197
Galieva A, Egorov A, Malogolovkin A, Brovin A, Karabelsky A. RNA-Seq Analysis of Trans-Differentiated ARPE-19 Cells Transduced by AAV9-AIPL1 Vectors. International Journal of Molecular Sciences. 2024; 25(1):197. https://doi.org/10.3390/ijms25010197
Chicago/Turabian StyleGalieva, Alima, Alexander Egorov, Alexander Malogolovkin, Andrew Brovin, and Alexander Karabelsky. 2024. "RNA-Seq Analysis of Trans-Differentiated ARPE-19 Cells Transduced by AAV9-AIPL1 Vectors" International Journal of Molecular Sciences 25, no. 1: 197. https://doi.org/10.3390/ijms25010197
APA StyleGalieva, A., Egorov, A., Malogolovkin, A., Brovin, A., & Karabelsky, A. (2024). RNA-Seq Analysis of Trans-Differentiated ARPE-19 Cells Transduced by AAV9-AIPL1 Vectors. International Journal of Molecular Sciences, 25(1), 197. https://doi.org/10.3390/ijms25010197