

The Implication of a Polymorphism in the Methylenetetrahydrofolate Reductase Gene in Homocysteine Metabolism and Related Civilisation Diseases


{kind=link}
{kind=link}
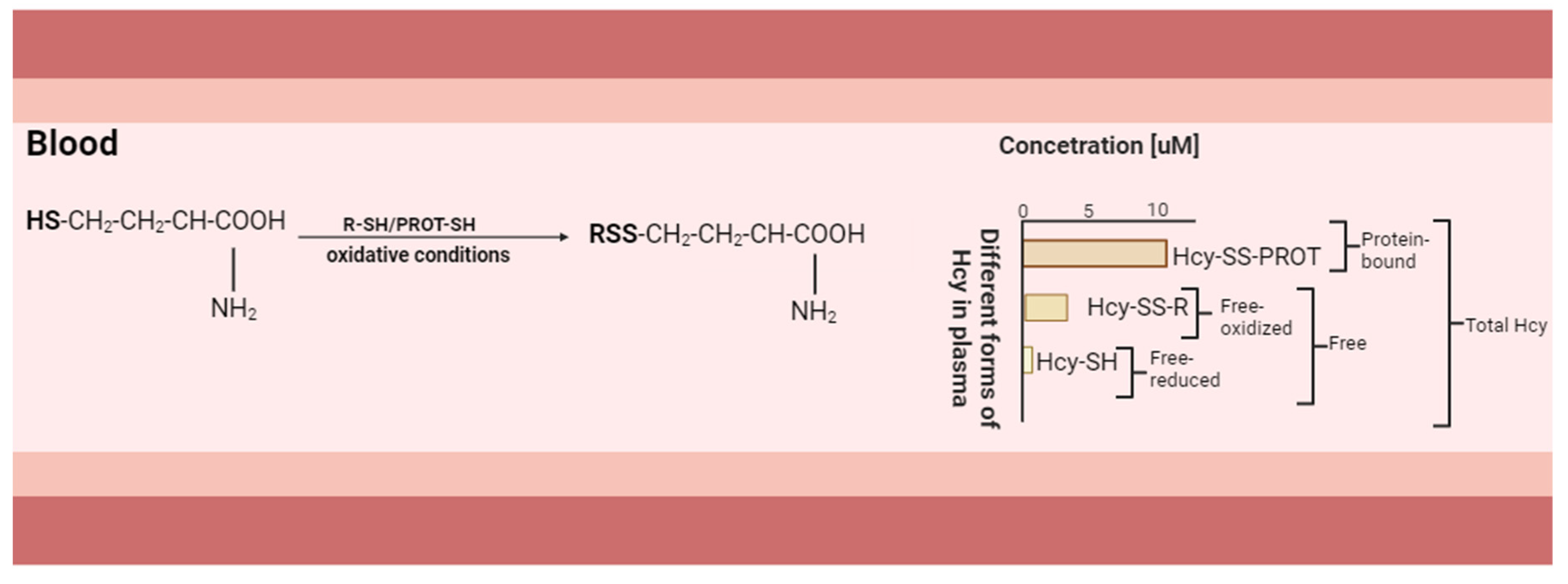{kind=link}
{kind=link}
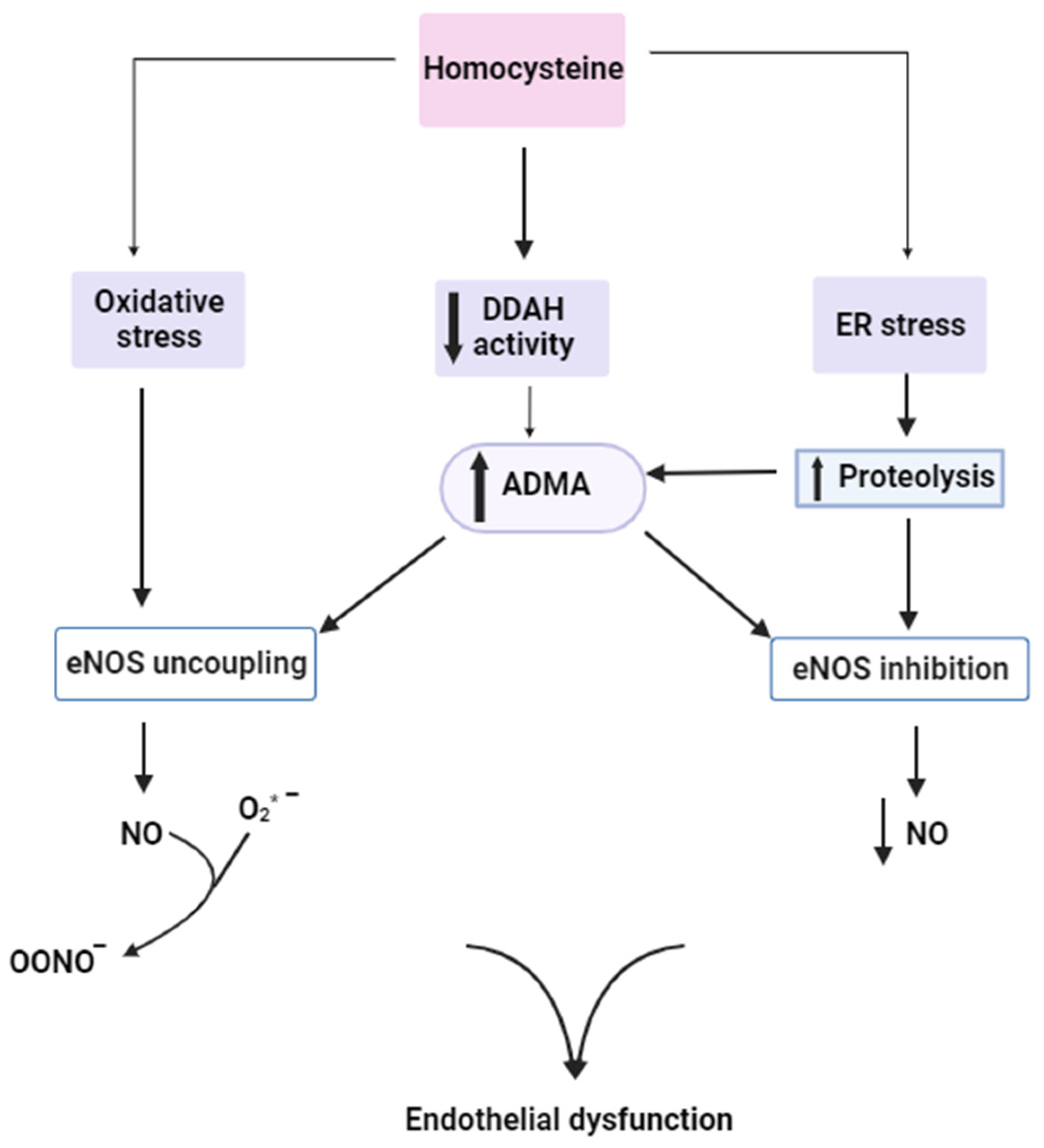{kind=link}
Abstract
1. Methylenetetrahydrofolate Reductase (MTHFR)
2. MTHFR Polymorphism
3. Regulation of MTHFR Activity
4. Folate, Vitamin B12, and Homocysteine Metabolism
5. MTHFR Gene Variants with Hyperhomocysteinemia and Cardiovascular Diseases
6. The Association of Variants of the MTHFR Gene with Obesity and Accompanying Disorders of Lipid and Carbohydrate Metabolism
7. Polymorphism of MTHFR and Its Relationship with Hyperhomocysteinemia, Oxidative Stress, and Lipoprotein Modification
8. Epigenetic Modifications and the C677T Polymorphism of the MTHFR Gene
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Center for Biotechnology Information (NCBI). 2023. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 8 October 2023).
- Homberger, A.; Linnebank, M.; Winter, C.; Willenbring, H.; Marquardt, T.; Harms, E.; Koch, H.G. Genomic structure and transcript variants of the human methylenertolate reductase gene. Eur. J. Hum. Genet. 2000, 8, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Goyette, P.; Pai, A.; Milos, R.; Frosst, P.; Tran, P.; Chen, Z.; Chan, M.; Rozen, R. Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR). Mamm. Genome 1998, 9, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, D.J.; Barbaux, S.; Kluijtmans, L.A.; Whitehead, A.S. The human and mouse methylenetetrahydrofolate reductase (MTHFR) genes: Genomic organization, mRNA structure and linkage to the CLCN6 gene. Gene 2000, 257, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Leclerc, D.; Chan, M.; Pai, A.; Hiou-Tim, F.; Wu, Q.; Goyette, P.; Artigas, C.; Milos, R.; Rozen, R. Multiple transcription start sites and alternative splicing in the methylenetetrahydrofolate reductase gene result in two enzyme isoforms. Mamm. Genome 2002, 13, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Swanson, D.A.; Liu, M.L.; Baker, P.J.; Garrett, L.; Stitzel, M.; Wu, J.; Harris, M.; Banerjee, R.; Shane, B.; Brody, L.C. Targeted disruption of the methionine synthase gene in mice. Mol. Cell. Biol. 2001, 21, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef]
- Mudd, S.H.; Uhlendorf, B.W.; Freeman, J.M.; Finkelstein, J.D.; Shih, V.E. Homocystinuria associated with decreased methylenetetrahydrofolate reductase activity. Biochem. Biophys. Res. Commun. 1972, 46, 905–912. [Google Scholar] [CrossRef]
- Kang, S.S.; Zhou, J.; Wong, P.W.; Kowalisyn, J.; Strokosch, G. Intermediate homocysteinemia: A thermolabile variant of methylenetetrahydrofolate reductase. Am. J. Hum. Genet. 1988, 43, 414–421. [Google Scholar] [PubMed]
- Kang, S.S.; Wong, P.W.; Bock, H.G.; Horwitz, A.; Grix, A. Intermediate hyperhomocysteinemia resulting from compound heterozygosity of methylenetetrahydrofolate reductase mutations. Am. J. Hum. Genet. 1991, 48, 546–551. [Google Scholar] [PubMed]
- Raza, S.T.; Abbas, S.; Ahmed, F.; Fatima, J.; Zaidi, Z.H.; Mahdi, F. Association of MTHFR and PPARγ2 gene polymorphisms in relation to type 2 diabetes mellitus cases among north Indian population. Gene 2012, 511, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Nefic, H.; Mackic-Djurovic, M.; Eminovic, I. The Frequency of the 677C>T and 1298A>C Polymorphisms in the Methylenetetrahydrofolate Reductase (MTHFR) Gene in the Population. Med. Arch. 2018, 72, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.; Kumar, P.; Rai, V. Global prevalence of MTHFR C677T gene polymorphism: A meta-analysis of population based studies. Indian J. Clin. Biochem. 2014, 29, 123. [Google Scholar]
- van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G.; Daubner, S.C. Modulation of methylenetetrahydrofolate reductase activity by S-adenosylmethionine and by dihydrofolate and its polyglutamate analogues. Adv. Enzym. Regul. 1982, 20, 123–131. [Google Scholar] [CrossRef][Green Version]
- Sumner, J.; Jencks, D.A.; Khani, S.; Matthews, R.G. Photoaffinity labeling of methylenetetrahydrofolate reductase with 8-azido-S-adenosylmethionine. J. Biol. Chem. 1986, 261, 7697–7700. [Google Scholar] [CrossRef]
- Yamada, K.; Chen, Z.; Rozen, R.; Matthews, R.G. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad. Sci. USA 2001, 98, 14853–14858. [Google Scholar] [CrossRef]
- Froese, D.S.; Kopec, J.; Rembeza, E.; Bezerra, G.A.; Oberholzer, A.E.; Suormala, T.; Lutz, S.; Chalk, R.; Borkowska, O.; Baumgartner, M.R.; et al. Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat. Commun. 2018, 9, 2261. [Google Scholar] [CrossRef]
- Yamada, K.; Strahler, J.R.; Andrews, P.C.; Matthews, R.G. Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 10454–10459. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ramsamooj, S.; Li, Q.; Johnson, J.L.; Yaron, T.M.; Sharra, K.; Cantley, L.C. Regulation of folate and methionine metabolism by multisite phosphorylation of human methylenetetrahydrofolate reductase. Sci. Rep. 2019, 9, 4190. [Google Scholar] [CrossRef] [PubMed]
- Patanwala, I.; King, M.J.; Barrett, D.A.; Rose, J.; Jackson, R.; Hudson, M.; Philo, M.; Dainty, J.R.; Wright, A.J.; Finglas, P.M.; et al. Folic acid handling by the human gut: Implications for food fortification and supplementation. Am. J. Clin. Nutr. 2014, 100, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.C. Folic acid and diseases—Supplement it or not? Rev. Assoc. Med. Bras. 2016, 62, 90–100. [Google Scholar] [CrossRef]
- EFSA. Panel on Dietetic Products, Nutrition and Allergies, Scientific Opinion on Dietary Reference Values for folate. EFSA J. 2014, 12, 3893. [Google Scholar]
- Palchetti, C.Z.; Paniz, C.; de Carli, E.; Marchioni, D.M.; Colli, C.; Steluti, J.; Pfeiffer, C.M.; Fazili, Z.; Guerra-Shinohara, E.M. Association between Serum Unmetabolized Folic Acid Concentrations and Folic Acid from Fortified Foods. J. Am. Coll. Nutr. 2017, 36, 572–578. [Google Scholar] [CrossRef]
- Tsang, B.L.; Devine, O.J.; Cordero, A.M.; Marchetta, C.M.; Mulinare, J.; Mersereau, P.; Guo, J.; Qi, Y.P.; Berry, R.J.; Rosenthal, J.; et al. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: A systematic review and meta-analysis of trials and observational studies. Am. J. Clin. Nutr. 2015, 101, 1286–1294. [Google Scholar] [CrossRef]
- Nishio, K.; Goto, Y.; Kondo, T.; Ito, S.; Ishida, Y.; Kawai, S.; Naito, M.; Wakai, K.; Hamajima, N. Serum folate and methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism adjusted for folate intake. J. Epidemiol. 2008, 18, 125–131. [Google Scholar] [CrossRef]
- Siri, P.W.; Verhoef, P.; Kok, F.J. Vitamins B6, B12, and folate: Association with plasma total homocysteine and risk of coronary atherosclerosis. J. Am. Coll. Nutr. 1998, 17, 435–441. [Google Scholar] [CrossRef]
- Bagley, P.J.; Selhub, J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc. Natl. Acad. Sci. USA 1998, 95, 13217–13220. [Google Scholar] [CrossRef]
- Hiraoka, M.; Kagawa, Y. Genetic polymorphisms and folate status. Congenit. Anom. 2017, 57, 142–149. [Google Scholar] [CrossRef]
- Lucock, M. Is folic acid the ultimate functional food component for disease prevention? Br. Med. J. 2004, 328, 211–214. [Google Scholar] [CrossRef]
- Hum, D.W.; MacKenzie, R.E. Expression of active domains of a human folate-dependent trifunctional enzyme in Escherichia coli. Protein Eng. 1991, 4, 493–500. [Google Scholar] [CrossRef]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef]
- Bailey, L.B.; Gregory, J.F., III. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: Metabolic significance, risks and impact on folate requirement. J. Nutr. 1999, 129, 919–922. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. Chapter 2—Antimetabolites. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Ankar, A.; Kumar, A. Vitamin B12 Deficiency; Updated 22 October 2022; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Baltaci, D.; Kutlucan, A.; Turker, Y.; Yilmaz, A.; Karacam, S.; Deler, H.; Ucgun, T.; Kara, J.H. Association of vitamin B12 with obesity, overweight, insulin resistance and metabolic syndrome, and body fat composition; primary care-based study. Med. Glas. 2013, 10, 203–210. [Google Scholar]
- Baltaci, D.; Kutlucan, A.; Öztürk, Ş.; Karabulut, I.; Yildirim, H.; Celer, A.; Celbek, G.; Kara, J. Evaluation of vitamin B12 level in middle-aged obese women with metabolic and nonmetabolic syndrome: Case-control study. Turk. J. Med. Sci. 2012, 42, 802–809. [Google Scholar]
- Alemán, G.; Tovar, A.R.; Torres, N. [Homocysteine metabolism and risk of cardiovascular diseases: Importance of the nutritional status on folic acid, vitamins B6 and B12]. Rev. Investig. Clin. 2001, 53, 141–151. [Google Scholar]
- Malinow, M.R. Hyperhomocyst(e)inemia. A common and easily reversible risk factor for occlusive atherosclerosis. Circulation 1990, 81, 2004–2006. [Google Scholar] [CrossRef]
- Baszczuk, A.; Kopczyński, Z. Hyperhomocysteinemia in patients with cardiovascular disease. Adv. Hyg. Exp. Med. 2014, 68, 579–589. [Google Scholar] [CrossRef]
- Tavakkoly Bazzaz, J.; Shojapoor, M.; Nazem, H.; Amiri, P.; Fakhrzadeh, H.; Heshmat, R.; Parvizi, M.; Hasani Ranjbar, S.; Amoli, M.M. Methylenetetrahydrofolate reductase gene polymorphism in diabetes and obesity. Mol. Biol. Rep. 2010, 37, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Upchurch, G.R., Jr.; Welch, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F., Jr.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017. [Google Scholar] [CrossRef] [PubMed]
- Gellekink, H.; den Heijer, M.; Heil, S.G.; Blom, H.J. Genetic determinants of plasma total homocysteine. Semin. Vasc. Med. 2005, 5, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Poddar, R.; Tipa, E.V.; Dibello, P.M.; Moravec, C.D.; Robinson, K.; Green, R.; Kruger, W.D.; Garrow, T.A.; Jacobsen, D.W. Homocysteine metabolism in cardiovascular cells and tissues: Implications for hyperhomocysteinemia and cardiovascular disease. Adv. Enzyme Regul. 1999, 39, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, A.; Houston, D.S. Induction of monocyte tissue factor expression by homocysteine: A possible mechanism for thrombosis. Blood 2000, 96, 966–972. [Google Scholar] [CrossRef]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hara, H.; Adachi, T. Effects of homocysteine on the binding of extracellular-superoxide dismutase to the endothelial cell surface. FEBS Lett. 2000, 486, 159–162. [Google Scholar] [CrossRef]
- Chang, L.; Geng, B.; Yu, F.; Zhao, J.; Jiang, H.; Du, J.; Tang, C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 2008, 34, 573–585. [Google Scholar] [CrossRef]
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.A.; de Assis, A.M.; Wyse, A.T. 1,25-Dihydroxyvitamin D(3) prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Nutr. Res. 2017, 38, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Navneet, S.; Cui, X.; Zhao, J.; Wang, J.; Kaidery, N.A.; Thomas, B.; Bollinger, K.E.; Yoon, Y.; Smith, S.B. Excess homocysteine upregulates the NRF2-antioxidant pathway in retinal Müller glial cells. Exp. Eye Res. 2019, 178, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Sibrian-Vazquez, M.; Escobedo, J.O.; Lim, S.; Samoei, G.K.; Strongin, R.M. Homocystamides promote free-radical and oxidative damage to proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Kredan, M.B.; Moat, S.J.; Hussain, S.A.; Powell, C.A.; Bellamy, M.F.; Powers, H.J.; Lewis, M.J. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: Role for superoxide anions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Finkelstein, J.D.; Refsum, H.; Ueland, P.M.; Malinow, M.R.; Lentz, S.R.; Jacobsen, D.W.; Brattström, L.; Wilcken, B.; Wilcken, D.E.; et al. Homocysteine and its disulfide derivatives: A suggested consensus terminology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1704–1706. [Google Scholar] [CrossRef]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef]
- Sacco, R.L.; Adams, R.; Albers, G.; Alberts, M.J.; Benavente, O.; Furie, K.; Goldstein, L.B.; Gorelick, P.; Halperin, J.; Harbaugh, R.; et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: Co-sponsored by the Council on Cardiovascular Radiology and Intervention: The American Academy of Neurology affirms the value of this guideline. Circulation 2006, 113, e409–e449. [Google Scholar]
- Naruszewicz, M. Homocysteina jako czynnik ryzyka chorób cywilizacyjnych; w jakich przypadkach konieczne jest jej oznaczanie? Chor. Serca I Naczyń 2008, 5, 156–158. [Google Scholar]
- Seo, H.; Oh, H.; Park, H.; Park, M.; Jang, Y.; Lee, M. Contribution of dietary intakes of antioxidants to homocysteine-induced low density lipoprotein (LDL) oxidation in atherosclerotic patients. Yonsei Med. J. 2010, 51, 526–533. [Google Scholar] [CrossRef]
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J. Clin. Investig. 1991, 88, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, M.; Shimada, K. Heparan sulfate proteoglycan of endothelial cells: Homocysteine suppresses anticoagulant active heparan sulfate in cultured endothelial cells. Rinsho Byori 1994, 42, 340–345. [Google Scholar] [PubMed]
- Hajjar, K.A. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J. Clin. Investig. 1993, 91, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, L.; Li, H.; Chen, G.; Qi, G.; Ma, X.; Jin, Y. Role of homocysteine in the development and progression of Parkinson’s disease. Ann. Clin. Transl. Neurol. 2020, 7, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Buysschaert, M.; Dramais, A.S.; Wallemacq, P.E.; Hermans, M.P. Hyperhomocysteinemia in type 2 diabetes: Relationship to macroangiopathy, nephropathy, and insulin resistance. Diabetes Care 2000, 23, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, N. Relationship between paraoxonase and homocysteine: Crossroads of oxidative diseases. Arch. ed. Sci. 2012, 8, 138–153. [Google Scholar]
- Jakubowski, H.; Goldman, E. Synthesis of homocysteine thiolactone by methionYL-TRNA synthetase in cultured mammalian cells. FEBS Lett. 1993, 317, 237–240. [Google Scholar] [CrossRef]
- Silla, Y.; Varshney, S.; Ray, A.; Basak, T.; Zinellu, A.; Sabareesh, V.; Carru, C.; Sengupta, S. Hydrolysis of homocysteine thiolactone results in the formation of Protein-Cys-S-S-homocysteinylation. Proteins 2019, 87, 625–634. [Google Scholar] [CrossRef]
- Exner, M.; Hermann, M.; Hofbauer, R.; Hartmann, B.; Kapiotis, S.; Gmeiner, B. Homocysteine promotes the LDL oxidase activity of ceruloplasmin. FEBS Lett. 2002, 531, 402–406. [Google Scholar] [CrossRef]
- Sengupta, S.; Wehbe, C.; Majors, A.K.; Ketterer, M.E.; DiBello, P.M.; Jacobsenet, D.W. Relative roles of albumin and ceruloplasmin in the formation of homocystine, homocysteine-cysteine-mixed disulfide, and cystine in circulation. J. Biol. Chem. 2001, 276, 46896–46904. [Google Scholar] [CrossRef] [PubMed]
- Capasso, R.; Sambri, I.; Cimmino, A.; Salemme, S.; Lombardi, C.; Acanfora, F.; Satta, E.; Puppione, D.L.; Perna, A.F.; Ingrosso, D. Homocysteinylated albumin promotes increased monocyte-endothelial cell adhesion and up-regulation of MCP1, Hsp60 and ADAM17. PLoS ONE 2012, 7, e31388. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. Nitric oxide: Discovery and impact on clinical medicine. J. R. Soc. Med. 1999, 92, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Arlouskaya, Y.; Sawicka, A.; Głowala, M.; Giebułtowicz, J.; Korytowska, N.; Tałałaj, M.; Nowicka, G.; Wrzosek, M. Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA) Concentrations in Patients with Obesity and the Risk of Obstructive Sleep Apnea (OSA). J. Clin. Med. 2019, 8, 897. [Google Scholar] [CrossRef]
- Matté, C.; Mackedanz, V.; Stefanello, F.M.; Scherer, E.B.; Andreazza, A.C.; Zanotto, C.; Moro, A.M.; Garcia, S.C.; Gonçalves, C.A.; Erdtmann, B.; et al. Chronic hyperhomocysteinemia alters antioxidant defenses and increases DNA damage in brain and blood of rats: Protective effect of folic acid. Neurochem. Int. 2009, 54, 7–13. [Google Scholar] [CrossRef]
- Li, Q.; Lancaster, J.R., Jr. A Conspectus of Cellular Mechanisms of Nitrosothiol Formation from Nitric Oxide. For. Immunopathol. Dis. Ther. 2012, 3, 183–191. [Google Scholar] [CrossRef]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: A meta-analysis. J. Am. Med. Assoc. 2002, 288, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Bostom, A.G.; Williams, R.R.; Ellison, R.C.; Eckfeldt, J.H.; Rosenberg, L.H.; Selhub, J.; Rozen, R. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation 1996, 93, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Li, Y.; Zhang, Z.; Sun, Z.; He, Y.; Li, R. Methylenetetrahydrofolate reductase and psychiatric diseases. Transl. Psychiatry 2018, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; De Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. Heart disease and stroke statistics—2010 update: A report from the American Heart Association. Circulation 2010, 121, e46–e215. [Google Scholar] [PubMed]
- Xuan, C.; Bai, X.Y.; Gao, G.; Yang, Q.; He, G.W. Association between polymorphism of methylenetetrahydrofolate reductase (MTHFR) C677T and risk of myocardial infarction: A meta-analysis for 8,140 cases and 10,522 controls. Arch. Med. Res. 2011, 42, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, Y.; Wu, S.; Deng, X.; Li, J.; Ning, N.; Hou, X. Meta-analysis of association between MTHFR C677T polymorphism and risk of myocardial infarction: Evidence from forty-four case-control studies. Int. J. Clin. Exp. Med. 2016, 9, 5630–5642. [Google Scholar]
- Dalen, J.E.; Alpert, J.S.; Goldberg, R.J.; Weinstein, R.S. The epidemic of the 20(th) century: Coronary heart disease. Am. J. Med. 2014, 127, 807–812. [Google Scholar] [CrossRef]
- Shan, J.G.; Xue, S. MTHFR C677T polymorphism and coronary artery disease risk in the Chinese population: A meta-analysis based on 33 studies. Int. J. Clin. Exp. Med. 2016, 9, 2822–2830. [Google Scholar]
- Hao, L.; Ma, J.; Stampfer, M.J.; Ren, A.; Tian, Y.; Tang, Y.; Willett, W.C.; Li, Z. Geographical, seasonal and gender differences in folate status among Chinese adults. J. Nutr. 2003, 133, 3630–3635. [Google Scholar] [CrossRef]
- Klerk, M.; Verhoef, P.; Clarke, R.; Blom, H.J.; Kok, F.J.; Schouten, F.G. MTHFR 677C-->T polymorphism and risk of coronary heart disease: A meta-analysis. J. Am. Med. Assoc. 2002, 288, 2023–2031. [Google Scholar] [CrossRef]
- Fan, S.; Yang, B.; Zhi, X.; Wang, Y.; Wei, J.; Zheng, Q.; Sun, G. Interactions of Methylenetetrahydrofolate Reductase C677T Polymorphism with Environmental Factors on Hypertension Susceptibility. Int. J. Environ. Res. Public. Health 2016, 13, 1243. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zeng, X.; Xie, Y.; Wu, H.; Zhao, P. Genetic polymorphisms of methylenetetrahydrofolate reductase C677T and risk of ischemic stroke in a southern Chinese Hakka population. Medicine 2018, 97, e13645. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, D.; Wong, K.S.; Wang, Y. Stroke and stroke care in China: Huge burden, significant workload, and a national priority. Stroke 2011, 42, 3651–3654. [Google Scholar] [CrossRef] [PubMed]
- Goracy, I.; Cyrylowski, L.; Kaczmarczyk, M.; Fabian, A.; Koziarska, D.; Goracy, J.; Ciechanowicz, A. C677T polymorphism of the methylenetetrahydrofolate reductase gene and the risk of ischemic stroke in Polish subjects. J. Appl. Genet. 2009, 50, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Q.; Lu, J.; Sun, H.; Zhang, J.S. Association of methylenetetrahydrofolate reductase (MTHFR) gene polymorphism with ischemic stroke in the E astern Chinese Han population. Genet. Mol. Res. 2015, 14, 4161–4168. [Google Scholar] [CrossRef] [PubMed]
- Alves-Silva, J.M.; Zuzarte, M.; Girão, H.; Salgueiro, L. The Role of Essential Oils and Their Main Compounds in the Management of Cardiovascular Disease Risk Factors. Molecules 2021, 26, 3506. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Carvalho, M.; Fernandes, A.P.; Sabino Ade, P.; Loures-Vale, A.A.; da Fonseca Neto, C.P.; Garcia, J.C.; Saad, J.A.; Sousa, M.O. Homocysteine and methylenetetrahydrofolate reductase in subjects undergoing coronary angiography. Arq. Bras. Cardiol. 2007, 88, 167–172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018; NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2020; pp. 1–8. [Google Scholar]
- Tremblay, A.; Pérusse, L.; Bouchard, C. Energy balance and body-weight stability: Impact of gene-environment interactions. Br. J. Nutr. 2004, 92 (Suppl. 1), S63–S66. [Google Scholar] [CrossRef]
- Lewis, S.J.; Lawlor, D.A.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Ebrahim, S.; Zacho, J.; Ness, A.; Leary, S.; Smith, G.D. The methylenetetrahydrofolate reductase C677T genotype and the risk of obesity in three large population-based cohorts. Eur. J. Endocrinol. 2008, 159, 35–40. [Google Scholar] [CrossRef]
- Wrzosek, M.; Ślusarczyk, K. Methylenetetrahydrofolate Reductase C677T Gene Variant in Relation to Body Mass Index and Folate Concentration in a Polish Population. Biomedicines 2022, 10, 3140. [Google Scholar] [CrossRef]
- Leal-Ugarte, E.; Peralta, V.; Meza-Espinoza, J.P.; Duran, J.; Macias-Gomez, N.; Bocanegra-Alonso, A.; Lara-Ramos, J. Association of the MTHFR 677C>T Polymorphism with Obesity and Biochemical Variables in a Young Population of Mexico. J. Med. Biochem. 2019, 38, 461. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, F.F.; Belini Junior, E.; Okumura, J.V.; Salvarani, M.; Bonini-Domingos, C.R.; Ruiz, M.A. The relationship between of ACE I/D and the MTHFR C677T polymorphisms in the pathophysiology of type 2 diabetes mellitus in a population of Brazilian obese patients. Arch. Endocrinol. Metab. 2018, 62, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Caputo, T.; Gilardi, F.; Desvergne, B. From chronic overnutrition to metaflammation and insulin resistance: Adipose tissue and liver contributions. FEBS Lett. 2017, 591, 3061–3088. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Kato, B.S.; Wilson, S.G.; Spector, T.D. Linkage of genes to total lean body mass in normal women. J. Clin. Endocrinol. Metab. 2007, 92, 3171–3176. [Google Scholar] [CrossRef][Green Version]
- Di Renzo, L.; Rizzo, M.; Iacopino, L.; Sarlo, F.; Domino, E.; Jacoangeli, F.; Colica, C.; Sergi, D.; De Lorenzo, A. Body composition phenotype: Italian Mediterranean Diet and C677T MTHFR gene polymorphism interaction. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2555–2565. [Google Scholar]
- Frelut, M.L.; Nicolas, J.P.; Guilland, J.C.; de Courcy, G.P. Methylenetetrahydrofolate reductase 677 C->T polymorphism: A link between birth weight and insulin resistance in obese adolescents. Int. J. Pediatr. Obes. 2011, 6, e312–e317. [Google Scholar] [CrossRef]
- Ong, K.K. Size at birth, postnatal growth and risk of obesity. Horm. Res. 2006, 65 (Suppl. 3), 65–69. [Google Scholar] [CrossRef]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Huang, T.; Yuan, G.; Zhang, Z.; Zou, Z.; Li, D. Cardiovascular pathogenesis in hyperhomocysteinemia. Asia Pac. J. Clin. Nutr. 2008, 17, 8–16. [Google Scholar] [PubMed]
- Berstad, P.; Konstantinova, S.V.; Refsum, H.; Nurk, E.; Vollset, S.E.; Tell, G.S.; Ueland, P.M.; Drevon, C.A.; Ursin, G. Dietary fat and plasma total homocysteine concentrations in 2 adult age groups: The Hordaland Homocysteine Study. Am. J. Clin. Nutr. 2007, 85, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Kucukhuseyin, O.; Kurnaz, O.; Akadam-Teker, A.B.; Isbir, T.; Bugra, Z.; Ozturk, O.; Yilmaz-Aydogan, H. The association of MTHFR C677T gene variants and lipid profiles or body mass index in patients with diabetic and nondiabetic coronary heart disease. J. Clin. Lab. Anal. 2013, 27, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.S. The epidemic of obesity and diabetes: Trends and treatments. Tex. Heart Inst. J. 2011, 38, 142–144. [Google Scholar] [PubMed]
- Li, Y.; Zhang, H.; Jiang, C.; Xu, M.; Pang, Y.; Feng, J.; Xiang, X.; Kong, W.; Xu, G.; Li, Y.; et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J. Biol. Chem. 2013, 288, 9583–9592. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, C.; Xu, G.; Wang, N.; Zhu, Y.; Tang, C.; Wang, X. Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes 2008, 57, 817–827. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Lunegova, O.; Kerimkulova, A.; Turdakmatov, N.; Sovkhozova, N.; Nabiev, M.; Isakova, Z.; Iusupova, É.; Moldokeeva, C.; Gotfrid, I.; Mirrakhimov, A.; et al. Association of C677T Gene Polymorphism of Methylenetetrahydrofolate Reductase with Insulin Resistance Among Kirghizes. Kardiologiia 2011, 51, 58–62. [Google Scholar]
- Schettini, M.A.S.; Passos, R.F.D.N.; Koike, B.D.V. Shift Work and Metabolic Syndrome Updates: A Systematic Review. Sleep. Sci. 2023, 16, 237–247. [Google Scholar] [CrossRef]
- Kheradmand, M.; Maghbooli, Z.; Salemi, S.; Sanjari, M. Associations of MTHFR C677T polymorphism with insulin resistance, results of NURSE Study (Nursing Unacquainted Related Stress Etiologies). J. Diabetes Metab. Disord. 2017, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Yang, B.; Fan, S.; Li, Y.; He, M.; Wang, D.; Wang, Y.; Wei, J.; Zheng, Q.; Sun, G. Additive interaction of MTHFR C677T and MTRR A66G polymorphisms with being overweight/obesity on the risk of type 2 diabetes. Int. J. Environ. Res. Public Health 2016, 13, 1243. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, P.; Prejbisz, A.; Kuryłowicz, A.; Baska, A.; Burchardt, P.; Chlebus, K.; Dzida, G.; Jankowski, P.; Jaroszewicz, J.; Jaworski, P.; et al. Metabolic syndrome—A new definition and management guidelines: A joint position paper by the Polish Society of Hypertension, Polish Society for the Treatment of Obesity, Polish Lipid Association, Polish Association for Study of Liver, Polish Society of Family Medicine, Polish Society of Lifestyle Medicine, Division of Prevention and Epidemiology Polish Cardiac Society, “Club 30” Polish Cardiac Society, and Division of Metabolic and Bariatric Surgery Society of Polish Surgeons. Arch. Med. Sci. 2022, 18, 1133–1156. [Google Scholar] [PubMed]
- Canale, M.P.; Manca di Villahermosa, S.; Martino, G.; Rovella, V.; Noce, A.; De Lorenzo, A.; Di Daniele, N. Obesity-related metabolic syndrome: Mechanisms of sympathetic overactivity. Int. J. Endocrinol. 2013, 2013, 865965. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.R.; Zhang, H.G.; Wang, Z.P.; Fu, S.J.; Yang, P.Q.; Ren, J.G.; Ning, Y.Y.; Hu, X.J.; Tian, L.H. C-reactive protein, vitamin B12 and C677T polymorphism of N-5,10-methylenetetrahydrofolate reductase gene are related to insulin resistance and risk factors for metabolic syndrome in Chinese population. Clin. Investig. Med. 2010, 33, E290–E297. [Google Scholar] [CrossRef]
- Ellingrod, V.L.; Miller, D.D.; Taylor, S.F.; Moline, J.; Holman, T.; Kerr, J. Metabolic syndrome and insulin resistance in schizophrenia patients receiving antipsychotics genotyped for the methylenetetrahydrofolate reductase (MTHFR) 677C/T and 1298A/C variants. Schizophr. Res. 2008, 98, 47–54. [Google Scholar] [CrossRef]
- Wang, J.; Xu, L.; Xia, H.; Li, Y.; Tang, S. Association of MTHFR C677T gene polymorphism with metabolic syndrome in a Chinese population: A case-control study. J. Int. Med. Res. 2018, 46, 2658–2669. [Google Scholar] [CrossRef]
- Rosano, G.M.; Vitale, C.; Marazzi, G.; Volterrani, M. Menopause and cardiovascular disease: The evidence. Climacteric 2007, 10 (Suppl. 1), 19–24. [Google Scholar] [CrossRef]
- Lee, C.C.; Kasa-Vubu, J.Z.; Supiano, M.A. Androgenicity and obesity are independently associated with insulin sensitivity in postmenopausal women. Metabolism 2004, 53, 507–512. [Google Scholar] [CrossRef]
- Lambrinoudaki, I.; Kaparos, G.; Papadimitriou, D.; Sergentanis, T.N.; Creatsa, M.; Alexandrou, A.; Logothetis, E.; Christodoulakos, G.; Kouskouni, E. Methylenetetrahydrofolate reductase C677T polymorphism is associated with central adiposity and increased androgenicity in healthy postmenopausal women. Eur. J. Endocrinol. 2008, 159, 233–241. [Google Scholar] [CrossRef]
- Fan, S.J.; Yang, B.Y.; Zhi, X.Y.; He, M.; Wang, D.; Wang, Y.X.; Wang, Y.N.; Wei, J.; Zheng, Q.M.; Sun, G.F. Are MTHFR C677T and MTRR A66G Polymorphisms Associated with Overweight/Obesity Risk? From a Case-Control to a Meta-Analysis of 30,327 Subjects. Int. J. Mol. Sci. 2015, 16, 11849–11863. [Google Scholar] [CrossRef] [PubMed]
- Real, J.T.; Martinez-Hervas, S.; Garcia-Garcia, A.B.; Chaves, F.J.; Civera, M.; Ascaso, J.F.; Carmena, R. Association of C677T polymorphism in MTHFR gene, high homocysteine and low HDL cholesterol plasma values in heterozygous familial hypercholesterolemia. J. Atheroscler. Thromb. 2009, 16, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Chen, Q.; Venners, S.A.; Zhong, G.; Hsu, Y.H.; Xing, H.; Wang, X.; Xu, X. Effect of simvastatin on plasma homocysteine levels and its modification by MTHFR C677T polymorphism in Chinese patients with primary hyperlipidemia. Cardiovasc. Ther. 2013, 31, e27–e33. [Google Scholar] [CrossRef] [PubMed]
- Villela, M.P.; Andrade, V.L.; Eccard, B.; Jordao, A.A.; Sertorio, J.T.; Tanus-Santos, J.E.; Silva, L.F.; Silveira, J.N.; Sandrim, V.C. Homocysteine and nitrite levels are modulated by MTHFR 677C>T polymorphism in obese women treated with simvastatin. Clin. Exp. Pharmacol. Physiol. 2014, 41, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Mojtabai, R. Body mass index and serum folate in childbearing age women. Eur. J. Epidemiol. 2004, 19, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, M.; Beydoun, H.; Kancherla, V. Association between body mass index and folate insufficiency indicative of neural tube defects risk among nonpregnant women of childbearing age in the United States, NHANES, 2007–2010. Birth Defects Res. 2020, 112, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.; Moskau, S.; Grigull, A.; Farmand, S.; Klockgether, T.; Smulders, Y.; Blom, H.; Zur, B.; Stoffel-Wagner, B.; Linnebank, M. Plasma folate levels are associated with the lipoprotein profile: A retrospective database analysis. Nutr. J. 2010, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Terruzzi, I.; Senesi, P.; Fermo, I.; Lattuada, G.; Luzi, L. Are genetic variants of the methyl group metabolism enzymes risk factors predisposing to obesity? J. Endocrinol. Invest. 2007, 30, 747–753. [Google Scholar] [CrossRef]
- Raghubeer, S.; Matsha, T.E. Methylenetetrahydrofolate (MTHFR), the One-Carbon Cycle, and Cardiovascular Risks. Nutrients 2021, 13, 4562. [Google Scholar] [CrossRef]
- de Oliveira, R.P.D.; da Silva, E.G.; de Faria Santos, K.; da Silva Santos, R.; da Silva Reis, A.A. The combined effects of GSTM1/GSTT1 and MTHFR C677T polymorphisms on the systemic arterial hypertension susceptibility: A genetic association study in Brazilian diabetic patients. Hum. Gene 2023, 35, 201138. [Google Scholar] [CrossRef]
- Abd-Elmawla, M.A.; Rizk, S.M.; Youssry, I.; Shaheen, A.A. Impact of Genetic Polymorphism of methylenetetrahydrofolate reductase C677T on Development of Hyperhomocysteinemia and Related Oxidative Changes in Egyptian Beta-Thalassemia Major PatiBeta. PLoS ONE 2016, 11, e0155070. [Google Scholar] [CrossRef] [PubMed]
- Pitsavos, C.; Panagiotakos, D.; Trichopoulou, A.; Chrysohoou, C.; Dedoussis, G.; Chloptsios, Y.; Choumerianou, D.; Stefanadis, C. Interaction between Mediterranean diet and methylenetetrahydrofolate reductase C677T mutation on oxidized low density lipoprotein concentrations: The ATTICA study. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 91–99. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; de Oliveira, J.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, L.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid. Med. Cell Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Takemoto, D.J. Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J. Biol. Chem. 2005, 280, 13682–13693. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Tian, D.C.; Li, Z.G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed]
- AnandBabu, K.; Sen, P.; Angayarkanni, N. Oxidized LDL, homocysteine, homocysteine thiolactone and advanced glycation end products act as pro-oxidant metabolites inducing cytokine release, macrophage infiltration and pro-angiogenic effect in ARPE-19 cells. PLoS ONE 2019, 14, e0216899. [Google Scholar] [CrossRef]
- Barathi, S.; Angayarkanni, N.; Pasupathi, A.; Natarajan, S.K.; Pukraj, R.; Dhupper, M.; Velpandian, T.; Muralidharan, C.; Sivashanmugham, M. Homocysteinethiolactone and paraoxonase: Novel markers of diabetic retinopathy. Diabetes Care 2010, 33, 2031–2037. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Moroni, C.; Vignini, A.; Nanetti, L.; Curatola, G. Effect of homocysteinylation of low density lipoproteins on lipid peroxidation of human endothelial cells. J. Cell Biochem. 2004, 92, 351–360. [Google Scholar] [CrossRef]
- McCully, K.S. Chemical pathology of homocysteine. I. Atherogenesis. Ann. Clin. Lab. Sci. 1993, 23, 477–493. [Google Scholar] [PubMed]
- Chwatko, G.; Boers, G.H.; Strauss, K.A.; Shih, D.M.; Jakubowski, H. Mutations in methylenetetrahydrofolate reductase or cystathionine beta-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007, 21, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.C.; Alves, M.C.; Augusto, E.M.; Abdalla, D.S.; Horst, M.A.; Cominetti, C. The MTHFR C677T Polymorphism Is Related to Plasma Concentration of Oxidized Low-Density Lipoprotein in Adolescents with Cardiovascular Risk Factors. J. Nutrigenet. Nutrigenom. 2015, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Mikael, L.G.; Genest, J., Jr.; Rozen, R. Elevated homocysteine reduces apolipoprotein A-I expression in hyperhomocysteinemic mice and in males with coronary artery disease. Circ. Res. 2006, 98, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Yang, X.; Wang, H. Hyperhomocysteinemia and high-density lipoprotein metabolism in cardiovascular disease. Clin. Chem. Lab. Med. 2007, 45, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Rampersaud, G.; Kauwell, G.; Hutson, A.; Cerda, J.; Bailey, L. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 2000, 72, 998–1003. [Google Scholar] [CrossRef]
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Tu, B.P. A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 2017, 66, 180–193.e8. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Marion, D.W.; Cornatzer, W.E.; Duerre, J.A. S-Adenosylmethionine and S-adenosylhomocystein metabolism in isolated rat liver. Effects of L-methionine, L-homocystein, and adenosine. J. Biol. Chem. 1980, 255, 10822–10827. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef]
- Stern, L.L.; Mason, J.B.; Selhub, J.; Choi, S.W. Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol. Biomark. Prev. 2000, 9, 849–853. [Google Scholar]
- Yadav, S.; Longkumer, I.; Joshi, S.; Saraswathy, K.N. Methylenetetrahydrofolate reductase gene polymorphism, global DNA methylation and blood pressure: A population based study from North India. BMC Med. Genom. 2021, 14, 59. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarembska, E.; Ślusarczyk, K.; Wrzosek, M. The Implication of a Polymorphism in the Methylenetetrahydrofolate Reductase Gene in Homocysteine Metabolism and Related Civilisation Diseases. Int. J. Mol. Sci. 2024, 25, 193. https://doi.org/10.3390/ijms25010193
Zarembska E, Ślusarczyk K, Wrzosek M. The Implication of a Polymorphism in the Methylenetetrahydrofolate Reductase Gene in Homocysteine Metabolism and Related Civilisation Diseases. International Journal of Molecular Sciences. 2024; 25(1):193. https://doi.org/10.3390/ijms25010193
Chicago/Turabian StyleZarembska, Emilia, Klaudia Ślusarczyk, and Małgorzata Wrzosek. 2024. "The Implication of a Polymorphism in the Methylenetetrahydrofolate Reductase Gene in Homocysteine Metabolism and Related Civilisation Diseases" International Journal of Molecular Sciences 25, no. 1: 193. https://doi.org/10.3390/ijms25010193
APA StyleZarembska, E., Ślusarczyk, K., & Wrzosek, M. (2024). The Implication of a Polymorphism in the Methylenetetrahydrofolate Reductase Gene in Homocysteine Metabolism and Related Civilisation Diseases. International Journal of Molecular Sciences, 25(1), 193. https://doi.org/10.3390/ijms25010193